Sulfobetaine-terminated PEG improves the qualities of an immunosensing surface†
Yukichi
Horiguchi
a,
Naoki
Nakayama
a,
Naoki
Kanayama
a and
Yukio
Nagasaki
*abc
aGraduate School of Pure and Applied Sciences, University of Tsukuba, Ten-noudai 1-1-1, Tsukuba, Ibaraki 305-8573, Japan
bMaster's School of Medical Sciences, University of Tsukuba, Ten-noudai 1-1-1, Tsukuba, Ibaraki 305-8573, Japan
cSatellite Laboratory of International Center for Materials Nanoarchitectonics (WPI-MANA), National Institute of Materials Science (NIMS), Ten-noudai 1-1-1, Tsukuba, Ibaraki 305-8573, Japan. E-mail: yukio@nagalabo.jp; Fax: +81-29-853-5749; Tel: +81-29-853-5749
First published on 8th January 2014
Abstract
Poly(ethylene glycol) (PEG) possessing a sulfobetaine (SB) moiety at one end and a pentaethylenehexamine (N6) at the other end (SB–PEG–N6) was newly synthesized as a blocking agent for immunosensing surfaces. The N6 moiety strongly coordinates on gold surfaces, facilitating the tethering of the PEG chain to the sensor chip surface, and leaves the SB moiety free. Non-specific adsorption of bovine serum albumin (BSA) was analyzed on the SB–PEG–N6 tethered surface and compared with the methoxy-PEG–N6 (M-PEG–N6) tethered surface using a surface plasmon resonance (SPR) sensor. Non-specific BSA adsorption decreased with decreasing PEG chain length on the SB–PEG tethered chain surface. Non-specific adsorption of BSA decreased as ionic strength increased on SB–PEG–N6 surfaces; this phenomenon was completely opposite to that observed with an M-PEG–N6 tethered chain surface. The results show that SB moieties located close to the gold surface perform well with regard to protein rejection. Actually, low-molecular weight alkane thiol SB (SB-SH) showed minimum BSA adsorption. To evaluate protein recognition efficacy on a PEGylated surface, an antibody (IgG) immobilized surface was then constructed on a gold sensor chip using SB–PEG–N6 as the blocking agent. The specific protein recognition efficacy of SB–PEG–N6/IgG co-immobilized surfaces was higher than that obtained using SB-SH/IgG co-immobilized surfaces. We conclude that SB-terminated PEG exhibits the optimal qualities of a blocking agent, as it possesses both high suppression efficacy of nonspecific protein adsorption and specific protein recognition ability.
Introduction
A functionalization of the surface to improve specific protein recognition efficacy is a desirable property of immunosensors. Suppression of nonspecific protein adsorption greatly improves immunosensing system functionality, because it reduces the background noise derived from various nonspecific proteins in biological fluids. As such, bovine serum albumin and gelatin are used as blocking agents to reduce the nonspecific background noise. Artificial polymers such as poly(2-methoxyethyl acrylate) (PMEA) and 2-methacryloyloxyethyl phosphorylcholine (MPC) polymers are also reported to be surface modifiers.1,2 PEG is able to suppress nonspecific protein adsorption when densely packed PEG layers are tethered to the surface of the sensor. The suppression effect is mainly derived from PEG's hydrophilicity, and the excluded volume effect due to the brush structure of PEG at the surface.3–5 Optimization of surface PEGylation can be achieved simply by changing the chain length, density, and terminal groups.6 For example, the excluded volume effect increases proportionally with PEG chain length. The highly non-fouling character of PEGylated surfaces can be obtained by optimizing the chain lengths and density on the surfaces.7 It is also reported that combinations of different length PEG chains tethered to the surface confer highly non-fouling characteristics.7,8 Furthermore, the biomolecular ligand–receptor binding efficacy of PEGylated surfaces is increased when PEGs and ligands are co-immobilized at high density on the surface.9 These observations underscore the utility of PEGylation techniques for enhancing the performance of sensors. Despite such improvements, there is still some background noise in the case of practical analysis using versatile real samples. Thus, in the cases where only trace amounts of the target sample are present, a completely non-fouling surface is demanded in order to differentiate signal from noise.Zwitterions, which are molecules containing both cationic and anionic properties, are widely employed as surface modifiers to improve the sensing performance of surfaces.10–12 Recently, zwitterion species such as phosphobetaine, sulfobetaine and carboxybetaine were shown to have an extremely non-protein fouling character when immobilized on a substrate surface.13–18 The characteristics of betaine that impart a non-fouling nature to surfaces are chemically distinct from those associated with PEG tethered surfaces. In the latter case, this is mainly governed by entropical factors associated with the PEG chain. Thus, a combination of both PEG-tethered chains and zwitterions on the same surface may enhance both non-protein fouling characteristics and specific protein recognition performance of sensors.
In this report, we designed a new heterobifunctional PEG, which possesses an SB at one end and an N6 moiety at the other chain end. This SB–PEG–N6 was used to modify a gold sensor chip. The N6 group at the end of the PEG chain strongly coordinates with the gold surface, which facilitates modification of the chip, and leaves an SB moiety free at the distal end. Here, we characterized the non-specific interaction of BSA at the SB–PEG tethered chain surface, and investigated the performance of an antibody/SB–PEG–N6 hybrid surface.
Materials and methods
Materials and devices
Tetrahydrofuran (THF) and methanesulfonyl chloride were purchased from Kanto Chemical Co., Ltd, Tokyo, Japan, and 2-(dimethylamino)ethanol from Wako Pure Chemical Industries, Ltd, Osaka, Japan. These chemicals were distilled by conventional methods. 1,3-Propanesultone, 2-propanol, benzene and acetone were purchased from Wako Pure Chemical Industries, Ltd, Japan and pentaethylenehexamine was purchased from Tokyo Chemical Industry Co., Ltd, Tokyo, Japan. N-(11-Mercaptoundecyl)-N,N-dimethyl-3-ammonio-1-propanesulfonate (sulfobetaine-3-undecanethiol, SB-SH) was purchased from Dojindo Laboratories, Kumamoto, Japan. These compounds were used as received. Potassium naphthalene was prepared as described previously.19 Recombinant C-reactive protein (CRP) (Oriental Yeast Co., Ltd, Tokyo, Japan) and anti-CRP (H-90) human from rabbit (Santa Cruz Biotechnology, Inc., TX, USA) were used in protein recognition experiments. Gel permeation chromatography (GPC) measurements were carried out using a TOSOH HLC-8120 equipped with a gel column (Superdex 75 10/300 GL) to evaluate the molecular weight. The 1H and 13C NMR spectra were measured with a JEOL EX400 spectrometer (JEOL Ltd, Japan) at 400 MHz and 100 MHz, respectively. MALDI-TOF MS spectra were measured with an ultrafleXtreme™ (Bruker Corporation, MA, USA). SPR measurements were carried out using BIAcore 1000 and BIAcore 3000 (GE Healthcare, Little Chalfont, UK).Synthesis of poly(ethylene glycol) possessing pentaethylenehexamine and sulfobetaine moieties at either end
A detailed procedure of SB–PEG–N6 synthesis is described in Scheme 1. One mmol of 2-(N,N-dimethylamino)ethanol was added to 30 mL of THF under a nitrogen atmosphere at room temperature (RT). Then 1 mmol of potassium naphthalene (in THF, 0.35 mol L−1) was added into the solution and stirred for 10 min. 110 mmol of ethylene oxide was added to the solution using a cooled syringe. The solution was stirred for 48 h under a nitrogen atmosphere at RT and then poured into cold 2-propanol to precipitate the polymer. Precipitates were next dissolved in THF, and the precipitation procedure was repeated twice to obtain a pure compound. The remaining 2-propanol was completely removed by evaporation. The residue (DMA–PEG–OH) was dissolved in benzene and lyophilized, with a corresponding yield of 80%. | ||
| Scheme 1 Synthesis of SB–PEG–N6. | ||
0.2 mmol of synthesized DMA–PEG–OH was mixed with 30 mL of dehydrated acetone solution containing 1,3-propanesultone (10 times molar equivalent of DMA–PEG–OH), then stirred for 2 h under a nitrogen atmosphere at RT. Acetone was removed by evaporation after the reaction, and the product was dissolved in methanol and precipitated using 1 L of cold 2-propanol. The precipitate was dissolved in methanol and the procedure was repeated 3 times for purification. We completely removed 2-propanol by using evaporation. The residue (SB–PEG–OH) was dissolved in benzene and lyophilized, with a corresponding yield of 95%.
The preparation of methanesulfonyl-terminated PEG–SB (SB–PEG–MS) was carried out using SB–PEG–OH prepared above according to our previously described method, in which methane sulfonylation of MeO–PEG–OH was performed.20 SB–PEG–OH (0.06 mmol), 30 mL of THF, 110 μL of triethylamine and 80 μL of methanesulfonyl chloride were mixed and stirred for 24 h under a nitrogen atmosphere at RT. The resultant salt was removed by suction filtration, and the filtrate was poured into diethyl ether to precipitate the polymer, which was then dissolved in THF. This precipitation procedure was repeated twice to obtain a pure product. After vacuum filtration, the product (SB–PEG–MS) was dissolved in benzene and lyophilized, with a corresponding yield of 93%.
0.05 mmol of SB–PEG–MS was dissolved in 15 mL of THF. The SB–PEG–MS solution was added slowly to 5 mL of pentaethylenehexamine solution (1 M in THF) and reacted for 72 h under a nitrogen atmosphere at RT, followed by pouring into cold 2-propanol to precipitate the polymer. The precipitates were then dissolved in THF, and the precipitation procedure was repeated 3 times to obtain a pure product. Finally, the product (SB–PEG–N6) was lyophilized with benzene, with a corresponding yield of 90%. The low molecular weight SB–PEG–N6 (molecular mass is 2.1 kDa) was also synthesized in a similar manner. Two kinds of methoxy-terminated PEG–N6 molecules (M-PEG–N6 (2 kDa, 4.4 kDa)) were prepared for control experiments.
Evaluation of the PEG immobilized gold surface using SPR
The SPR sensor chip consisting of a plain gold surface was washed with piranha solution before modification of the surface. For PEG modification of the gold surface, the sensor chip was placed in an SPR instrument (BIAcore 1000 or BIAcore 3000). 50 mM PBS (100 mM NaCl, pH 9.0) was used as running buffer. The flow rate was 10 μL min−1. SB–PEG–N6, M-PEG–N6 or SB-SH solution (0.02–0.5 mM) was flowed for 30 min and excess blocking agents were washed with 2 M NaCl flow (30 s, 4 times). The concentration of NaCl in running buffer was varied within the range 10–1000 mM. Nonspecific protein adsorption on the sensor chip surface was evaluated using BSA. For 30 min, 1 mg mL−1 of BSA was applied, after which the adsorbed BSA was evaluated using SPR.Evaluation of protein adsorption by PEG/antibody co-immobilized gold surface using SPR
The SPR sensor chip with a plain gold surface was washed with piranha solution before modification of the surface. For PEG modification of the gold surface, the sensor chip was placed in an SPR instrument (BIAcore 1000 or BIAcore 3000). 50 mM PBS (150 mM NaCl, pH 7.4) solution was used as running buffer and the flow rate was 5 μL min−1. 0.02 μM of anti-CRP was applied for 3 min, and 200 μM of a blocking agent (SB–PEG–N6 (4.4 kDa) or SB-SH) was flowed for 30 min. The prepared co-immobilized surface was stored for 60 min in the presence of flowing running buffer in order to stabilize the response. One micromolar of CRP containing buffer was flowed for 30 min. The amount of CRP binding was evaluated by measurement of the SPR angle shift.Results and discussion
Synthesis of SB–PEG–N6
Synthesis of SB–PEG–N6 was carried out according to the reaction in Scheme 1. The SB–PEG–N6 obtained was evaluated by 1H-NMR, 13C-NMR, GPC and MALDI-TOF MS after purification. Fig. 1a shows the 1H-NMR spectrum of the purified SB–PEG–N6 in CDCl3. The peaks observed at 3.9, 3.1, 2.9, and 2.2 ppm were attributed to protons in the SB moiety. Peaks attributable to the SB moiety were also observed in the 13C-NMR spectrum (64, 63.3, 62, 51, 47.6, and 19 ppm, Fig. 1b). The peaks attributable to CH2 in the N6 moiety were observed at 2.75–2.83 ppm and at 53, 48.5–49, 45.6 ppm in the 1H- and 13C-NMR spectra, respectively. The peaks of the MS moiety of SB–PEG–MS (4.29–4.32 ppm, and 3.01 ppm as an intermediate of SB–PEG–N6, shown in Fig. S3†) almost disappeared from the spectrum (inset of Fig. 1a) because of the conversion of SB–PEG–MS to SB–PEG–N6 (≈ 98%). The molecular weight of the polymer was determined by GPC analysis (Fig. 2). The number average molecular weight, weight-average molecular weight, and polydispersity index were 4.4 kDa, 4.6 kDa and 1.05 respectively. Fig. 3 shows the mass spectrum of SB–PEG–N6 derived from MALDI-TOF, using 2,5-dihydroxybenzoic acid (DHB) as the matrix. Repeated peaks of SB–PEG–N6 were clearly observed at 44.08 intervals, which is the same as the ethylene oxide unit (CH2CH2O). The molecular mass of the SB and N6 moieties are 195.27 and 232.37, respectively. The total amounts of each peak agree well with the following equation:| 195.27 + 44.08(n + 1) + 232.37 |
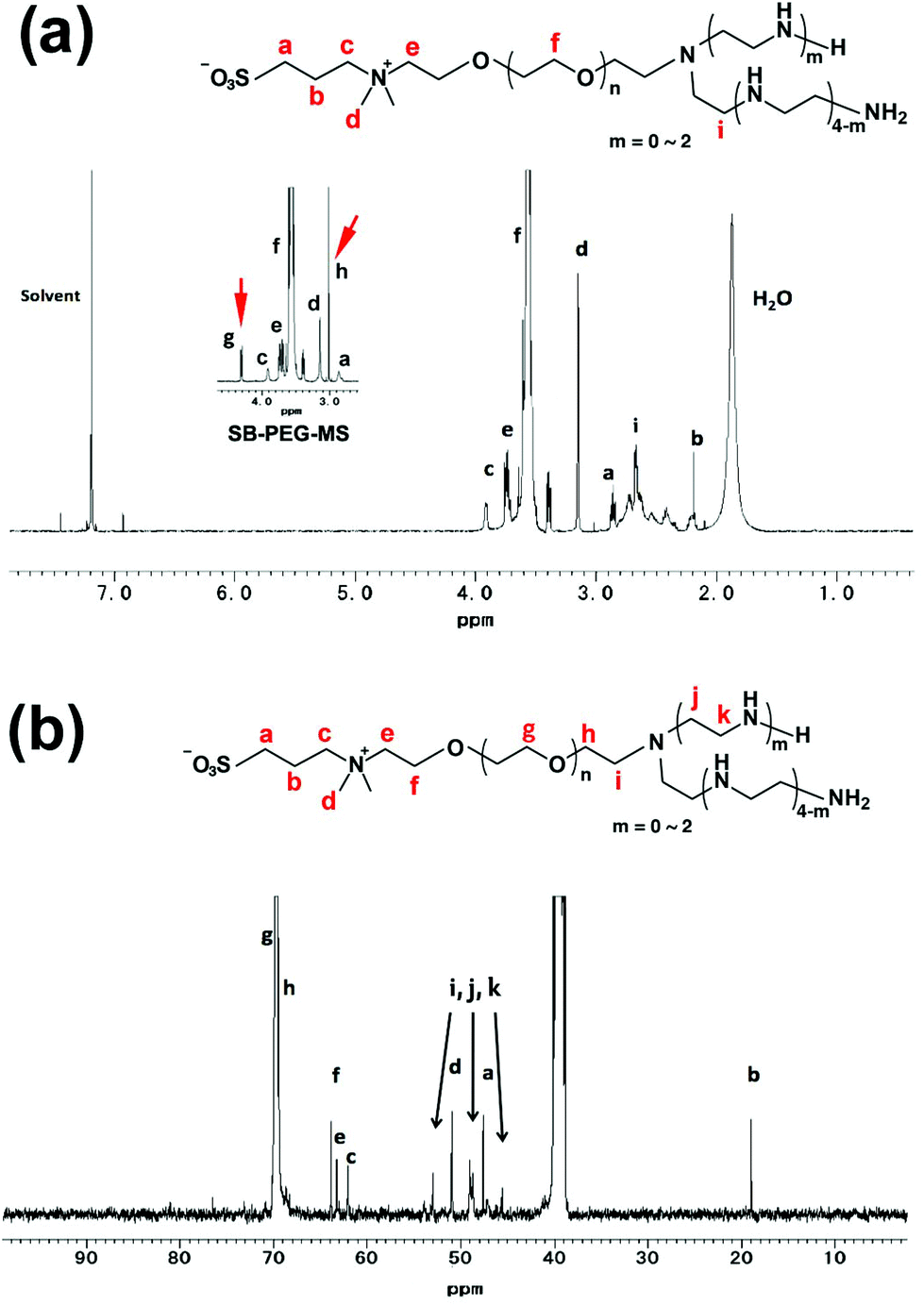 | ||
| Fig. 1 (a) The 1H-NMR spectrum of SB–PEG–N6 (400 MHz, 10 mg mL−1, CDCl3, RT). The inset figure is the 1H-NMR spectrum of SB–PEG–MS. Peaks g and h are attributed to the MS moiety of SB–PEG–MS (4.32 and 3.00 ppm). The complete 1H-NMR spectrum of SB–PEG–MS was shown in Fig. S3a.† (b) The 13C-NMR spectrum of SB–PEG–N6 (100 MHz, 100 mg mL−1, DMSO, RT). Peaks with arrows attributable to CH2 in the N6 moiety were observed at 53, 48.5–49 and 45.6 ppm. | ||
 | ||
| Fig. 2 The evaluation of molecular weight synthesized SB–PEG–N6 by GPC elution time. The number average molecular weight, weight-average molecular weight and its polydispersity index were 4.4 kDa, 4.6 kDa and 1.05, respectively. | ||
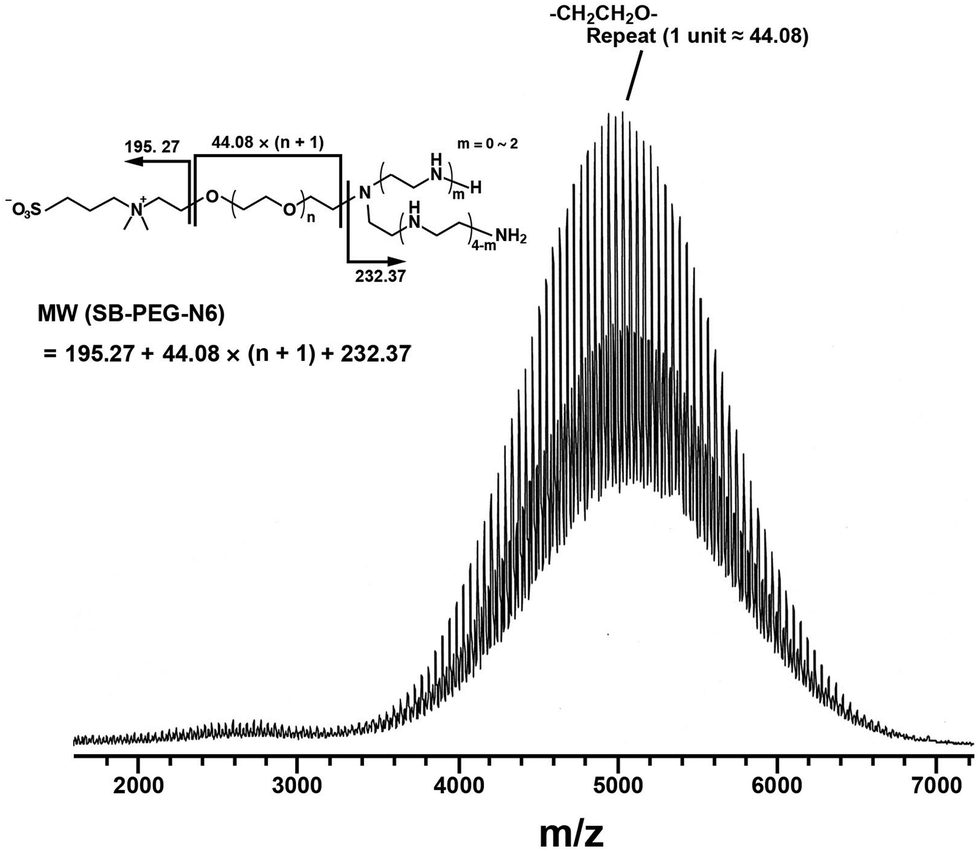 | ||
| Fig. 3 MALDI-TOF MS spectrum of SB–PEG–N6. Repeated peaks of SB–PEG–N6 due to different numbers of ethylene oxide units (–CH2CH2O–) were observed. The average of the oxide unit is 44.08. The molecular masses of an SB moiety and an N6 moiety are 195.27 and 232.37, respectively. The mass spectrum peaks are almost identical to the sum of an SB moiety, an N6 moiety, and multiple ethylene oxide units. The calculated peaks and the measured peaks of SB–PEG–N6 are shown in Table 1. | ||
This indicates that the polymer possesses one SB and one N6 moiety in each molecule. For example, the mass of the largest peak in the figure is 5055.96, which is consistent with n = 104 and an error of 0.08 (Table 1). The average molecular weight and its polydispersity index determined from this mass spectrum were 4.4 kDa and <1.05, respectively (the mass spectra of PEG standards and its standard curve are shown in Fig. S4 and S5†). The calculated molecular mass from the mass spectra agreed well with that from the GPC. These data clearly indicate that SB–PEG–N6 with a molecular mass of 4.4 kDa was obtained quantitatively. The data from a similar quantitative analysis of other synthesized polymers are listed in Table 2.
| n | Molecular mass | n | Molecular mass | ||
|---|---|---|---|---|---|
| Calculated peak | Observed peak | Calculated peak | Observed peak | ||
| 86 | 4262.60 | 4262.36 | 99 | 4835.64 | 4835.48 |
| 87 | 4306.68 | 4306.63 | 100 | 4879.72 | 4879.59 |
| 88 | 4350.76 | 4350.82 | 101 | 4923.80 | 4923.73 |
| 89 | 4394.84 | 4394.64 | 102 | 4967.88 | 4967.75 |
| 90 | 4438.92 | 4438.37 | 103 | 5011.96 | 5011.86 |
| 91 | 4483.00 | 4482.92 | 104 | 5056.04 | 5055.96 |
| 92 | 4527.08 | 4526.90 | 105 | 5100.12 | 5100.05 |
| 93 | 4571.16 | 4570.96 | 106 | 5144.20 | 5144.04 |
| 94 | 4615.24 | 4615.05 | 107 | 5188.28 | 5188.16 |
| 95 | 4659.32 | 4659.12 | 108 | 5232.36 | 5232.27 |
| 96 | 4703.40 | 4703.21 | 109 | 5276.44 | 5276.53 |
| 97 | 4747.48 | 4747.29 | 110 | 5320.52 | 5320.47 |
| 98 | 4791.56 | 4791.44 | 111 | 5364.60 | 5364.53 |
| M n (kDa) | M w (kDa) | PDI | |
|---|---|---|---|
| SB–PEG–N6 (4.4k) | 4.4 | 4.6 | 1.05 |
| SB–PEG–N6 (2.1k) | 2.1 | 2.2 | 1.04 |
| M-PEG–N6 (4.4k) | 4.4 | 4.6 | 1.05 |
| M-PEG–N6 (2.0k) | 2.0 | 2.1 | 1.05 |
Construction of PEG-immobilized gold surfaces
We have reported previously that a terminal pentaethylenehexamine group favors strong immobilization of PEG chains on bare gold surfaces under alkaline conditions.21 Coordination of an amino lone pair on the gold surface plays an important role. Thus, the SB–PEG–N6 we synthesized is suitable for construction of tethered chains on the gold surface of the sensor. In order to determine the optimal conditions for modification, we examined the performance of chips modified under various conditions. Fig. 4a shows the SPR sensorgrams of chips modified with SB–PEG–N6 formed using different PEG concentrations. The amount of PEG was increased to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μg mL−1 and the SPR signal response reached a plateau at 1000 μg mL−1 (Fig. 4b). Complete removal of excess immobilized PEG was confirmed by 2 M of NaCl solution. The SB–PEG–N6 chains were strongly adsorbed onto the gold surface by coordination of the lone pair of the amino group.22,23 Although a thiol-terminated PEG is commonly used to form gold–sulfur bonds on gold surfaces, such linkages can be dissociated by oxidation of the thiol group.24–26 Additionally, thiol-terminated compounds on gold surfaces can also be exchanged by other thiol-containing compounds.27 By contrast, the modification of gold surfaces by N6-terminated PEG is extremely strong due to the presence of multivalent gold–nitrogen bonds.21
000 μg mL−1 and the SPR signal response reached a plateau at 1000 μg mL−1 (Fig. 4b). Complete removal of excess immobilized PEG was confirmed by 2 M of NaCl solution. The SB–PEG–N6 chains were strongly adsorbed onto the gold surface by coordination of the lone pair of the amino group.22,23 Although a thiol-terminated PEG is commonly used to form gold–sulfur bonds on gold surfaces, such linkages can be dissociated by oxidation of the thiol group.24–26 Additionally, thiol-terminated compounds on gold surfaces can also be exchanged by other thiol-containing compounds.27 By contrast, the modification of gold surfaces by N6-terminated PEG is extremely strong due to the presence of multivalent gold–nitrogen bonds.21
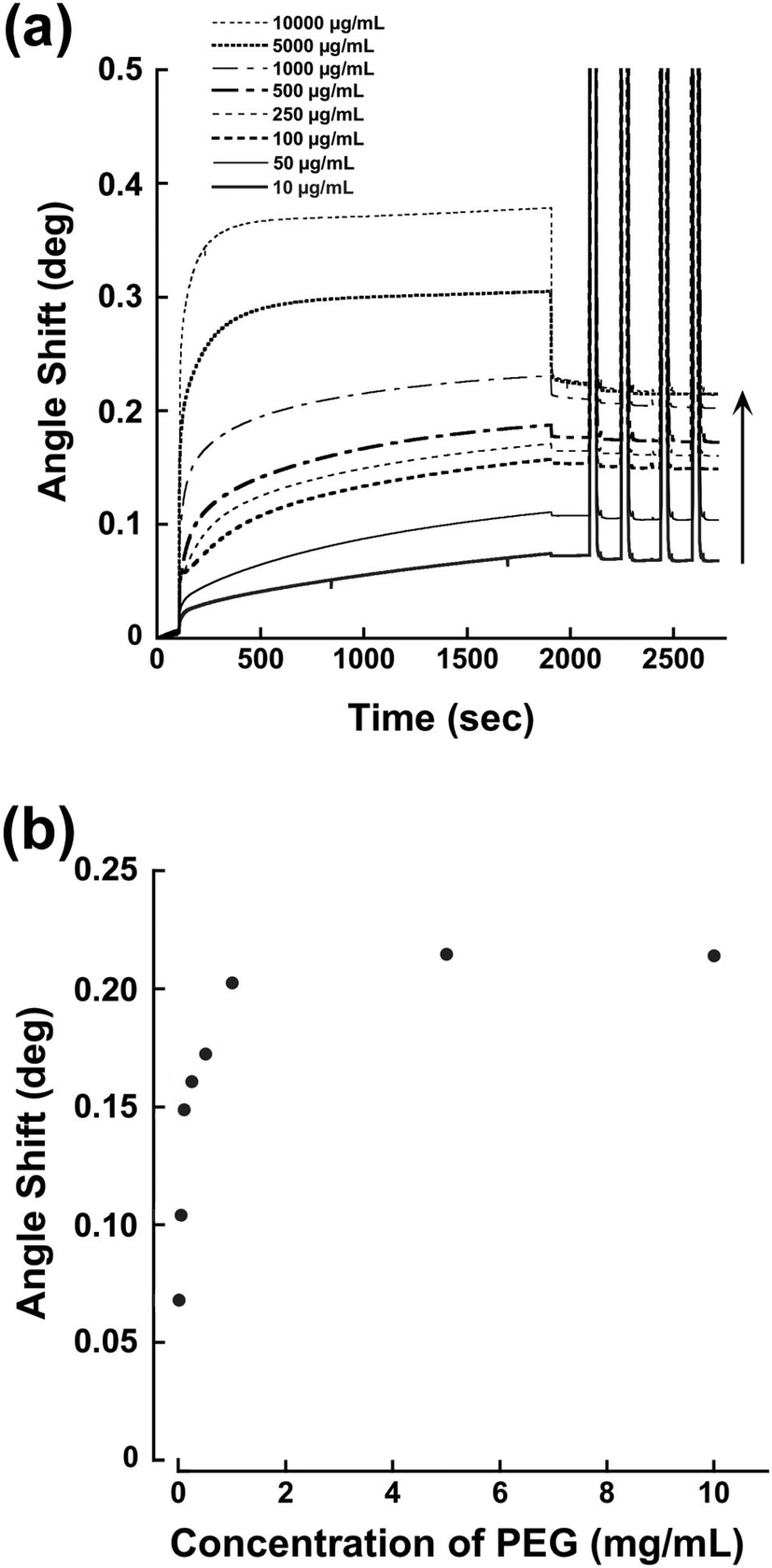 | ||
| Fig. 4 (a) Immobilization of different concentrations of SB–PEG–N6 on gold surfaces of SPR sensor chips. After the immobilization, excess SB–PEG–N6 was removed by 2 M NaCl solution flow (30 s, 4 times). (b) Scatter plot converted from (a) between the PEG concentration and the angle shift after the immobilization processes. | ||
Non-specific adsorption of BSA on SB–PEG–N6 immobilized surface
The suppression effects of BSA adsorption on SB–PEG–N6 immobilized surfaces were evaluated by SPR measurements. Fig. 5 shows the amount of nonspecific adsorption of BSA on gold sensor chips modified by SB–PEG–N6 (4.4 kDa), SB–PEG–N6 (2.1 kDa), M-PEG–N6 (4.4 kDa), M-PEG–N6 (2.0 kDa) or SB-SH as a function of the coating concentration (which is proportional to surface PEG density8). These molecules were immobilized under pH = 9.0 because alkaline conditions are suitable for M-PEG–N6 and SB–PEG–N6 to immobilize onto the gold surface via gold–nitrogen bonds.28 When the concentrations of blocking agents were increased during modification, the amount of adsorbed BSA decreased. This shows that effective modification of the substrate surface by PEG homologs prevents nonspecific BSA adsorption. In the case of M-PEG–N6, longer PEG chains had a greater suppressive effect on BSA adsorption by the immobilized surface, although the PEG chain density was not fully optimized under the present conditions. As mentioned above, the excluded volume effect depends on PEG chain length, meaning that longer rather than shorter PEG chains are better. On the other hand, the suppression effect of SB–PEG–N6 (2.1 kDa) was higher than that of SB–PEG–N6 (4.4 kDa) in the entire range of the present experiments. This may be due to the density of SB on the surfaces. The chain density of shorter PEG is higher than that of longer PEG, because the excluded volume effect is different.8 As a result, the densely packed SB formed on surfaces when shorter SB–PEG–N6 is used may lead to a high blocking effect, but not a strong exclusive volume effect of the PEG chain. To confirm this phenomenon, the suppression effect of commercially available SB-SH was also evaluated, which is known to give a much higher density on the substrate surface.29 The SB-SH modified surface resulted in a much lower non-specific adsorption of BSA when compared with the other samples, indicating that a high amount of SB on the surface suppresses the adsorption of BSA. It is reported that the density of alkanethiol chains on the gold surface is 4.5 chains per nm2,29 and the maximum density of PEG 5k is 0.44 chain per nm2.8 Because the spatial distribution of PEG chains exists on the surface, the chain density is one-tenth compared with alkanethiol. Thus, unwanted adsorption was greatly suppressed by a densely packed zwitterion layer on the surfaces. We note that these effects were concentration-dependent indicating that the results are reliable. | ||
| Fig. 5 The amount of non-specific adsorption of BSA on blocking agent modified sensor chips. The modification time was 30 min at each concentration of blocking agents (10 μL min−1). SB–PEG–N6 (4.4 kDa, filled squares), SB–PEG–N6 (2.1 kDa, filled circles), M-PEG–N6 (4.4 kDa, open squares), M-PEG–N6 (2.0 kDa, open circles) and SB-SH (× marks) were applied. | ||
To obtain further information about the properties of synthesized SB–PEG–N6, the BSA adsorption measurements were taken in the presence of different NaCl concentrations. As the NaCl concentration increased, the amount of adsorbed BSA on the M-PEG–N6 surface tended to increase. In contrast, the amount of adsorbed BSA on the SB–PEG–N6 surface decreased with increasing NaCl concentration (Fig. 6a). It is reported that the thickness of the PEG layer decreases with increasing salt concentration in aqueous media;30 thus, the PEG brush shrinks as the NaCl concentration is increased. This may explain the increase in nonspecific protein adsorption on the M-PEG–N6 modified surface under a high salt environment, since the entropically repulsive force of PEG chains on the surface is reduced under these conditions. It is rather surprising that the non-specific adsorption of BSA decreased with increasing NaCl concentration, despite the fact that SB–PEG–N6-immobilized surfaces were prepared under the same conditions. Note that the total SB amount on the sensor chip surface was constant regardless of the salt conditions, indicating that this phenomenon might be influenced by the distribution of zwitterions within a few nanometers from the surface. When PEG chains shrink in the presence of high NaCl concentrations, SBs on the surface become closer to the surface, and also closer to each other. This may explain the decrease in non-specific BSA adsorption.
 | ||
| Fig. 6 The suppressive effect of SB–PEG–N6 (filled circles) and M-PEG–N6 (filled squares) on the BSA adsorption of immobilized gold surfaces at various NaCl concentrations. (a) The amount of unwanted adhesion after BSA solution flow (10 μL min−1, 30 min, *p = 0.017). (b) The amount of unwanted adhesion of FBS solution flow (10 μL min−1, 30 min, *p = 0.031). | ||
The same experiments in fetal bovine serum (FBS) were also evaluated under each NaCl concentration (Fig. 6b), and the nonspecific adsorption of FBS was effectively suppressed compared with M-PEG–N6 immobilized surfaces.
Specific protein recognition efficacy of SB–PEG–N6/antibody co-immobilized surfaces
As shown above, SB-terminated PEG immobilized surfaces decrease the unwanted irreversible adsorption of proteins on the gold surfaces compared with the methoxy-terminated PEG. However, the direct immobilization of SB-SH on the gold surface showed the most effective performance in terms of protein rejection. To explore sensing ability, a hybrid surface of antibody and modification agents was constructed on the gold sensor chip. Fig. 7a shows SPR sensorgrams for the construction of SB–PEG–N6/anti-CRP IgG (SB–PEG–N6/IgG) and SB-SH/anti-CRP IgG (SB-SH/IgG) co-immobilized surfaces. To construct the hybrid surfaces, 0.02 μM of the anti-CRP solution was applied to the surfaces, and the remaining spaces on the gold surfaces were filled by SB–PEG–N6 or SB-SH. After the preparation of co-immobilized surfaces, protein recognition efficacy was evaluated by injection of 1 μM of CRP.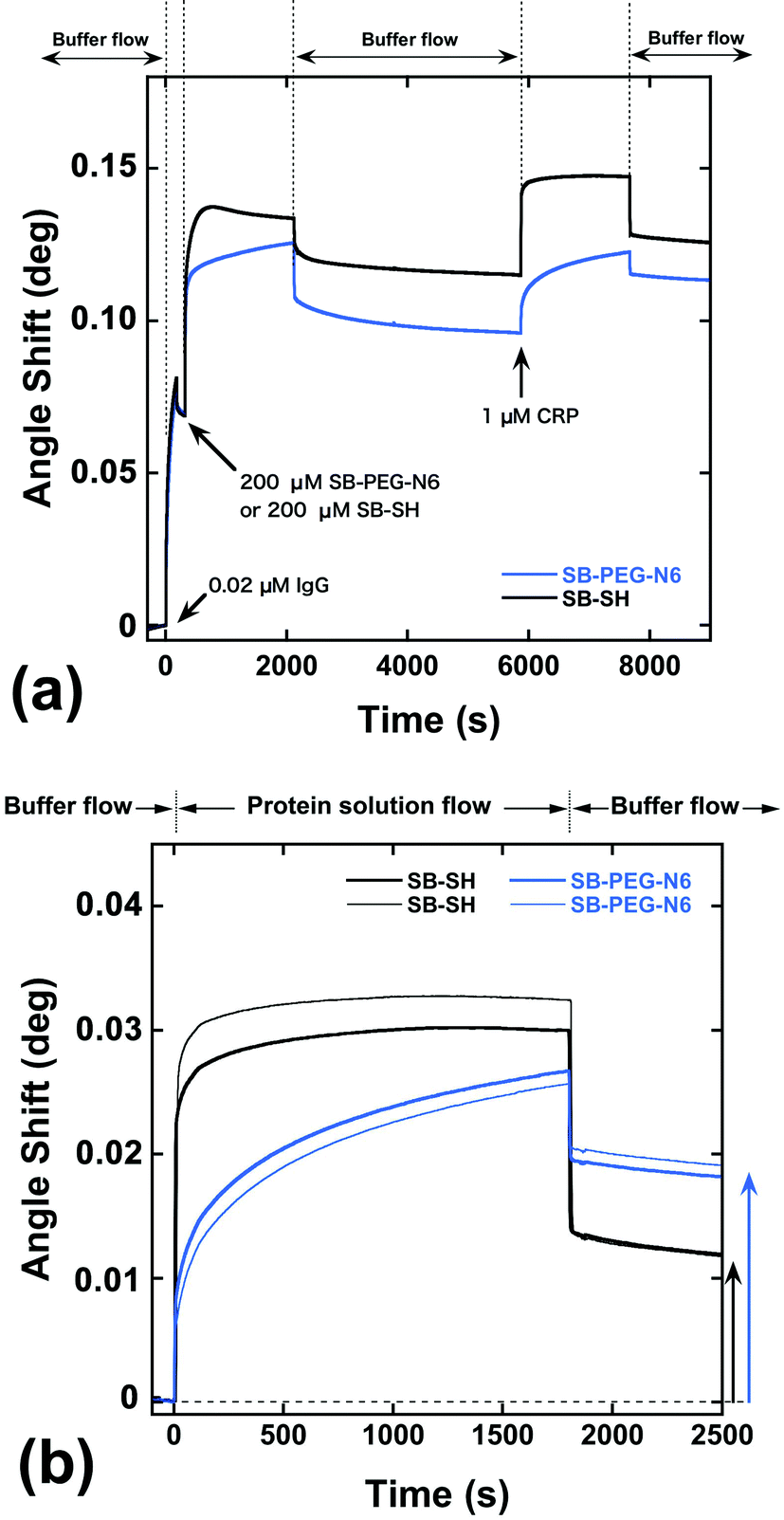 | ||
| Fig. 7 (a) SPR sensorgrams of the construction of SB–PEG–N6/IgG (blue line) and SB-SH/IgG co-immobilized gold surfaces. The first changes in the SPR angle shift were caused by the injection of 0.02 μM anti-CRP IgG. Then, 200 μM of SB–PEG–N6 or SB-SH containing buffer were applied to the surfaces. After the preparation of co-immobilized surfaces, 1 μM of CRP was added. (b) SPR sensorgrams monitoring the CRP adsorption process. The baselines were fixed after the SB–PEG–N6/IgG or SB-SH/IgG co-immobilized surfaces were prepared. One micromolar buffer containing CRP was applied to the surfaces for 0–1800 s. The angle shift derived from absorbed protein was calculated after exchange buffer containing CRP to running buffer (arrows). | ||
Fig. 7b shows the SPR sensorgrams focused on the CRP adsorption process on hybrid surfaces, or more specifically, the SB–PEG–N6/IgG or SB-SH/IgG co-immobilized sensor chip surfaces. After 30 min of 1 μM CRP flow, the angle shift by adsorption of CRP on SB-SH/IgG co-immobilized surfaces was 0.0118 ± 0.0001°, while the angle shift by adsorption of CRP on SB–PEG–N6/IgG co-immobilized surfaces was 0.0186 ± 0.0005°. Note that the antibody immobilization processes in these experiments were identical (Fig. 7a). Alternatively, we have confirmed that the immobilized antibodies thus treated were strongly immobilized and were not removed by SB-SH or protein treatment during the modification process (see Fig. S6–S8†). Although SB-SH modified surfaces can highly suppress nonspecific adsorption, the CRP adsorption signal is lower than for SB–PEG–N6 modified surfaces. It is reported that the PEG chain not only possesses blocking properties, but also assists molecular recognition efficacy of co-immobilized biomolecules such as antibodies and oligonucleotides.9,31 In the case of SB–PEG–N6/IgG, the CRP recognition efficacy of IgG surrounded by a PEG layer might be increased because the orientation of IgG is controlled by the PEG layer itself. As a result, the CRP recognition efficacy of the SB–PEG–N6/IgG surface is higher than that of SB-SH/IgG co-immobilized surfaces. Collectively, the data regarding SB–PEG–N6 show that the combination of PEG and SB is effective as a blocking agent for the construction of high-performance surfaces for immunosensing.
Conclusions
Material design to improve the biocompatibility of surfaces is a critical factor in immunosensor development, because the suppression of unwanted surface protein adsorption is required for the construction of high-performance sensor surfaces. The suppression of non-specific protein adsorption on the M-PEG–N6 immobilized surface tended to be lower with increasing NaCl concentration because the PEG layer thickness decreases with increasing salt concentration in aqueous media. On the other hand, the suppressive effect of SB–PEG–N6 on nonspecific protein adsorption on the sensor surface tended to increase with increasing NaCl concentration. This increase is due to the distribution of SB on the surface, as the thickness of the PEG layer decreases. The SB moieties of SB–PEG–N6 are close to the surface and lead to the formation of the dense SB layer. This may in turn lead to an increased suppressive effect on non-specific protein adsorption.From the perspective of specific protein recognition efficacy, an SB–PEG–N6/IgG co-immobilized surface is better than SB-SH/IgG co-immobilized surfaces, because the orientation of IgG appears to be influenced by the PEG layer. This indicates that PEG not only has blocking properties, but also modulates the characteristics of biomolecules. Thus, SB-terminated PEG is a high-performance blocking agent that possesses the advantages of PEG and SB in a single molecule.
Acknowledgements
This research was supported partly by a Grant-in-Aid for Scientific Research on Innovative Areas (Soft Interface # 20106011) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.References
- K. Ishihara, R. Aragaki, T. Ueda, A. Watenabe and N. Nakabayashi, J. Biomed. Mater. Res., 1990, 24, 1069–1077 CrossRef CAS PubMed.
- M. Tanaka, T. Motomura, M. Kawada, T. Anzai, K. Yuu, T. Shiroya, K. Shimura, M. Onishi and M. Akira, Biomaterials, 2000, 21, 1471–1481 CrossRef CAS.
- N. D. Winblade, H. Schmökel, M. Baumann, A. S. Hoffman and J. A. Hubbell, J. Biomed. Mater. Res., 2001, 59, 618–631 CrossRef PubMed.
- S. Nagaoka and A. Nakao, Biomaterials, 1990, 11, 119–121 CrossRef CAS.
- G. Tamura, Y. Shinohara, A. Tamura, Y. Sanada, M. Oishi, I. Akiba, Y. Nagasaki, K. Sakurai and Y. Amemiya, Polym. J., 2012, 44, 240–244 CrossRef CAS.
- D. Miyamoto, M. Oishi, K. Kojima, K. Yoshimoto and Y. Nagasaki, Langmuir, 2008, 24, 5010–5017 CrossRef CAS PubMed.
- K. Yoshimoto, T. Hirase, J. Madsen, S. P. Armes and Y. Nagasaki, Macromol. Rapid Commun., 2009, 30, 2136–2140 CrossRef CAS PubMed.
- K. Uchida, H. Otsuka, M. Kaneko, K. Kataoka and Y. Nagasaki, Anal. Chem., 2005, 77, 1075–1080 CrossRef CAS.
- K. Yoshimoto, M. Nishio, H. Sugasawa and Y. Nagasaki, J. Am. Chem. Soc., 2010, 132, 7982–7989 CrossRef CAS PubMed.
- H. Takahashi, Y. Niidome, T. Niidome, K. Kaneko, H. Kawasaki and S. Yamada, Langmuir, 2006, 22, 2–5 CrossRef CAS PubMed.
- M. Kojima, K. Ishihara, A. Watanabe and N. Nakabayashi, Biomaterials, 1991, 12, 121–124 CrossRef CAS.
- Y. Iwasaki, K. Kurita, K. Ishihara and N. Nakabayashi, J. Biomater. Sci., Polym. Ed., 1997, 8, 151–163 CrossRef PubMed.
- R. E. Holmlin, X. Chen, R. G. Chapman, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 2841–2850 CrossRef CAS.
- J. Yuan, C. Mao, J. Zhou, J. Shen, S. C. Lin, W. Zhu and J. L. Fang, Polym. Int., 2003, 52, 1869–1875 CrossRef CAS.
- J. Yuan, Q. Hou, B. Liu, J. Shen and S. Lin, Colloids Surf., B, 2004, 36, 19–26 CrossRef PubMed.
- Z. Zhang, M. Zhang, S. Chen, T. A. Horbett, B. D. Ratner and S. Jiang, Biomaterials, 2008, 29, 4285–4291 CrossRef CAS PubMed.
- Y. Chang, S.-H. Shu, Y.-J. Shih, C.-W. Chu, R.-C. Ruaan and W.-Y. Chen, Langmuir, 2010, 26, 3522–3530 CrossRef CAS PubMed.
- H. Kitano, H. Suzuki, K. Matsuura and K. Ohno, Langmuir, 2010, 26, 6767–6774 CrossRef CAS PubMed.
- Y. Akiyama, A. Harada, Y. Nagasaki and K. Kataoka, Macromolecules, 2000, 33, 5841–5845 CrossRef CAS.
- Y. Akiyama, H. Otsuka, Y. Nagasaki, M. Kato and K. Kataoka, Bioconjugate Chem., 2000, 11, 947–950 CrossRef CAS PubMed.
- Y. Nagasaki, H. Kobayashi, Y. Katsuyama, T. Jomura and T. Sakura, J. Colloid Interface Sci., 2007, 309, 524–530 CrossRef CAS PubMed.
- R. D. Felice and A. Selloni, J. Chem. Phys., 2004, 120, 4906–4914 CrossRef PubMed.
- Y. Nagasaki, Sci. Technol. Adv. Mater., 2010, 11, 054505 CrossRef.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086 CrossRef CAS.
- E. Cooper and G. J. Leggett, Langmuir, 1998, 14, 4795–4801 CrossRef CAS.
- J. P. Bearinger, S. Terrettaz, R. Michel, N. Tirelli, H. Vogel, M. Textor and J. A. Hubbell, Nat. Mater, 2003, 2, 259–264 CrossRef CAS PubMed.
- M. J. Hostetler, S. J. Green, J. J. Stokes and R. W. Murray, J. Am. Chem. Soc., 1996, 118, 4212–4213 CrossRef CAS.
- K. Yoshimoto, M. Nozawa, S. Matsumoto, T. Echigo, S. Nemoto, T. Hatta and Y. Nagasaki, Langmuir, 2009, 25, 12243–12249 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- J. Janzen, X. Song and D. E. Brooks, Biophys. J., 1996, 70, 313–320 CrossRef CAS.
- K. Yoshimoto, S. Matsumoto, R. Asakawa, K. Uchida, T. Ishii and Y. Nagasaki, Chem. Lett., 2007, 36, 1444–1445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60212e |
| This journal is © The Royal Society of Chemistry 2014 |