Pharmacokinetics and transgene expression of implanted polyethylenimine-based pDNA complexes†‡
Laura
Rose
a,
Parvin
Mahdipoor
b,
Cezary
Kucharski
b and
Hasan
Uludağ
*abc
aDepartment of Biomedical Engineering, Faculties of Medicine & Dentistry and Engineering, University of Alberta, Edmonton, Alberta, T6G 2G6 Canada
bDepartment of Chemical & Materials Engineering, Faculty of Engineering, University of Alberta, Edmonton, Alberta, T6G 2G6 Canada. E-mail: huludag@ualberta.ca; Fax: +(780) 492-2881; Tel: +(780) 492-8809
cFaculty of Pharmacy & Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, T6G 2G6 Canada
First published on 28th November 2013
Abstract
Locally delivered plasmid DNA (pDNA) is currently pursued as gene-based therapy for regenerative medicine, but important information on in situ pDNA pharmacokinetics and transgene expression is lacking in animal models. To investigate pDNA pharmacokinetics in implants, low molecular weight (2 kDa) polyethylenimine (PEI) and linoleic acid substituted 2 kDa PEI (PEI-LA) were used for pDNA delivery in gelatin sponges. An efficient pDNA extraction method combined with quantitative PCR (qPCR) was found to give equivalent quantitation of naked and polymer-bound pDNA, making it suitable to assess pDNA polyplexes in implants. Naked pDNA implanted in a rat subcutaneous model was >98% lost after 24 hours whereas PEI and PEI-LA delivered pDNA remained intact in implants for 2 and 4 weeks, respectively. Using a plasmid expressing DsRed as a reporter gene, mRNA and protein expression was observed only for PEI-LA despite the extended retention and cellular uptake of PEI complexes. The in vivo data were in agreement with in vitro results showing that only PEI-LA was an effective transfection agent even though both PEI and PEI-LA complexes were internalized by the cells. Dose dependence was observed for mRNA expression, with a 20 μg dose giving faster onset and higher expression levels compared to a 5 μg pDNA dose. The mRNA expression after PEI-LA mediated delivery was sustained for at least 4 weeks and a significant correlation between pDNA retention in sponges and mRNA expression was observed. In addition to establishing a promising gene carrier for gene delivery, these studies provided important information about the retention and transgene expression by implanted non-viral carriers.
1. Introduction
Gene delivery is an exciting prospect for regenerative medicine especially for bone tissue repair,1 based on its ability to deliver a continuous supply of a therapeutic protein, in place of protein delivery schemes where high protein doses are associated with undesirable and serious side effects.2 The envisioned paradigm is that a biomaterial scaffold impregnated with either naked plasmid DNA (pDNA) or a carrier/pDNA complex is implanted at the desired repair site. Infiltrating host cells take up the administered pDNA, leading to local production of a protein to induce effective bone formation. This approach has been previously demonstrated for bone regeneration with both viral3,4 and non-viral carriers,5,6 although many virus-based vectors show reduced or abolished efficacy in immunocompetent animals.7–9 Several studies have employed pDNA without a carrier,10–13 but higher doses of pDNA (500–1000 μg) were required in this case, indicating the low efficiency of naked pDNA delivery. More tailored non-viral carriers have been successfully applied to ectopic and intraosseous models, and resulted in significant bone regeneration.6,14–16 The development of new non-viral carriers, however, is impeded by an uncertain correlation between in vitro and in vivo performance characteristics of the delivery systems.5,17,18 This is further compounded by a lack of important information on the pharmacokinetics of locally administered pDNA and corresponding transgene expression in these gene-activated matrix models.The pharmacokinetics and pharmacodynamics of current gene delivery systems have been recently reviewed, but with a focus on intravenous administration of delivery systems.19 Only a few studies reported in situ gene expression following pDNA delivery5,6,20 but pharmacokinetics of the implanted pDNA was missing in these studies. There is a fairly poor understanding of specific events once the complexes are locally delivered in implants, including how long the pDNA remains at the local implant site, how long the pDNA is expressed, and the role of a delivery system on transgene expression. More detailed studies on the onset and duration of mRNA expression would be helpful to understand differences in functional response. Such information might also allow investigations into correlations between in vitro and in vivo performance of carriers, especially in light of conflicting efficacy results from similar carriers in bone regeneration models.6,14 The pharmacokinetic studies for intravenous delivery have typically employed modified pDNA to allow for tracking of both complexes and naked pDNA using either fluorescent21 or radioactive labels.22 The presence of such a label, however, does not guarantee the presence of pDNA or whether the pDNA is still intact. Other options include Southern blots23 or agarose gels,24 but these approaches are likely to detect only naked pDNA that is not bound to a carrier, which is usually impermeable into electrophoretic gels. Real-time PCR is a more precise way to quantify pDNA, but challenges remain in the amplification of pDNA bound to non-viral carriers, since this technique is incapable of amplifying polymer-bound pDNA. A recent method described an extraction method with an anionic polymer to allow amplification of bound pDNA in circulation,25 which could provide a more reliable assessment of in situ levels of total pDNA, including the bound pDNA fraction.
This study was conducted to determine the local kinetics of pDNA delivered with polymeric carriers. A low molecular weight (2 kDa) polyethylenimine modified with linoleic acid (PEI-LA) was employed for in vivo pDNA delivery, given its superior performance over the native 2 kDa PEI.18 Unlike high molecular weight (25 kDa) PEI, which is highly effective but also toxic, unmodified PEI of this low molecular weight is considered ineffective for in vitro delivery of pDNA26 and siRNA27,28 alike. We set out to conduct thorough gene expression studies following in vivo pDNA delivery using PEI and PEI-LA, and present studies in this paper on the pharmacokinetics of pDNA, including a method of extracting pDNA from complexes to allow PCR amplification of polymer-bound pDNA. We specifically sought to identify the role of the carrier, duration of pDNA retention at local implant sites and the onset and length of mRNA expression.
2. Materials and methods
2.1. Materials
Dulbecco's modified Eagle media (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, trypsin/EDTA, and DNase/RNase free water were obtained from Life Technologies (Grand Island, NY). Absorbable gelatin sponges (Gelfoam) were obtained from Pharmacia & Upjohn (Walkersville , MD). The pCAG-DsRed plasmid was purchased from Addgene (Cambridge, MA), while the gWiz plasmid was purchased from Aldevron (Fargo, ND). The Cy5 pDNA labelling kit was from Mirus (Madison, WI). Branched PEI (2 kDa), poly-aspartic acid (2 kDa), poly-acrylic acid (2 kDa), Tris-HCl, Proteinase K, Hoechst 33258, heparin sodium salt, and sucrose were purchased from Sigma (St Louis, MO). Poly-L-aspartic acid sodium salt was from Alamanda Polymers (Huntsville, AL). Phenylmethanesulphonylfluoride (PMSF) was from BioShop (Burlington, ON). DNeasy Blood & Tissue kit, RNeasy kit, RNA Later RNA stabilization reagent, and DNase I were all obtained from Qiagen (Hilden, Germany). Ketamine was obtained from Bayer HealthCare (Toronto, ON), while Xylazine was obtained from Wyeth (Guelph, ON). Tris-hydroxypropylphosphine (THP) was from EMD Millipore (San Diego, CA). Shandon Cryomatrix was from Thermo Scientific (Pittsburgh, PA), Hank's balanced salt solution (HBSS) from BioWhittaker (Walkersville, MD). Primers and FAM-labelled probes with Zen and Iowa fluorescent black quenchers were obtained from IDT Technologies (Ottawa, ON). 2 kDa PEI modified with linoleic acid (PEI-LA; 4 LA substitutions per PEI) was prepared as previously described.262.2. Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 with unlabelled gWiz for use in in vitro and in vivo delivery studies. The DsRed expressing plasmid pCAG-DsRed2 was used for transfection.29 Sponges for animal studies were loaded with either gWiz, gWiz-Cy5 or pCAG-DsRed. The plasmids were loaded with PEI or PEI-LA in a total volume of 150 μL with the pDNA dose described in each of the studies (see figure legends). Blank sponges loaded with saline alone were included as controls.
000 cells were counted for each sample, and analysed for Cy5 fluorescence (λex = 640 nm, λem = 670 nm) or DsRed fluorescence (λex = 561 nm, λem = 586 nm) using Hoechst (λex = 355 nm, λem = 450 nm) to distinguish cells from debris. Each study group contained three replicates.
000 cells were counted for each sample and analysed for Cy5 fluorescence (λex = 640 nm, λem = 670 nm) using Hoechst (λex = 355 nm, λem = 450 nm) to distinguish cells from debris. Cells were also imaged with a Zeiss LSM 710 laser scanning confocal microscope to investigate the intracellular presence of complexes. The other half of the explants were used for histological analysis; the explant was fixed for 24 hours in 3.7% formalin at 4 °C and then for another 24 hours in 30% sucrose as a cryoprotectant. The samples were then frozen at −20 °C in Shandon Cryomatrix, sections and stained with DAPI. The cryosections were imaged with the fluorescent microscope FSX100 (Olympus).
:12 with unlabelled gWiz for use in in vitro and in vivo delivery studies. The DsRed expressing plasmid pCAG-DsRed2 was used for transfection.29 Sponges for animal studies were loaded with either gWiz, gWiz-Cy5 or pCAG-DsRed. The plasmids were loaded with PEI or PEI-LA in a total volume of 150 μL with the pDNA dose described in each of the studies (see figure legends). Blank sponges loaded with saline alone were included as controls.
000 cells were counted for each sample, and analysed for Cy5 fluorescence (λex = 640 nm, λem = 670 nm) or DsRed fluorescence (λex = 561 nm, λem = 586 nm) using Hoechst (λex = 355 nm, λem = 450 nm) to distinguish cells from debris. Each study group contained three replicates.
000 cells were counted for each sample and analysed for Cy5 fluorescence (λex = 640 nm, λem = 670 nm) using Hoechst (λex = 355 nm, λem = 450 nm) to distinguish cells from debris. Cells were also imaged with a Zeiss LSM 710 laser scanning confocal microscope to investigate the intracellular presence of complexes. The other half of the explants were used for histological analysis; the explant was fixed for 24 hours in 3.7% formalin at 4 °C and then for another 24 hours in 30% sucrose as a cryoprotectant. The samples were then frozen at −20 °C in Shandon Cryomatrix, sections and stained with DAPI. The cryosections were imaged with the fluorescent microscope FSX100 (Olympus).
3. Results
3.1. In vitro pDNA uptake and transfection
The pDNA uptake was investigated in 293T cells using gWiz-Cy5. Cells exposed to PEI-LA/gWiz-Cy5 complexes gave high fluorescence (Fig. 1A) whereas the background from PEI-LA/gWiz complexes and naked gWiz-Cy5 was much lower as expected. In a follow-up study, non-treated cells and cells treated with unlabelled gWIZ complexes (PEI/gWiz and PEI-LA/gWiz) gave low levels of autofluorescence, whereas cells treated with PEI/gWiz-Cy5 and PEI-LA/gWiz-Cy5 gave high levels of fluorescence (Fig. 1B-i). Approximately 63 and 68% of the cells were positive for gWiz-Cy5 in the case of PEI and PEI-LA, respectively (not shown). A DsRed expressing plasmid (pCAG-DsRed) was used to evaluate transfection efficiency (Fig. 1B-ii). Cells transfected with PEI/gWiz and PEI-LA/gWIZ complexes gave no DsRed expression as expected. Some DsRed positive cells were observed (∼2%, not shown) leading to a small increase in DsRed fluorescence for PEI/pCAG-DsRed transfected cells (433 au for PEI/pCAG-DsRed vs. 107 au for PEI/gWiz). For PEI-LA/pCAG-DsRed transfected cells, however, the fluorescence increased to 2.6 × 104 au (Fig. 1B-ii), corresponding to ∼65% DsRed-positive cell population (not shown).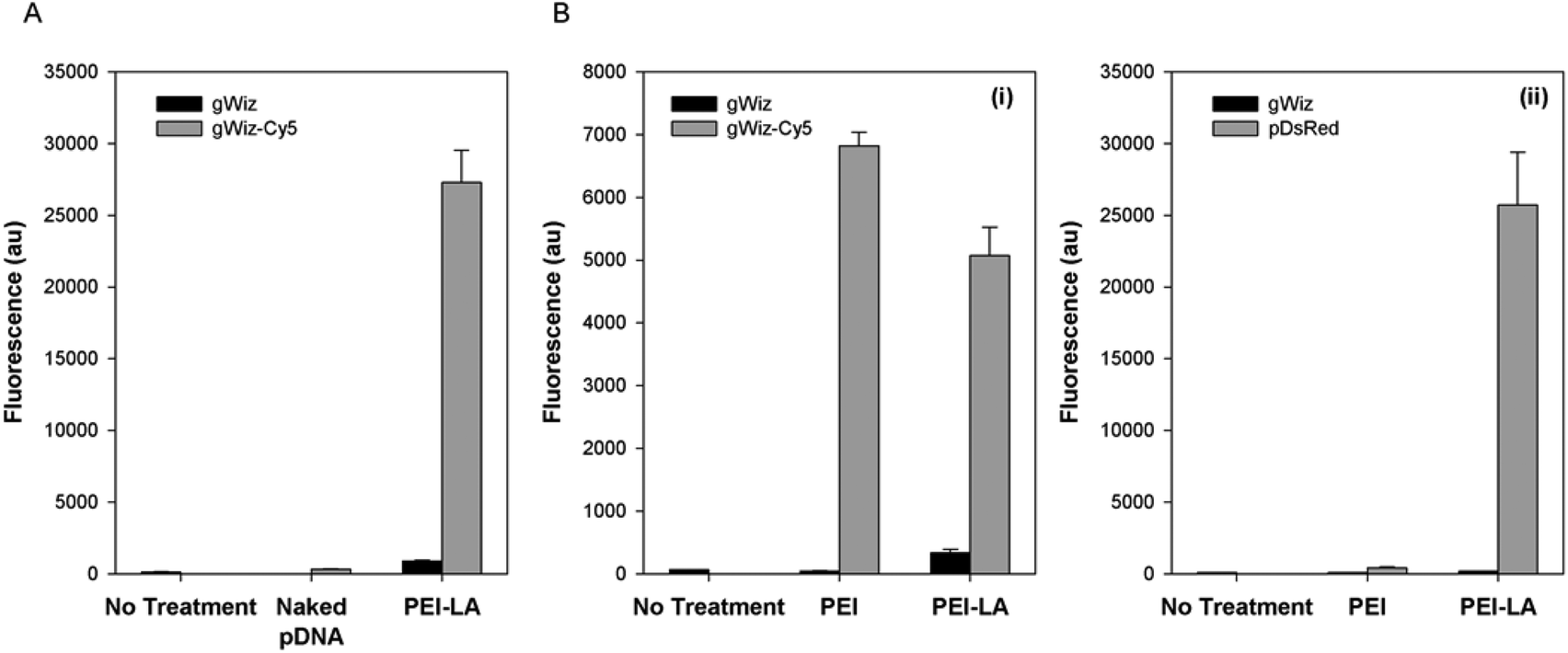 | ||
| Fig. 1 pDNA uptake and transfection in vitro. 293T cells were used to investigate pDNA uptake with naked and PEI-LA complexes (A) and to compare PEI and PEI-LA for uptake (B-i) and transfection (B-ii). For the uptake, cells were exposed to the indicated pDNA or complexes for 24 hours, and then analysed for Cy5 content using flow cytometry. For transfection, cells were exposed to the indicated complexes for 24 hours, and then harvested for flow cytometry after two days for analysis of DsRed expression. | ||
3.2. Pharmacokinetics of Cy5-labelled pDNA
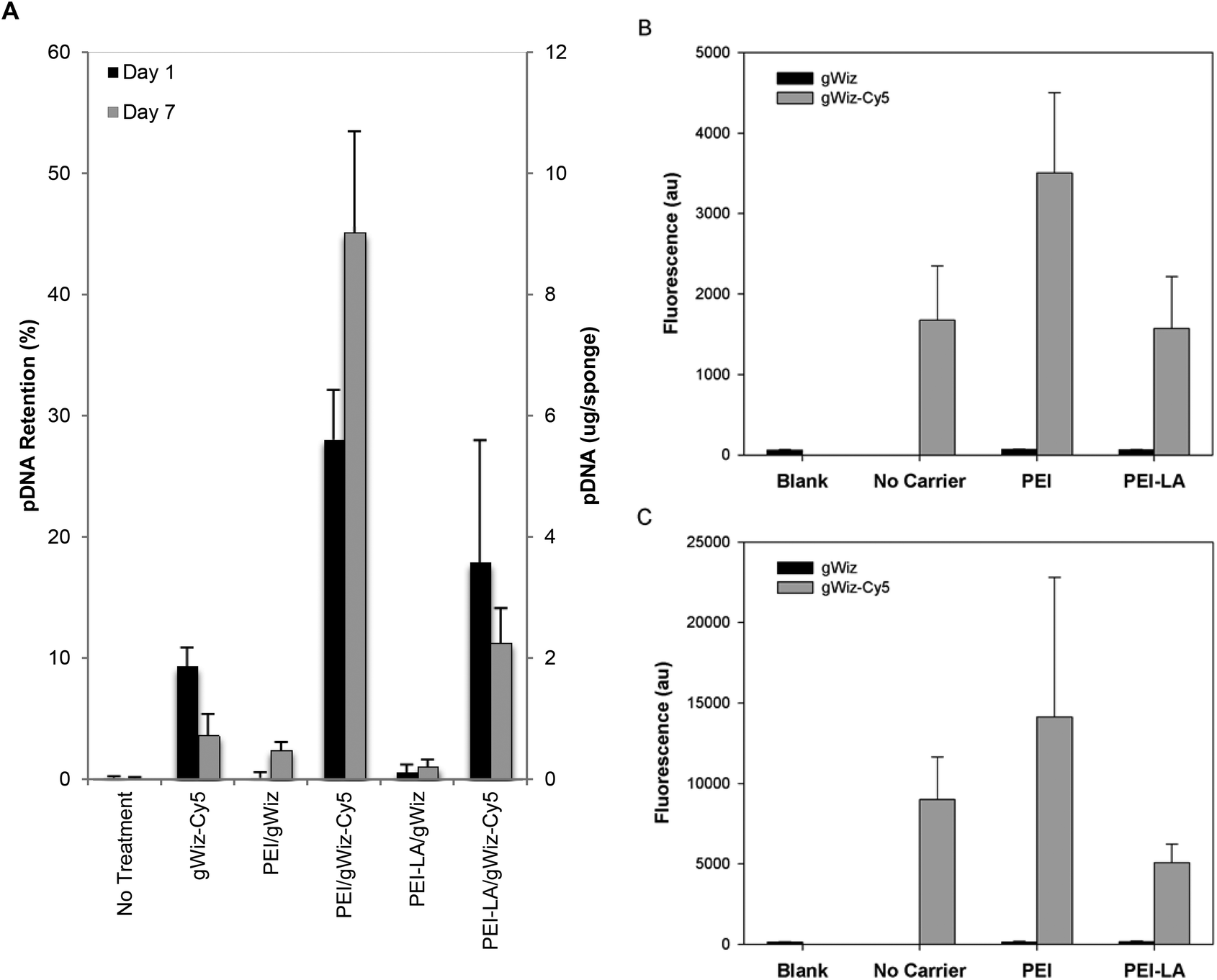 | ||
| Fig. 2 Pharmacokinetics of pDNA assessed with gWiz-Cy5. Sponges were loaded with indicated gWIZ and gWIZ-Cy5 complexes of PEI and PEI-LA, and implanted subcutaneously in rats. Harvested sponges were assessed for Cy5 fluorescence 1 and 7 days after implantation (A). Cells extracted from the Day 1 (B) and Day 7 (C) harvested sponges were analysed for Cy5 fluorescence with flow cytometry and the mean (±SD) fluorescence/cell are shown. | ||
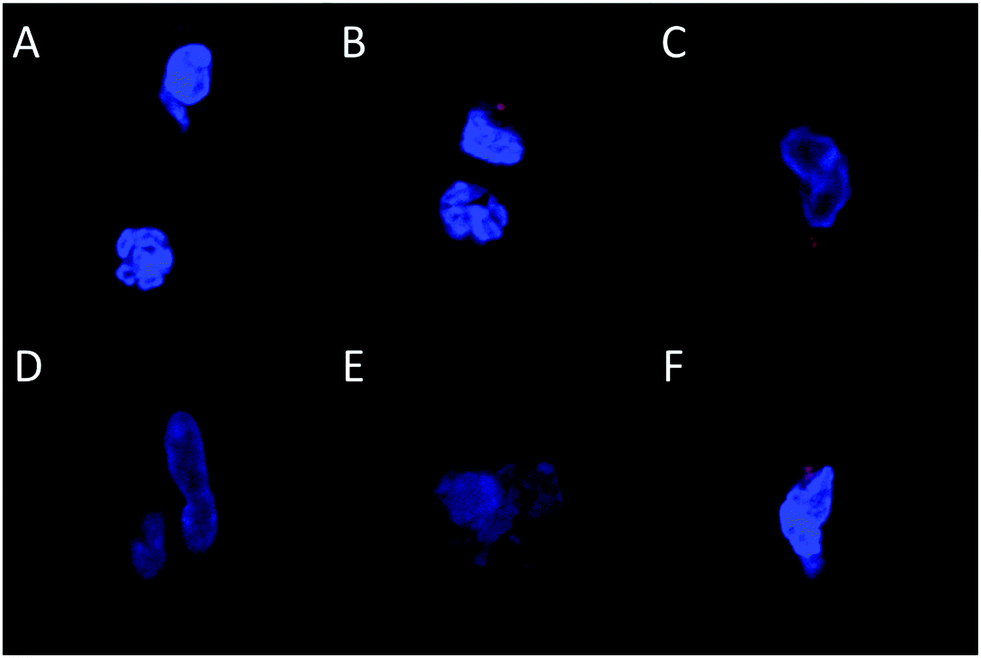 | ||
| Fig. 3 Confocal microscopy of cells recovered from implants. Cells were harvested from sponges explanted on Day 1 (A–C) or Day 7 (D–F) and fixed cells were analysed with confocal microscopy (representative pictures shown). Sponges were loaded with naked gWiz-Cy5 (A and D), PEI/gWiz-Cy5 complexes (B and E), or PEI-LA/gWiz-Cy5 complexes (C and F). Blue: Hoechst, Red: gWIZ-Cy5. | ||
3.3. Method development for PCR based detection of pDNA in sponges
A qPCR-based method was investigated in order to determine the intact amount of pDNA in implants. Standard curves were constructed from naked pCAG-DsRed and PEI-LA/pCAG-DsRed complexes either in solution or absorbed onto sponges. Using a commercial kit designed for pDNA extraction from blood and tissue, a linear standard curve was obtained for naked pDNA but no relationship was obtained for the PEI-LA/pDNA complexes (ESI 2A‡). For the PEI-LA complexes, the cycle thresholds for all points of the standard curve were 23–24 in our set-up, which was similar to the background levels (i.e., no pDNA). Polymer binding therefore seems to abolish the feasibility of amplification by PCR. To allow amplification in PEI-LA/pDNA complexes, heparin was used to dissociate the complex,20 but this did not also allow amplification of the PEI-LA/pDNA complexes (ESI 3‡) and even interfered with the amplification of naked pDNA.A second extraction method was undertaken where samples are incubated with anionic poly-acrylic acid. We used poly-acrylic acid in place of poly-L-aspartic acid employed in the original procedure25 since PCR amplification with poly-L-aspartic acid interfered with the ROX dye used as a passive reference (data not shown). Linear standard curves were obtained for both naked and PEI-LA-bound pDNA in this method (ESI 2B‡). Separate calibration curves were generated for naked and polymer bound pDNA, and due to the slight divergence of the curves at higher pDNA doses, treatment groups were run with a standard curve containing the same type of pDNA (i.e. either naked or specific polymer bound pDNA).
Finally, to account for the possibility of aberrant amplification from implanted sponge contents, standard curves were generated in a background of explanted sponges. Standard curves from 1-day implanted sponges had higher CT compared to un-implanted sponges (ESI 4‡). Sponges implanted for 7 days also gave higher CT compared to sponges implanted for 1 day. To determine the reason for this shift, pDNA standard curves were spiked with blood components. The addition of serum had minimal effect on the amplification of pDNA (ESI 5A‡) while the addition of clotted blood exhibited a dose-dependence shift in CT (ESI 5B‡). Based on this shift, we estimate that an implant with ∼50% whole blood would lead to approximately 1 CT decrease, giving an apparent reduction of half pDNA dose. The total error for explanted sponges, however, is likely less than this value since the bloodiest implants had an estimated ∼30% blood content.
3.4. Comparison of pDNA pharmacokinetics and expression by qPCR
Sponges implanted with naked pCAG-DsRed or PEI-LA/pCAG-DsRed, complexes were harvested after 1 or 7 days to measure pDNA retention with qPCR (Fig. 4A). No pDNA was detected in blank sponges (i.e., the signal below the detection limit of 0.01 μg per sponge on Day 1 and 0.3 μg per sponge on Day 7). A minimal amount of pDNA was detected in both naked pDNA groups on Day 1: 0.06 μg of pCAG-DsRed left from the 5 μg implant dose and 0.1 μg from the 20 μg implant dose. This represents 1.3% and 0.6% retention for the corresponding groups. All explants from 5 μg and 20 μg naked pCAG-DsRed groups on Day 7 gave signals below the detection limit of the PCR assay (∼0.3 μg per explant). For the 5 μg dose of PEI-LA/pCAG-DsRed, an average of 6 μg of pDNA/sponge was detected after a 1 day implantation and an average of 5 μg after 7 days. For the 20 μg dose of PEI-LA/pCAG-DsRed, PCR calibration curves indicated ∼31 and 39 μg of pDNA/sponge were detected after 1 and 7 days respectively. These values were seemingly equivalent or higher than the implant dose (see the Discussion section).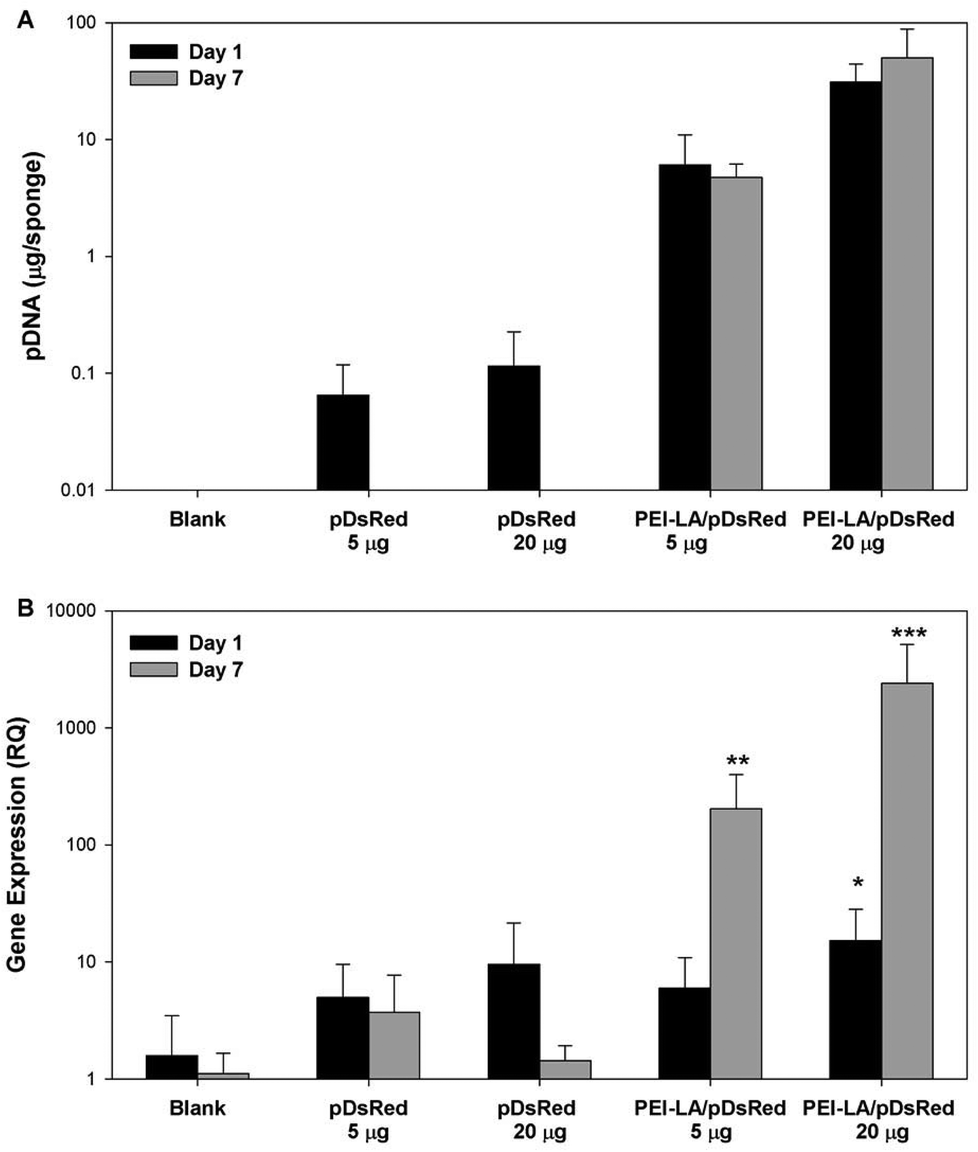 | ||
| Fig. 4 Retention and expression of naked and PEI-LA-bound pCAG-DsRED. The plasmid retention was quantitated by qPCR and using blank implants spiked with either free pCAG-DsRED or PEI-LA/pCAG-DsRED complexes to create standard curves (A). qPCR was used to measure mRNA expression of DsRed relative to beta-actin in explants harvested after 1 or 7 days (B). The mRNA results were analysed with a Kruskal–Wallis test. *p < 0.05, **p < 0.01, ***p < 0.001 compared to blank implants for each time point. | ||
Sponges implanted with naked pCAG-DsRed and PEI-LA/pCAG-DsRed complexes were investigated for DsRed mRNA expression. Low levels of DsRed mRNA were found 1 day after implantation when most pDNA were present. Only the high (20 μg) dose of PEI-LA/pCAG-DsRed complexes led to significant (p < 0.05 vs. Blank) levels of DsRed mRNA on Day 1 (Fig. 4B). At 7 days, both the 5 μg (p < 0.01) and 20 μg (p < 0.001) dose of PEI-LA/pCAG-DsRed complexes led to high, dose-dependent DsRed mRNA expression.
3.5. Long-term pCAG-DsRED delivery by PEI and PEI-LA
The long-term (4 weeks) efficiency of in vivo gene delivery with unmodified and LA-modified PEI was next investigated. Explanted sponges were imaged for DsRed fluorescence, and normalized against the blank (saline) sponges at corresponding time points (Fig. 5A). No significant increase in DsRed fluorescence was observed for PEI/pCAG-DsRED at any time point. For PEI-LA/pCAG-DsRED implants, increased fluorescence (p < 0.01) was observed for sponges harvested at 1, 2, and 4 weeks. Although PEI-LA/pCAG-DsRED sponges on week 3 had comparable fluorescence to PEI-LA/pCAG-DsRED sponges from other time points, the background fluorescence for blank sponges was higher for this time point, leading to a lack of significant difference. The measured fluorescence is likely an underestimation of DsRed content in sponges, since the presence of blood significantly attenuated the DsRed fluorescence (ESI 7‡). Sponges were processed for histology to confirm the presence of transfected cells. No DsRed was observed in the DAPI-stained sections of blank (Fig. 5B) or PEI/pCAG-DsRED (Fig. 5C). Regions of DsRed fluorescence, however, were observed with PEI-LA/pCAG-DsRed samples (Fig. 5D).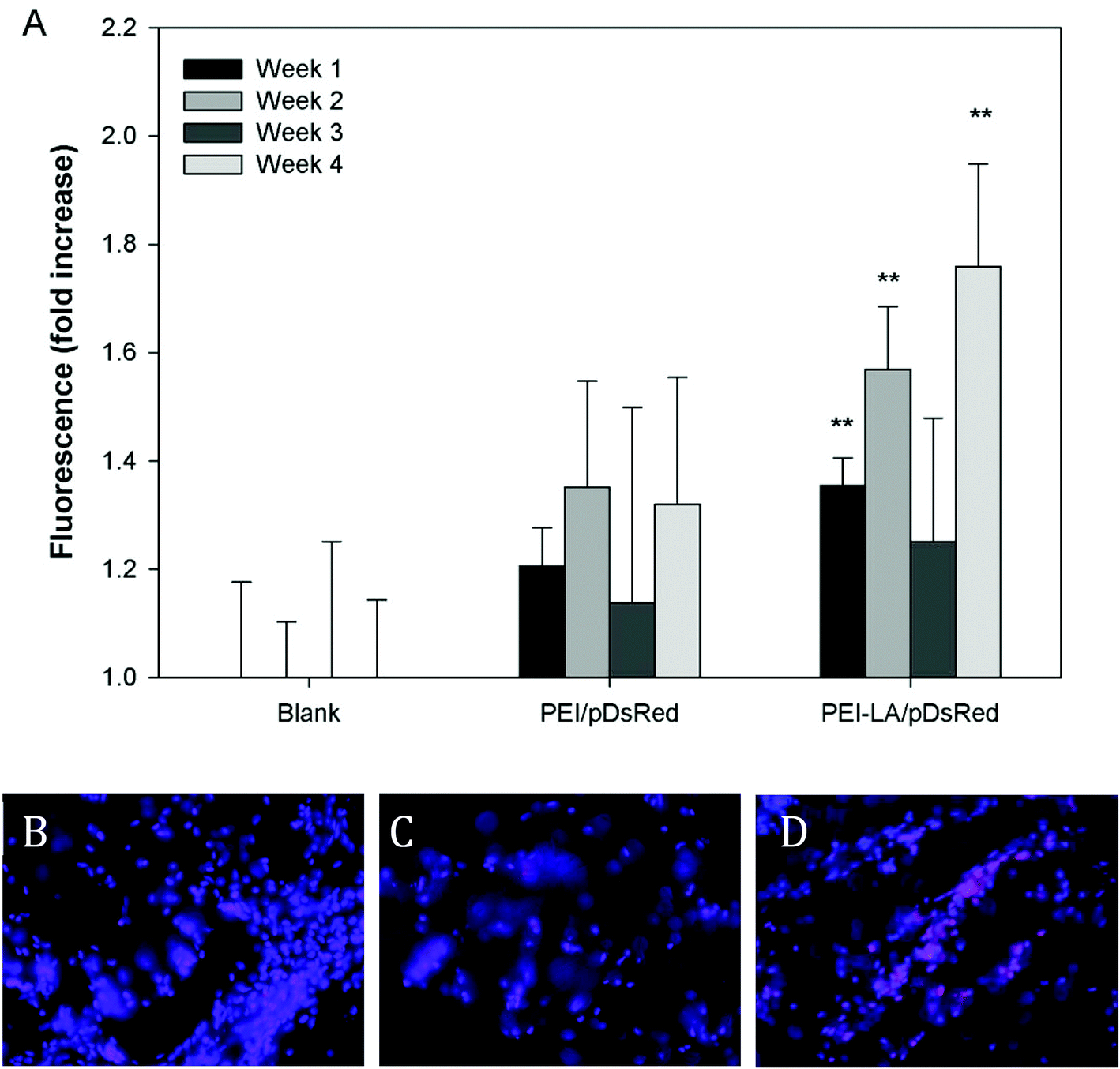 | ||
| Fig. 5 DsRed quantitation by ex vivo fluorescence imaging. Sponges were harvested after 1 to 4 weeks for DsRed imaging and the measured fluorescence was normalized against the mean fluorescence of blank sponges from the same harvest time (A). PEI/pDsRed and PEI-LA/pDsRed sponges were compared against blank sponges for each time point using a Kruskall–Wallis test, followed by the Dunn post-hoc test. **p < 0.01 compared to blank sponges for each time point. Sponges harvested after one week were frozen in cryomatrix and sectioned. Images show representative sections of blank sponges (B), PEI/pCAG-DsRed soaked sponges (C), and PEI-LA/pCAG-DsRed soaked sponges (D) from week 1. Blue: DAPI, Pink: DsRed. | ||
Using the qPCR methodology, no pDNA was detected in any of the blank implants again (Fig. 6A). The pDNA was detected in both the PEI and PEI-LA delivered pCAG-DsRed groups after 1 week, with 14 and 3 μg pDNA/sponge respectively. The PEI/pCAG-DsRed group had only ∼0.06 μg pDNA/sponge after 2 weeks, with pDNA being below the limit of detection after 3 and 4 weeks. The amount of pDNA in PEI-LA/pCAG-DsRed sponges was significantly higher; 4, 0.6, and 0.1 μg per sponge for week 2, 3, and 4, respectively. As before, there was no DsRed mRNA in any of the blank sponges, and no differences were found between blank sponges and PEI/pCAG-DsRed sponges (Fig. 6B). The PEI-LA/pCAG-DsRed containing sponges, however, gave increased DsRed expression at all four weekly time points (p < 0.01 for week 1, 3 and 4; p < 0.05 for week 2), with maximal expression appearing at 2 and 4 weeks. A relationship between mRNA expression and pDNA retention was investigated by pooling implants containing PEI-LA and PEI complexes from all time points; a significant correlation between mRNA expression and pCAG-DsRED retention was found for PEI-LA implants (p < 0.01), but not for PEI implants (Fig. 7).
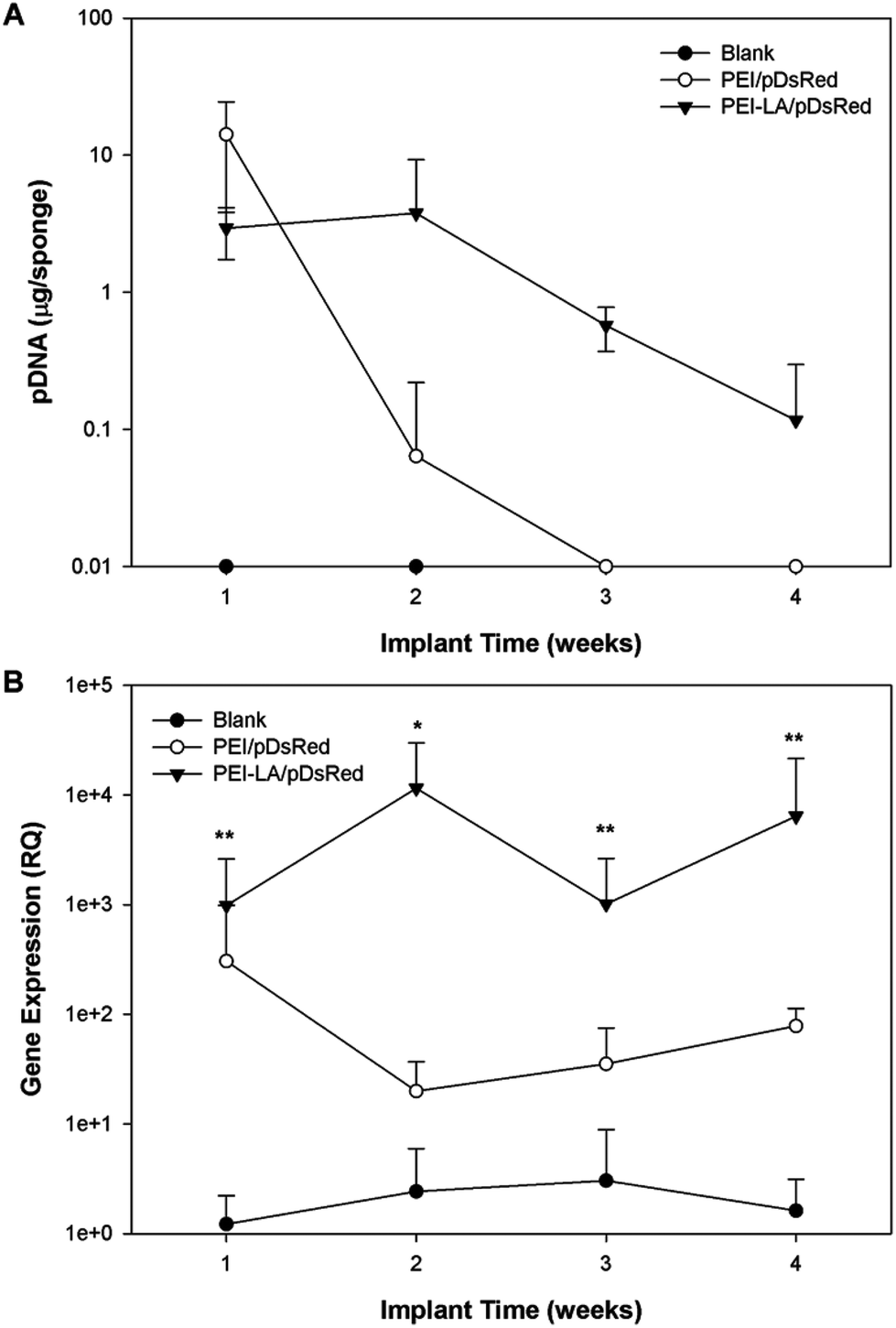 | ||
| Fig. 6 Long-term pDNA pharmacokinetics and mRNA expression. The amount of pDNA remaining on sponges was quantified with qPCR (A). The mRNA expression was also analysed using qPCR (B). The mRNA levels at each time point were compared against blank sponges with a Kruskal–Wallis test at each time point. *p < 0.05 and **p < 0.01. | ||
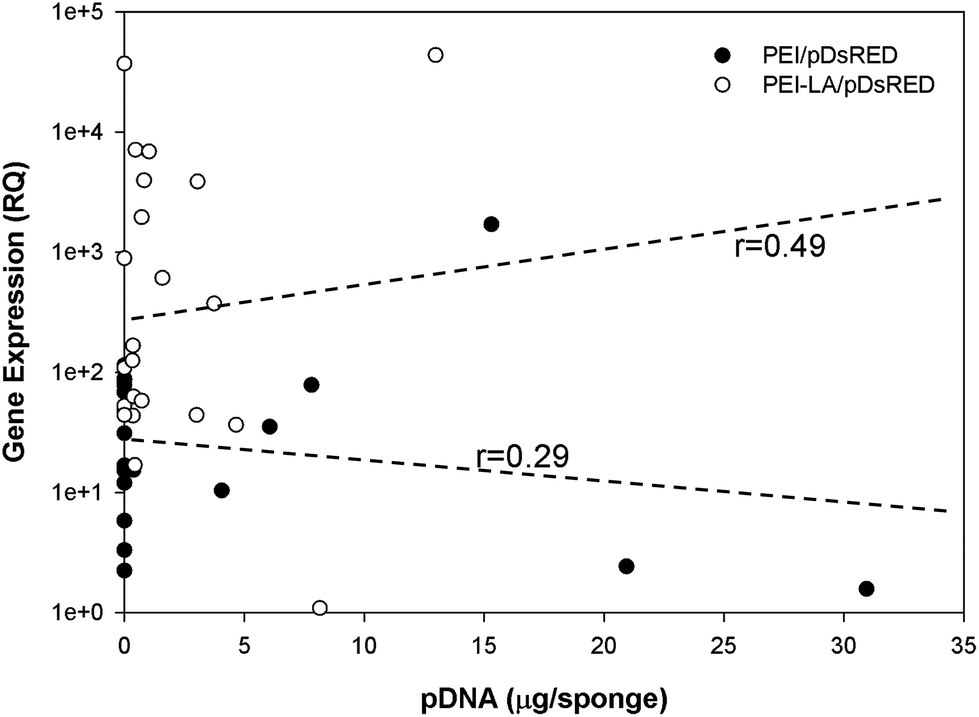 | ||
| Fig. 7 Correlation between gene expression and pDNA retention. Graph shows the correlation between DsRED mRNA expression (in relative quantity) and pDNA retention (in μg per sponge) for each individual implant. PEI/pCAG-DsRED and PEI-LA/pCAG-DsRED implants were pooled from all harvest points between week 1 and week 4. | ||
4. Discussion
Investigations into pDNA pharmacokinetics and mRNA expression dynamics are paramount to provide information for development of biomaterial carriers intended for in vivo use. Such studies will depend on a reliable method to quantitate pDNA and mRNA levels in implants. Commercially available kits are usually designed for extraction of naked pDNA and, although low concentrations of heparin were compatible with PCR,30 the high amount required to fully dissociate polymeric complexes impeded PCR amplification in this study. Besides the method described in this paper, only one other competitive qPCR-based method has been described where pDNA was quantitatively detected in polymeric complexes using an internal standard (IS) as a competitor molecule.31 In this method, a second plasmid template (IS) that produced a different size PCR product is amplified alongside the target plasmid. Standard curves with varying amounts of IS can be constructed to provide a relationship between concentration of the target pDNA and the ratio of amplification between the target and IS. In the study that compared uptake and intracellular trafficking of naked to that of PEI-bound pDNA,31 the generated standard curves allowed the authors to perform pharmacokinetics of pDNA bound to complexes, but the curves for naked and bound pDNAs were very different. This method would therefore be error-prone in biological samples: pDNA is eventually released from the complex during transfection, and therefore a portion of pDNA in a sponge sample would be naked pDNA. This contrasts our method where the standard curves for naked and bound pDNA are similar, so that we could generate the total quantity of pDNA in implants.Our qPCR-based method, which was modified from a previous report,25 was found to be suitable for quantifying pDNA on the sponge, but not differentiating between the naked and bound pDNA. This method, however, might still be associated with error due to the dynamic nature of tissue in sponges, where cell invasion and vascularization occurred as a function of time. There was a range of hematomas in sponges harvested at different times, whether they were blank or pDNA-containing sponges. The variability was evident within each study group (i.e., the same type of implant) so that this variability might reflect differences in the physiology of individual rats. Blood clots impeded the qPCR reaction in our hands and other studies have also reported qPCR interference due to both heme and leukocytes in blood.30,32 The original extraction protocol from Zhou et al.25 was applied to complexes in blood samples but it was possible that a systematic error was introduced to all samples given the same ‘background’ interference. The immune response to our complexes was investigated to ensure that blood in our studies was not enriched with activated leukocytes. Levels of tumor necrosis factor-α, interleukin-6, and interleukin-2 in implants showed no increase with implantation of complexes (ESI 8‡), ruling out the activated leukocytes as a source of error for qPCR. Another source of error may be due to a lack of uniform distribution of pDNA in sponges, particularly for the PEI-LA and PEI complexes. This variability might be due to initial loading differences or due to release from implants, which was expected to deplete pDNA from the periphery. Since we sample only a small fraction of sponges (15–50% depending on implant mass), it is possible to erroneously calculate the retained dose due to heterogeneous sampling. Despite these unresolved issues, the qPCR method remains a promising way to measure the amount of pDNA in sponge.
There was a good agreement between the Cy5- and qPCR-based assessments of pDNA pharmacokinetics for polymer complexes. Retention studies using gWIZ-Cy5 and qPCR gave similar results with respect to initial ability of PEI and PEI-LA to retain pDNA in implants. Both PEI and PEI-LA complexes showed similar retention after 1 and 7 days of implantation with both methods, even though PEI appeared to provide higher localization initially. Assessment by gWIZ-Cy5, however, gave results that were not in agreement with the qPCR-based methodology specifically for naked pDNA implants. Due to lack of DsRed mRNA expression, we hypothesize that the majority of naked pDNA implanted were degraded (or lost) within one day, as indicated by the qPCR method. It was considered likely that the gWIZ-Cy5 assessment was obscured by the liberated Cy5 label, which highlights the benefit of qPCR to ensure detection of intact pDNA. This finding is in agreement with previous systemic delivery studies that found naked pDNA to be rapidly degraded with a half-life on the scale of minutes.21 While some bone regeneration studies found a beneficial effect due to locally delivered pDNA without a carrier, the dose required for this effect was generally high5,10–12 or multiple smaller doses were required for an effect.13 Unlike our study, naked pDNA and mRNA expression were detected after 6 weeks in one study,5 but that study used a 1000 μg pDNA dose as compared to our 5 and 20 μg doses (i.e., such excessive doses may exceed the amount of pDNA that can be degraded in a short period of time). It is possible that difference in biomaterial scaffold used as a vehicle for pDNA implantation may alter the half-life of the pDNA payload, with changes in scaffold's integrity potentially altering the clearance of pDNA, either naked or polymer-bound, from the site. The sponges in our study showed a gradual decrease in size over the course of the four-week implantation period (size ultimately reducing to <half). It is likely that this is indicative of sponge degradation, which may have accelerated release or clearance of DNA from the implantation site. It remains unclear whether the therapeutic effect reported with different scaffolds was due to production of the protein or the foreign body/immune response to implanted pDNA. The beneficial effect of the naked pDNA may be due to a general immunogenic response, since unmethylated cytosine–guanine dinucleotides (CpG motifs) of pDNA are known to be immunogenic.33
The pDNA delivered with PEI and PEI-LA both showed prolonged retention in implants. The pDNA delivered by PEI was detected for up to 2 weeks and pDNA from PEI-LA complexes remained in implants for at least 4 weeks. This difference in retention could be due to a difference in intracellular fate of pDNA. Both in vitro and in vivo, this low molecular weight PEI gave comparable cellular uptake of pDNA but negligible transfection as compared to PEI-LA delivery. The rate-limiting step for 2 kDa PEI complexes appeared to be endosomal escape,34 and so that the pDNA contained within PEI complexes might be eventually destroyed and no longer detectable. In contrast, the PEI-LA complexes led to effective transgene expression, so that pDNA was likely in the nucleus where it was less likely to be destroyed. The agreement between the transfection results from an immortalized cell line and an animal model is particularly encouraging. Although the absolute transfection efficiency observed here in vitro and in vivo is likely different, intracellular trafficking appears to be a limited factor for unmodified PEI.
A correlation was found between mRNA expression and pDNA retention for PEI-LA implants, but not for PEI implants. This is in line with the lack of expression from the PEI complexes. An even stronger correlation would be expected with a better sampling strategy, as there should be a delay between pDNA presence and mRNA expression, and implants that retained higher pDNA at initial time points would have led to higher mRNA expression at a later time point. This, however, would have required multiple samplings from the same sponge while still being implanted, which was not possible with our current methodology. Nevertheless, the observed correlation is a testament to the stronger mRNA expression by the PEI-LA/pCAG-DsRed complexes during the study period. In addition to long duration of expression, mRNA was also expressed very quickly: with a 20 μg dose of pDNA in PEI-LA complexes, increased levels of DsRed mRNA were detected within 24 hours of implantation. The lower 5 μg dose was also effective, but increased mRNA expression was not observed until the second time point (7 days post-implantation). The subcutaneous model employed for these studies may be less biologically active than an intraosseous site, but these studies are intended to provide preliminary information about pDNA retention and mRNA expression for non-viral delivery systems. The length and onset of transgene expression is especially encouraging for bone regeneration therapies, where weeks of expression and fast onset would be beneficial. Since transgene expression was possible at four weeks, future studies will investigate when transgene expression diminishes, as extended production of bone inducing proteins is eventually not needed. The correlation between the presence of pDNA and mRNA expression also calls for gene delivery approaches that maximize pDNA retention in implants as a means to enhance transgene expression. With the availability of the current qPCR-based methodology, we envision more detailed investigations of pDNA pharmacokinetics in implanted sponges to be pursued in the future.
5. Conclusions
A polymeric carrier for gene delivery is expected to display two important functions, namely to protect pDNA from degradation in the extracellular environment and to navigate cellular barriers to allow protein expression. We distinguished between the pDNA uptake and successful intracellular trafficking with the comparison of PEI and PEI-LA carriers in this study. While both carriers appeared to protect pDNA in implants initially, the delivered pDNA remained detectable for a prolonged time (up to 4 weeks) with the PEI-LA carrier. Similarly, expression of a DsRED reporter gene was observed within 24 hours of implantation with the more effective PEI-LA, whose pharmacokinetics profile corresponded closely to the transgene expression profile up to 4 weeks. A relatively weak but nevertheless significant correlation between pDNA retention of transgene expression was observed. The methodology described here and the unique carriers characterized should facilitate gene delivery efforts in tissue regeneration and repair.References
- L. Rose and H. Uludag, J. Bone Miner. Res., 2013, 28, 2245 CrossRef CAS PubMed.
- E. J. Carragee, E. L. Hurwitz and B. K. Weiner, Spine J., 2011, 11, 471 CrossRef PubMed.
- Y. Chen, K. Luk, K. Cheung, R. Xu, M. Lin, W. Lu, J. Leong and H. Kung, Gene Ther., 2003, 10, 1345 CrossRef CAS PubMed.
- W. W. Hu, Z. Wang, S. J. Hollister and P. H. Krebsbach, Gene Ther., 2007, 14, 891 CrossRef CAS PubMed.
- J. Bonadio, E. Smiley, P. Patil and S. Goldstein, Nat. Med., 1999, 5, 753 CrossRef CAS PubMed.
- K. Itaka, S. Ohba, K. Miyata, H. Kawaguchi, K. Nakamura, T. Takato, U. Chung and K. Kataoka, Mol. Ther., 2007, 15, 1655 CrossRef CAS PubMed.
- T. Alden, D. Pittman, F. Hankins, E. Beres, J. Engh, S. Das, S. Hudson, K. Kerns, D. Kallmes and G. Helm, Hum. Gene Ther., 1999, 10, 2245 CrossRef CAS PubMed.
- Q. Kang, M. Sun, H. Cheng, Y. Peng, A. Montag, A. Deyrup, W. Jiang, H. Luu, J. Luo, J. Szatkowski, P. Vanichakarn, J. Park, Y. Li, R. Haydon and T. He, Gene Ther., 2004, 11, 1312 CrossRef CAS PubMed.
- J. Li, H. Li, T. Sasaki, D. Holman, B. Beres, J. Dumont, D. Pittman, G. Hankins and G. Helm, Gene Ther., 2003, 10, 1735 CrossRef CAS PubMed.
- C. Bright, Y. S. Park, A. N. Sieber, J. P. Kostuik and K. W. Leong, Spine, 2006, 31, 2163 CrossRef PubMed.
- J. Fang, Y. Y. Zhu, E. Smiley, J. Bonadio, J. Rouleau, S. Goldstein, L. McCauley, B. Davidson and B. Roessler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5753 CrossRef CAS.
- F. Geiger, H. Betram, I. Berger, H. Lorenz, O. Wall, C. Eckhardt, H. G. Simank and W. Richter, J. Bone Miner. Res., 2005, 20, 2028 CrossRef CAS PubMed.
- K. Osawa, Y. Okubo, K. Nakao, N. Koyama and K. Bessho, J. Gene Med., 2010, 12, 937 CrossRef CAS PubMed.
- Y. C. Huang, C. Simmons, D. Kaigler, K. G. Rice and D. J. Mooney, Gene Ther., 2005, 12, 418 CrossRef CAS PubMed.
- M. Oda, S. Kuroda, H. Kondo and S. Kasugai, J. Biomed. Mater. Res., Part B, 2009, 90, 101 Search PubMed.
- I. Ono, T. Yamashita, H. Y. Jin, Y. Ito, H. Hamada, Y. Akasaka, M. Nakasu, T. Ogawa and K. Jombow, Biomaterials, 2004, 25, 4709 CrossRef CAS PubMed.
- D. Li, W. Wang, R. Guo, Y. Y. Qi, Z. R. Gou and C. Y. Gao, Chin. Sci. Bull., 2012, 57, 435 CrossRef CAS.
- L. C. Rose, C. Kucharski and H. Uludag, Biomaterials, 2012, 33, 3363 CrossRef CAS PubMed.
- Z. P. Parra-Guillen, G. Gonzales-Aseguinolaza, P. Berraondo and I. F. Troconiz, Pharm. Res., 2010, 27, 1487 CrossRef CAS PubMed.
- L. Rose, H. M. Aliabadi and H. Uludag, Gelatin coating to stabilize the transfection ability of nucleic acid polyplexes, Acta Biomater., 2013, 9(7), 7429–7438 CrossRef CAS PubMed.
- B. E. Houk, R. Martin, G. Hochhaus and J. A. Hughes, Pharmacokinetics of plasmid DNA in the rat, Pharm. Res., 2001, 18, 67–74 CrossRef CAS.
- L. Yu, H. Suh, J. J. Koh and S. W. Kim, Systemic administration of TerplexDNA system: pharmacokinetics and gene expression, Pharm. Res., 2001, 18, 1277–1283 CrossRef CAS.
- D. Lew, S. E. Parker, T. Latimer, A. M. Abai, A. Kuwahararundell, S. G. Doh, Z. Y. Yang, D. Laface, S. H. Gromkowski and G. J. Nabel, et al. , Hum. Gene Ther., 1995, 6, 553 CrossRef CAS PubMed.
- A. R. Thierry, P. Rabinovish, B. Peng, L. C. Mahan, J. L. Bryant and R. C. Gallo, Gene Ther., 1997, 4, 226 CAS.
- Q. H. Zhou, C. Wu, D. S. Manickam and D. Oupicky, Pharm. Res., 2009, 26, 1581 CrossRef CAS PubMed.
- A. Neamnark, O. Suwantong, R. K. Bahadur, C. Y. Hsu, P. Supaphol and H. Uludag, Mol. Pharm., 2009, 6, 1798 CrossRef CAS PubMed.
- H. M. Aliabadi, P. Mahdipoor and H. Uludag, Cancer Gene Ther., 2013, 20, 169 CrossRef CAS PubMed.
- H. M. Aliabadi, B. Landry, P. Mahdipoor, C. Y. Hsu and H. Uludag, Eur. J. Pharm. Biopharm., 2012, 81, 33 CrossRef CAS PubMed.
- I. R. Wickersham, D. C. Lyon, R. J. Barnard, T. Mori, S. Finke, K. K. Conzelmann and J. A. Young, Neuron, 2007, 53, 639 CrossRef CAS PubMed.
- M. Yokota, N. Tatsumi, O. Nathalang, T. Yamada and I. Tsuda, J. Clin. Lab. Anal., 1999, 13, 133 CrossRef CAS.
- Y. K. Oh, D. Suh, J. M. Kim, H. G. Choi, K. Shin and J. J. Ko, Gene Ther., 2002, 9, 1627 CrossRef CAS PubMed.
- P. Morata, M. I. Queipo-Ortuno and J. de Dios Colmenero, J. Clin. Microbiol., 1998, 36, 2443 CAS.
- Y. Takahashi, M. Nishikawa and Y. Takakura, Front. Biosci., 2012, 4, 133 Search PubMed.
- C. M. Varga, N. C. Tedford, M. Thomas, A. M. Klibanov, L. G. Griffith and D. A. Lauffenburger, Gene Ther., 2005, 12, 1023 CrossRef CAS PubMed.
Footnotes |
| † We dedicate this manuscript to Prof. Michael V. Sefton on the occasion of his 65th birthday. Prof. Sefton, with his independent spirit and unique insight, has been a guiding light in our scientific endeavours. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60200a |
| This journal is © The Royal Society of Chemistry 2014 |