The synergistic effect of a hybrid graphene oxide–chitosan system and biomimetic mineralization on osteoblast functions
D.
Depan
a,
T. C.
Pesacreta
b and
R. D. K.
Misra
*a
aBiomaterials and Biomedical Engineering Research Laboratory, Center for Structural and Functional Materials, Institute of Materials Research and Innovation, University of Louisiana at Lafayette, P.O. Box 44130, Lafayette, LA 70504-4130, USA. E-mail: dmisra@louisiana.edu; Fax: +(337) 482-1220; Tel: +(337) 482-6430
bBiology Department and Microscopy Center, University of Louisiana at Lafayette, P.O. Box 42451, Lafayette, LA 70504-2451, USA
First published on 30th October 2013
Abstract
Graphene oxide and chitosan are promising materials for tissue regeneration. The present study explores a novel biomimetic mineralization route employing a graphene oxide (GO)–chitosan (CS) conjugate as a template material for the biomineralization of hydroxyapatite (HAP). Structural and morphological studies involving X-ray diffraction, Fourier transform infrared spectroscopy, and electron microscopy indicated that extensive mineralization occurred in the CS–GO conjugate system because of strong electrostatic interactions between the functional groups (carboxyl groups of GO and amino groups of CS) and calcium ions in the simulated body fluid (SBF). The combination of chitosan–graphene oxide conjugate and biomineralization was advantageous in favorably modulating cellular activity (osteoblast functions: cell attachment, proliferation, actin, vinculin and fibronectin expression). It is concluded that biomineralized hydroxyapatite in the HAP–CS–GO system induced homogeneous spatial osteoblastic cell growth and quantitatively (e.g. area) and qualitatively (e.g. mineral-to-matrix ratio) increased mineralization in relation to the HAP–GO system. The data underscore that covalent linkage of HAP to chitosan influences osteoblastic cell differentiation, mineralization, and cell growth. The proposed system and the revelation of fundamental insights merit consideration in tissue engineering.
1. Introduction
There is significant interest in developing new biomaterial systems or enhancing the capabilities of existing biomaterials for biomedical applications. Hydroxyapatite (Ca10(PO4)6(OH)2; HAP) is the major constituent of natural bone with excellent biocompatibility and bioactivity.1,2 As a consequence, it has been widely explored for hard tissue engineering. An important and recognized weakness of hydroxyapatite is poor fracture toughness. Much effort has been made to eliminate this weakness by compounding with other engineering materials including polymers.3–5In this regard, an ideal material is one that provides mechanical integrity, and does not compromise the inherent bioactivity of hydroxyapatite. Carbonaceous materials with high chemical inertness are recognized to exhibit good biocompatibility.6 In this regard, graphene oxide is particularly attractive.7,8 Additionally, the functional side groups (hydroxyl, carboxyl and epoxides) bound to the surface of GO are expected to enhance the interfacial interaction between GO and the polymer matrix. Graphene oxide is proven to be biocompatible in vivo.9 Thus, by combining remarkable mechanical properties, large aspect ratio, and superior electrical conductivity,10 two-dimensional GO nanosheets are expected to provide strength–toughness combination to the GO–HAP system without decreasing its bioactivity. In this regard, the addition of GO to chitosan was shown to enhance the elastic modulus ∼200 times.11 Graphene oxide also promoted adhesion and proliferation of osteoblasts.12
Thus, combining hydroxyapatite (HAP) and GO is an attractive option for bone tissue engineering. However, the traditional approach of compounding of GO and HAP does not render the synthesis of a homogenous material. Recently, HAP and graphite nanosheets were combined using a spark plasma sintering process.13 In this approach, it is difficult to control the hierarchical structure because of the use of high temperatures. In contrast, biomimetic mineralization, involving the synthesis of organic–inorganic hybrid materials, is potentially attractive because of its similarity to materials of biological origin. The bioinspired process facilitates the design of advanced functional materials.14
More recently, multicomponent systems involving the use of three constituents (HAP–chitosan–PMMA/collagen/gelatin) were explored for bone tissue engineering.15–18 These systems indicated enhanced cell proliferation.19 Motivated by the superior biological response of multicomponent systems, we innovatively combined the characteristics and benefits of GO (presence of –COOH functional groups), chitosan (biocompatibility, cellular porous structure), and HAP (excellent biocompatibility and constituent of bone) for bone tissue engineering. This was accomplished by conjugating GO with chitosan via a covalent linkage followed by mineralization of HAP on the hybrid GO–chitosan system. The bioinspired surface induced nucleation of HAP in a simulated physiological environment. Furthermore, CS–GO exhibited significantly higher capability for HAP mineralization in contrast to pristine GO.
2. Experimental procedure
2.1. Materials
Low molecular weight CS (50 kDa, 80% degree of deacetylation) and chemical reagents including 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), N-hydroxyl succinimide (NHS), sodium hydroxide, and acetic acid were obtained from Aldrich, USA. Graphene oxide (GO) was obtained from Global Nanotech, Mumbai, India. The lateral width and thickness of GO are ∼2 μm and 1.2 nm, respectively. Alpha minimum essential medium (α-MEM) and fetal bovine serum (FBS) used were obtained from GIBCO, Invitrogen Corporation, USA. Penicillin–streptomycin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 IU–10000 μg ml−1), trypsin–EDTA (0.25% trypsin/0.53 mM EDTA) in Hank's buffered salt solution, phosphate buffer saline (PBS) without calcium and magnesium were obtained from American Type Cell Culture Collection (ATCC, Manassas, USA).
000 IU–10000 μg ml−1), trypsin–EDTA (0.25% trypsin/0.53 mM EDTA) in Hank's buffered salt solution, phosphate buffer saline (PBS) without calcium and magnesium were obtained from American Type Cell Culture Collection (ATCC, Manassas, USA).
2.2. Fabrication of chitosan–graphene oxide: conjugation of graphene with chitosan
Conjugation of graphene oxide (GO) with chitosan was accomplished via a covalent mechanism. First, carboxyl (–COOH) groups of GO were activated by EDC and NHS (Fig. 1). Briefly, a GO dispersion was prepared by sonicating GO for 2 h in ultrapure water. This solution was stable for weeks, confirming a homogenous dispersion of GO nanosheets in water. Next a solution of 0.03 M EDC and 0.06 M NHS was added to GO dispersion with continuous stirring for 1 h in order to activate the carboxyl (–COOH) groups of GO. The pH of the resulting solution was maintained at 7.0 using dilute sodium hydroxide. The activated GO solution was then added drop-wise to the CS solution (1% w/v in acetic acid) and heated at 60 °C for 30 min and allowed to cool to room temperature, followed by drying on a glass Petri plate at room temperature. After drying, small sections were cut from the film and neutralized using 1% (w/v) NaOH and subsequent washing with distilled water. The concentration of GO in CS was 3% (w/v). The above obtained solution of CS–GO was dried and cast as membranes and cut at 1 cm × 1 cm prior to mineralization. The details of the chemical conjugation of GO and CS are described in our earlier work.8 | ||
| Fig. 1 Schematic illustration of chemical conjugation of graphene oxide (GO) with chitosan (CS) and subsequent mineralization of hydroxyapatite on CS–GO in simulated body fluid (SBF) for 21 days. | ||
2.3. Biomineralization of hydroxyapatite on graphene oxide (GO) and chitosan–graphene oxide (CS–GO)
Pristine GO and CS–GO hybrids were used for biomineralization of hydroxyapatite (HAP). A simulated body fluid (5×) solution was used for in vitro mineralization in 50 cm3 Nalgene polypropylene centrifuge tubes. This concentration was chosen to trigger homogenous nucleation and growth of hydroxyapatite nanoparticles on the surface of GO and CS–GO. The centrifuge tubes were incubated in a thermostatic bath at 37 °C for 21 days. The composition of SBF was NaCl, KCl, K2HPO4, CaCl2, MgCl2, NaHCO3, Na2SO4, and (Na2O)3SiO2. The concentrations of these reagents were as follows: Na+ (141 mM), K+ (5 mM), Ca2+ (2.5 mM), Mg2+ (1 mM), Cl− (131 mM), HCO3− (5 mM), SO42− (1 mM), and HPO42− (1 mM). These reagents were dissolved in deionized water and the pH (7.25) was maintained by adding 1 M HCl and tris (hydroxymethyl) aminomethane. After the incubation period of 21 days, the samples were subjected to a thorough washing with deionized water consisting of several centrifugation and ultrafiltration cycles, followed by drying at room temperature.2.4. Structural and chemical characterization
The chemical composition and the structural analysis of the biomineralized hydroxyapatite were characterized by Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD), and scanning electron microscopy (SEM). In the case of SEM, the samples were sputter-coated with a thin film of gold to render them conducting for examination in a scanning electron microscope (SEM) (Hitachi S-3000).To study conjugation and composition, Fourier transform infrared (FT-IR) spectra were recorded using a JASCO FT-IR-480 spectrophotometer in the range between 4000 and 400 cm−1, with a resolution of 2 cm−1 using a KBr pellet method. A Rigaku D/max X-ray diffractometer (CuKα; 40 kV; 120 mA; λ = 1.5405; rate 2° min−1) was used for structural characterization.
2.5. Cell culture
Fluorescence microscopy was used to study the proliferation of pre-osteoblasts with the nucleic acid staining dye, Hoechst 33342 (bisbenzimide trihydrochloride), at a predetermined incubation time. The samples seeded with cells were rinsed with PBS and incubated with the dye (10 μg ml−1 of PBS) and observed using a fluorescence microscope (Nikon, ECLIPSE E-600 FN) with UV excitation and emission of 346/442. The cell nuclei appeared as blue fluorescence after staining with Hoechst 33342.
The protocol was as follows: pre-osteoblasts seeded on bare GO, HAP–GO, and HAP–CS–GO were cultured for 2 days and used for immunostaining. The cell-seeded samples were washed twice with PBS, fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 for 5 min and blocked with 0.1% bovine serum albumin (BSA) for 30 min. Subsequently, immunocytochemistry staining was performed for studying the expression level of fibronectin, actin, and vinculin.
Fibronectin was immunostained (labeled) with diluted mouse monoclonal antibody against fibronectin (1:800) (Sigma-Aldrich, St. Louis, MO, USA) followed by rabbit–antimouse FITC conjugated secondary antibody at a working dilution of 1:200 (Sigma-Aldrich, St. Louis, MO, USA). Then, cell nuclei were stained with DAPI (Chemicon International, Inc., Temecula, CA, USA) at a working dilution of 1:1000 for 5 min. The actin cytoskeleton and focal adhesion contacts were stained with a focal adhesion staining kit (Chemicon International, Inc., Temecula, CA, USA). Actin and vinculin double staining was performed by incubating cells in diluted primary antibody (anti-vinculin) in blocking solution (1:200) for 1 h and subsequently labeled with diluted secondary antibody (1:100) (goat–anti-mouse FITC conjugate) (Chemicon International, Inc., Temecula, CA, USA), for 45 min, and simultaneous incubation with diluted TRITC-conjugated phalloidin (1:400).20 Finally, cell nuclei were labeled with DAPI as described above. After immunostaining, samples were mounted on a glass slide using an anti-fade mounting solution while covering with a cover slip and examined by fluorescence microscopy (Leica DM 6000B and Leica DM 600M).
Quantitative analysis of the expression level of proteins (actin and vinculin) was carried out in terms of pixel-based density of the recorded micrographs using image analysis software (Image Pro). The pixel density was calculated in two different ways, cell-based expression (total pixels per cell number) and cell area-based expression (total pixels per total cell area).
Mineralization was also assessed using FTIR. On day 21, the cells were washed with PBS and distilled water and total cell and matrix lysate collected in distilled water using a cell scraper. The lysate was homogenized and 10 ml of lysate was dried and mixed with KBr for FTIR analysis. The absorbance of infrared light to the sample was recorded in the range of 600–4000 cm−1 at a resolution of 4 cm−1. The number of scans was 64.
The mineral-to-matrix ratio was quantified as the area ratio of the phosphate peak at 900–1200 cm−1 to the amide peak at 1590–1700 cm−1.21,22
3. Results and discussion
3.1. Structural analysis of hybrid HAP–CS–GO
The conjugation of graphene oxide (GO) with chitosan (CS) was carried out via amidation reaction involving the carboxyl (–COOH) groups of graphene oxide and amine (–NH2) groups of chitosan, and is schematically illustrated in Fig. 1. The mineralization of hydroxyapatite (HAP) on the surface of graphene oxide and chitosan–graphene oxide was obtained by soaking the constituents in 5× SBF having ionic concentration of human blood plasma.23The biomineralization process in a multicomponent or hybrid material system depends on the availability of functional groups.24–26 Graphene oxide influences biomineralization of HAP because of the functional groups (primarily –COOH, –OH and epoxy) on GO. Similarly, the presence of the functional group in chitosan (–NH2) influences the deposition of HAP.
FTIR is an appropriate technique to monitor the deposition of HAP on GO–CS. FTIR spectra of the mineralized HAP after 21 days are presented in Fig. 2a, which shows a high intensity of PO43− bands. The band at 980 cm−1 is the symmetric stretching mode ν1 (PO43−), 1110 and 1037 cm−1 bands correspond to the vibration mode ν3 (PO43−), while 603 and 566 cm−1 bands are associated with bending modes ν4 (PO43−).27 FTIR spectroscopy data confirm the nucleation of HAP in the CS–GO conjugate and also suggests that the combination of both CS and GO increases the mineralization of HAP, when the FTIR data of HAP–CS–GO are compared with HAP–GO.
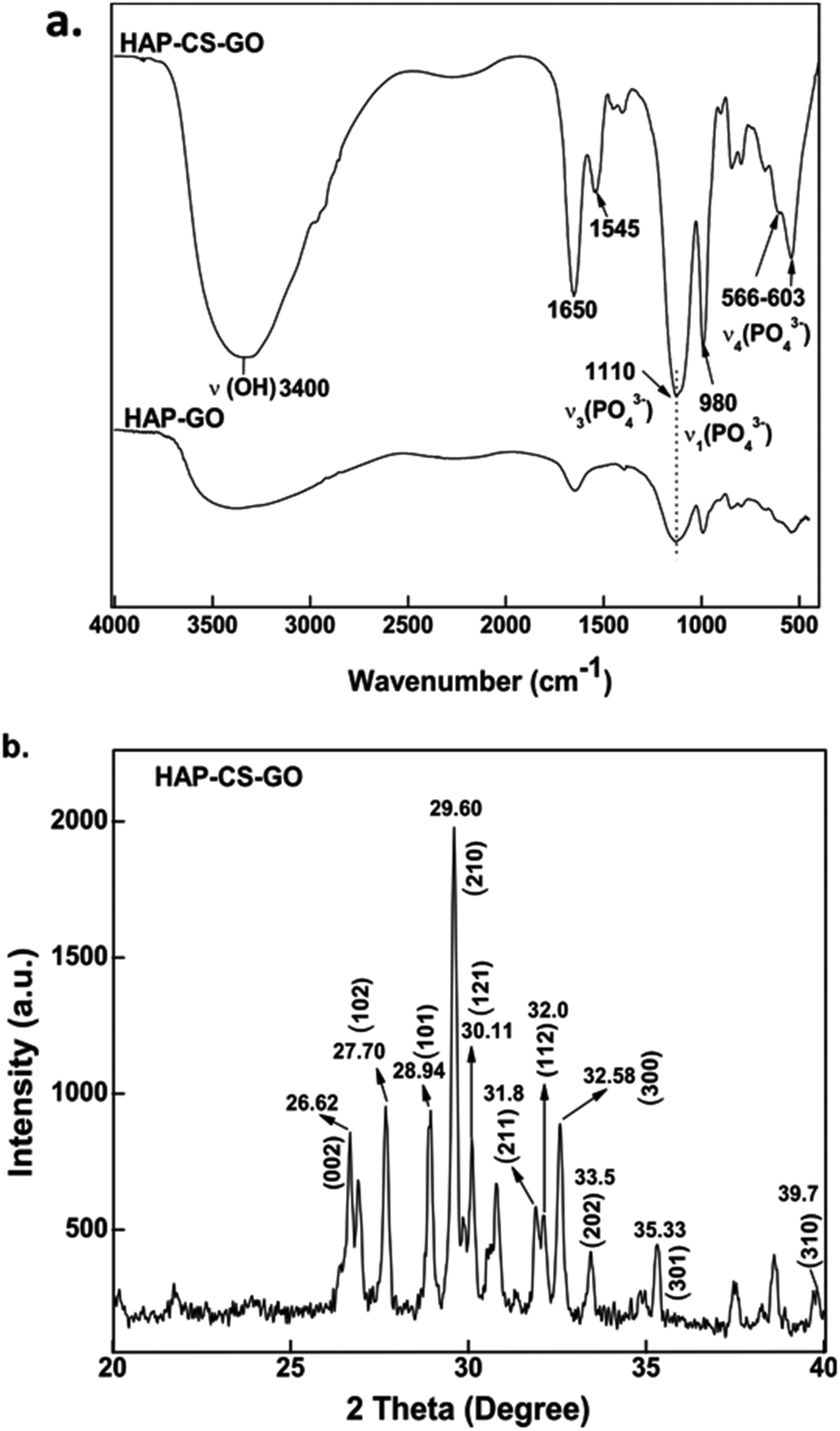 | ||
| Fig. 2 (a) Fourier transform infrared (FTIR) spectra obtained for hydroxyapatite (HAP) biomineralized onto chitosan–graphene oxide (CS–GO) and (b) wide-angle X-ray diffraction pattern of the biomineralized HAP on CS–GO system. | ||
The FTIR data were further corroborated by XRD. The XRD pattern recorded for HAP–CS–GO shows characteristic diffraction peaks of the hexagonal phase of HAP (JCPDS card, no. 09-0432), where the peaks at 31.8°, 32.0°, 33.5°, 29.60°, 28.94°, and 30.11° correspond to the (211), (300), (202), (002), (101), and (121) diffracted planes of HAP, respectively.
Further evidence for this conjecture is supported by morphological analysis. The change in the microstructure induced by biomineralization (21 days in SBF), as imaged via SEM, is presented in Fig. 3. A near uniform dispersion of mineralized HAP was observed for pure GO, while in the case of CS–GO system, a very fine dispersion of HAP particles was observed together with a few agglomerates of size ∼2 μm, and the nucleation of HAP was less uniform. The underlying reason is the atomistically flat surface of GO (AFM: roughness ∼1.3 nm). The fine dispersed particles of HAP in the case of CS–GO system is attributed to strong electrostatic interactions between NH2, –COOH, and Ca2+, which effectively triggers widespread nucleation of HAP. We conclude that the functional groups associated with GO and CS–GO enabled efficient interaction between the surface and mineral ions, and consequently promoted the nucleation of the high density of HAP particles.
 | ||
| Fig. 3 Scanning electron micrographs of hydroxyapatite (HAP) biomineralized on graphene oxide (GO) and chitosan–graphene oxide (CS–GO). | ||
3.2. Cell culture studies
 | ||
| Fig. 4 Fluorescence micrographs illustrating difference in initial cell attachment of pre-osteoblasts grown on pure GO, HAP–GO, and HAP–CS–GO. Cells were stained with nucleic acid dye, Hoechst 33342 (bisbenzimide hydrochloride, 1 mg ml−1) after 1 h and 24 h of culture period. A higher cell density with abundant cellular extensions is observed on pre-osteoblasts cultured on HAP–CS–GO compared to pure GO and HAP–GO, particularly after 24 h, signifying superior cytocompatibility of HAP–CS–GO. | ||
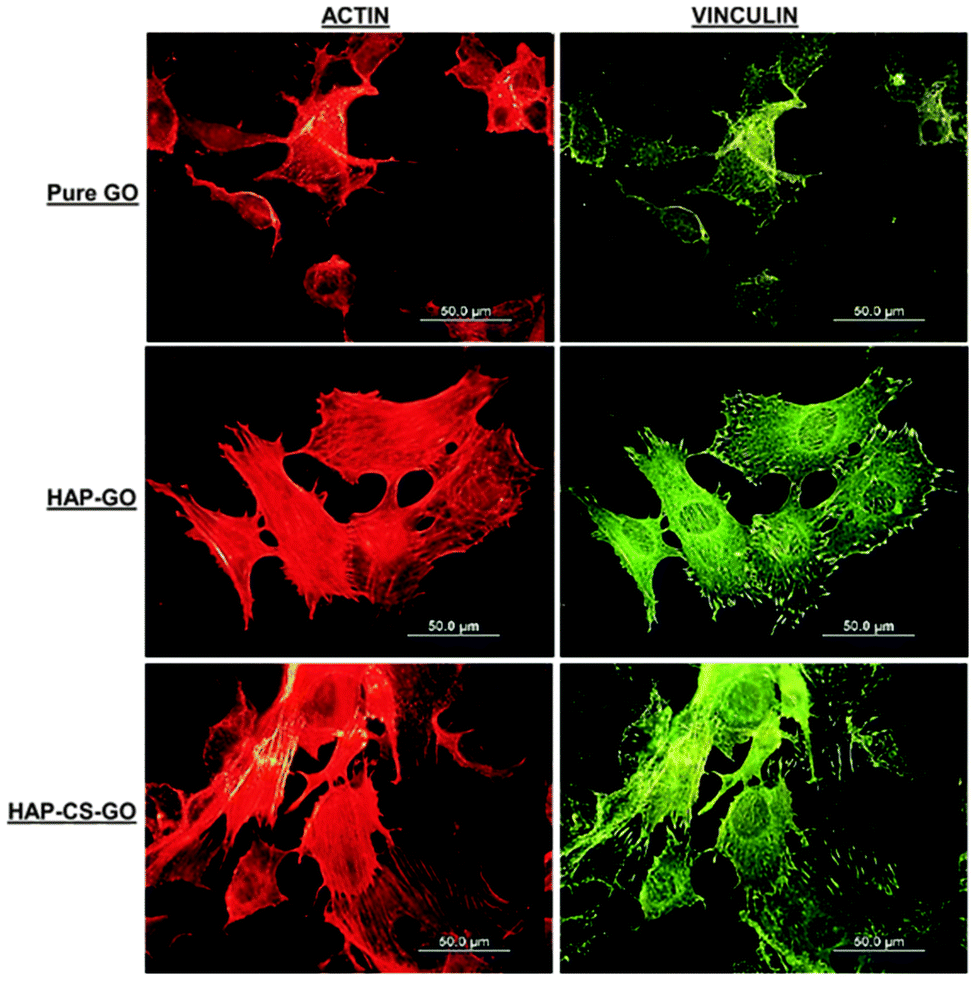 | ||
| Fig. 5 Micrographs illustrating organization and assessment of focal adhesion contacts and actin cytoskeleton of pre-osteoblasts after 2 day culture on pure GO, HAP–GO, and HAP–CS–GO. Vinculin shows a large number of focal contact sites in pre-osteoblasts grown on the surface of HAP–CS–GO compared to HAP–GO and pure GO. Note the higher number of focal adhesion points corresponds well with a high number of actin stress fibers on HAP–CS–GO. The scale bar in all the images is 50 μm. | ||
The data presented in Fig. 5 clearly suggest that biomineralization led to a well-organized actin cytoskeleton such that proliferation and spreading of osteoblasts was promoted on HAP–GO and HAP–CS–GO substrates. Osteoblasts on pure GO were slow to spread such that the cytoskeleton was less organized with a reduced level of expression and density of actin stress fibers and vinculin focal adhesion contacts. Cell spreading can be elicited by many factors. Here it correlates with biocompatibility and the positive aspects of the constituents of biomaterial constructs. Based on the observations presented in Fig. 4 and 5, CS certainly appears to have a positive impact on cellular activity in conjunction with HAP.
Calcium ions are advantageous in promoting cell growth.30,31 In contrast, high phosphate ion concentration inhibits cellular activity.32 Our data (Fig. 2 and 3) suggest that CS was instrumental in promoting mineralization of HAP on GO surfaces, such that CS and HAP had a synergistic effect on osteoblast function. In a manner similar to the behavior of actin and vinculin, the fibronectin (FN) expression of pre-osteoblasts in the case of HAP–CS–GO system was characterized by a distinct network-like pattern with highly proliferated cells present as cell clusters (Fig. 6). One of the earliest extracellular matrix molecules produced by osteoblasts following proliferation is fibronectin (FN). Fibronectin plays an important role in osteoblast morphogenesis, differentiation, and mineralization.33 The distribution pattern of FN is indicative of osteoblast's ability to synthesize, mineralize, and organize the extracellular matrix, irrespective of the nature of the system (GO, HAP–GO, HAP–CS–GO) (Fig. 6).
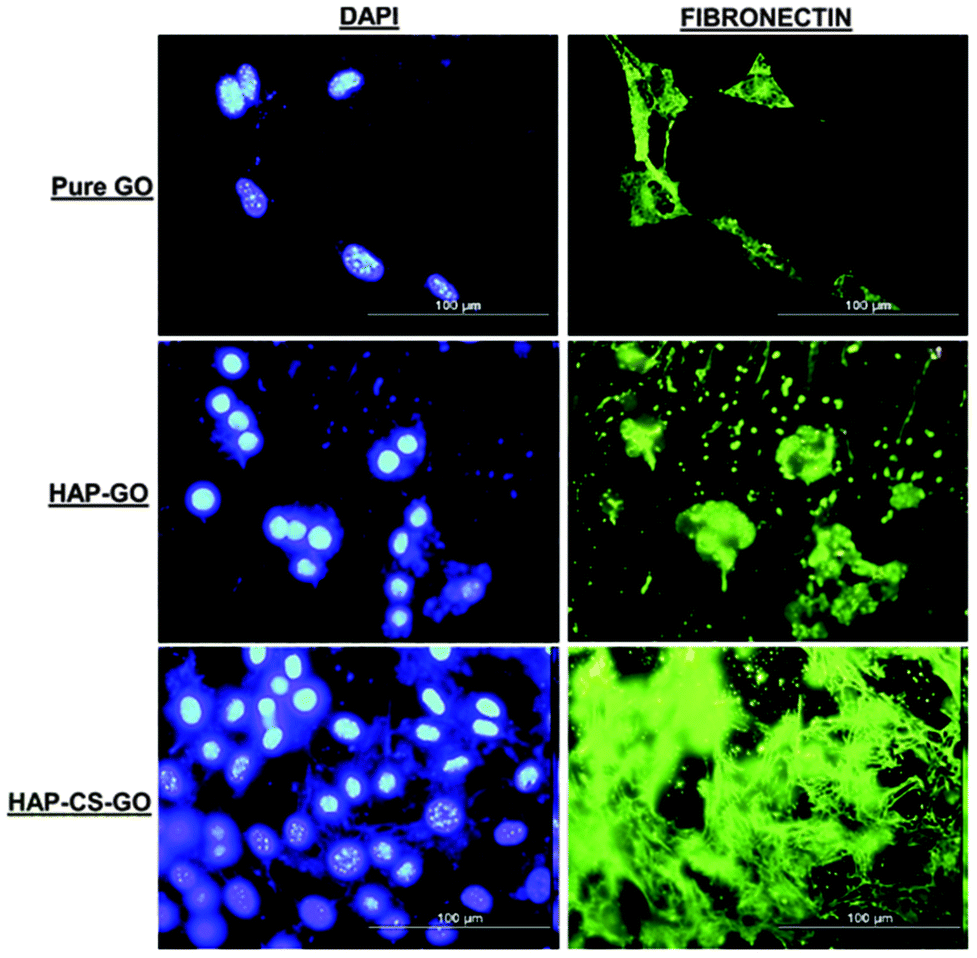 | ||
| Fig. 6 Fluorescence micrographs illustrating immunocytochemistry of fibronectin expression (green) and cell nuclei (blue) of pre-osteoblasts after 2 day culture on pure GO, HAP–GO, and HAP–CS–GO. A distinct network distribution with proliferated cells present as clusters with higher fluorescence intensity was observed for pre-osteoblasts grown on the surface of HAP–CS–GO after labeling cell-nuclei with DAPI. The scale bar in all the images is 100 μm. | ||
After 2 days of cell culture, FN was observed in all the three systems. However, the expression of FN in the HAP–CS–GO system was significantly greater in comparison to other systems such as PMMA-HAP, reported in the literature34 implying that CS is a potential material for bone tissue engineering. A highly organized extracellular matrix, rich in non-collagenous components, is necessary in the synthesis of extracellular matrix protein for subsequent mineralization of the matrix. Our observations underscore the impact of CS in the composite HAP–CS–GO system. Conversely, FN is shown to play a critical role in the formation of HAP crystals and biomineralization.35 Thus, HAP may interact with FN in a synergistic manner to provide osteoblasts with inductive signals to promote ECM and mineralization.
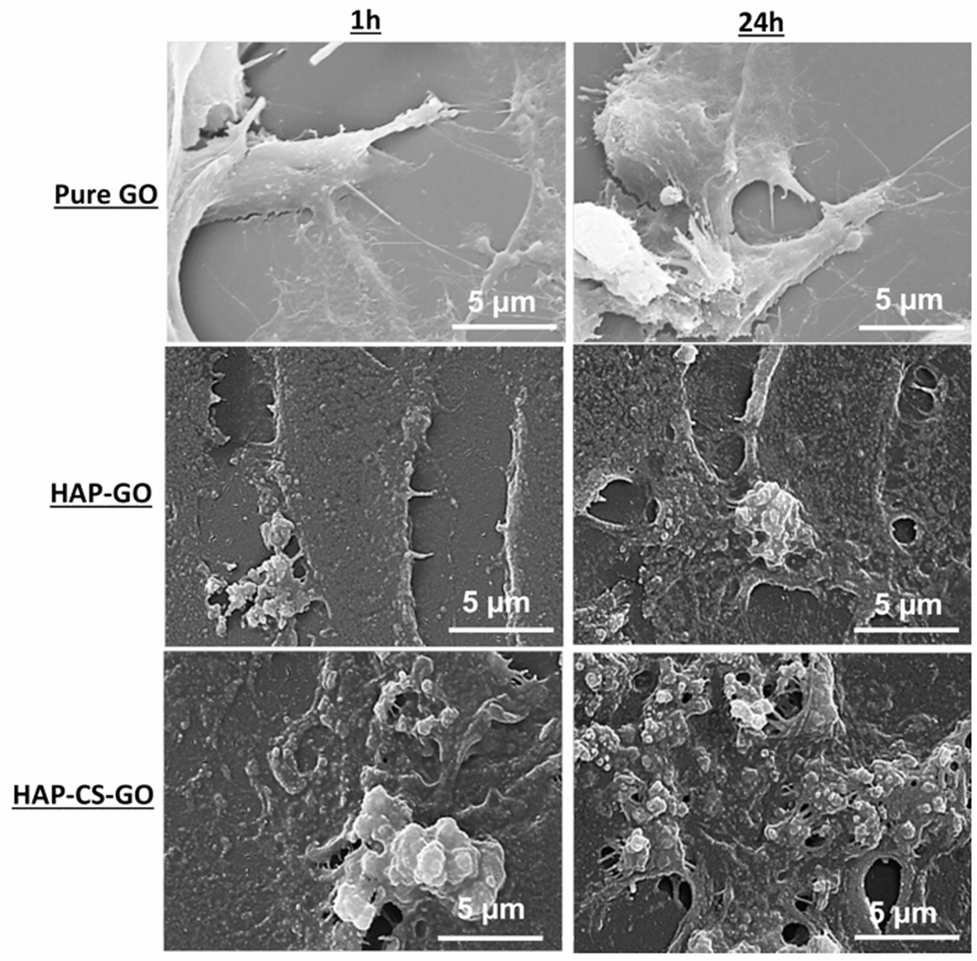 | ||
| Fig. 7 Scanning electron micrographs illustrating morphology of pre-osteoblasts seeded on pure GO, HAP–GO, and HAP–CS–GO after 1 h and 24 h. Pre-osteoblasts cultured on HAP–CS–GO had extensive spreading (sheet-like) and greater extracellular matrices than cells grown on HAP–GO. | ||
The merging of cells and ECM in the HAP–GO system and to a significantly greater extent in the HAP–CS–GO system is indicative of cellular activity in the respective mineralized samples. Furthermore, the cells developed polygonal cytoplasmic processes extending 20–30 μm. These cytoplasmic extensions are regions of dense cartilaginous layers that comprise a network or bundles of actin-containing micro-filaments, which enable movement of penetrating cells along the substratum and facilitate cell–cell communication for differentiation to occur.36,37 In summary, the HAP–CS–GO system exhibited significantly greater development of cytoplasmic processes, such that a confluent layer was formed. These results are significant and provide the first evidence of growth of pre-osteoblasts in HAP biomineralized CS–GO systems. Given that self-organization and mineralization of cartilage and bone are of significant interest, the current study provides new insights with regard to the applicability of GO and CS in cartilage and bone tissue engineering.
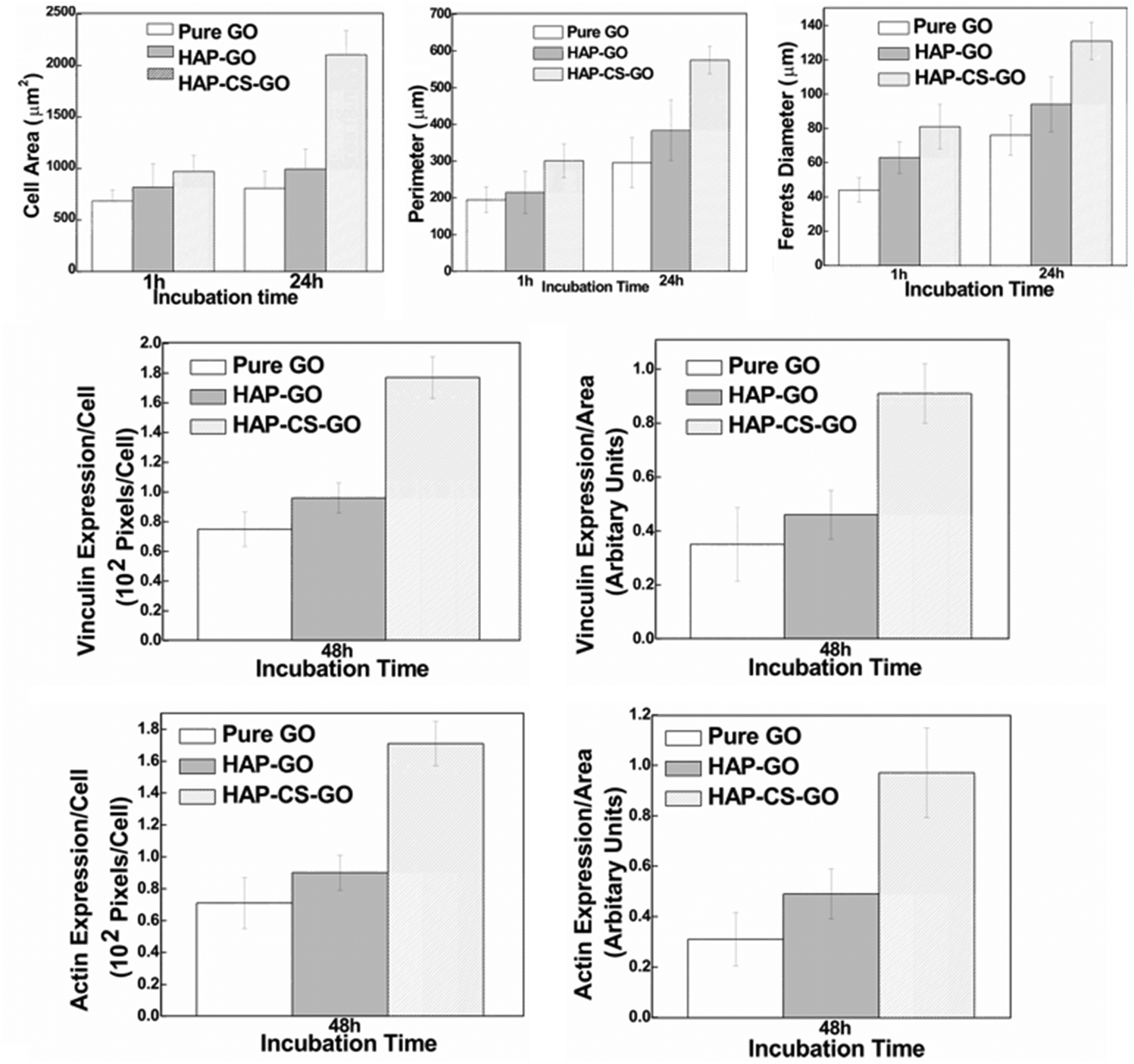 | ||
| Fig. 8 Cytomorphometric evaluation of various parameters (cell area, perimeter, Feret's diameter) using representative micrographs presented in Fig. 7 and evaluating level of proteins (actin and vinculin, Fig. 6) by densitometry approach using the image analysis software (NIH). Data are (mean ± sd (n = 5) p < 0.05), indicating a statistical difference between pure GO, HAP–GO, and HAP–CS–GO. | ||
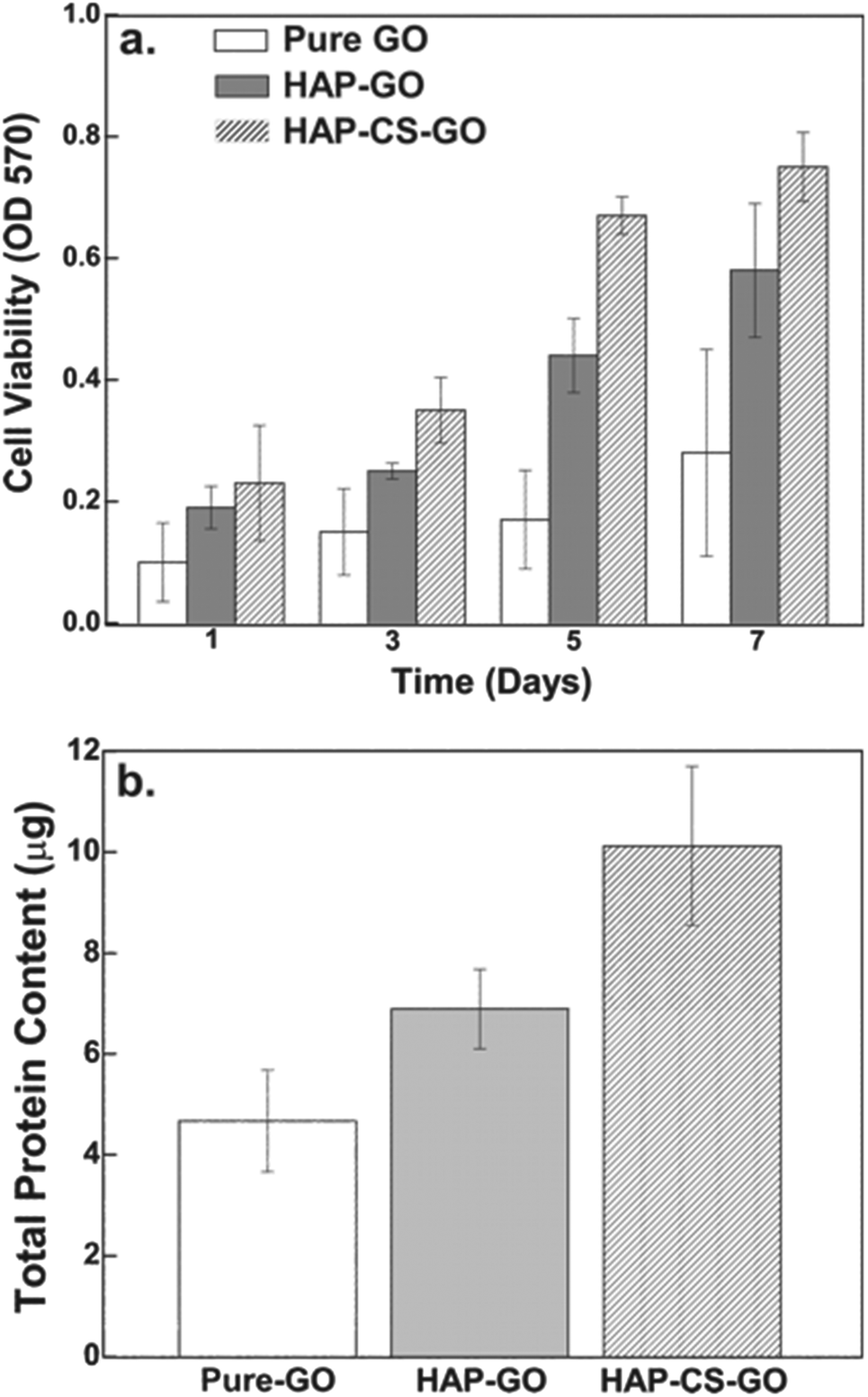 | ||
| Fig. 9 (a) Histogram representing mitochondrial activity on pure GO, HAP–GO, and HAP–CS–GO, as a function of time, measured by MTT assay. (b) Quantitative assessment of proteins from pre-osteoblasts incubated on pure GO, HAP–GO, and HAP–CS–GO by Mini Bradford protein assay. Histograms indicate higher total protein content in cells grown on HAP–CS–GO as compared to pure GO and HAP–GO. | ||
Similarly, the total protein content as determined by Mini Bradford assay was significantly greater for the HAP–CS–GO system as compared to the HAP–GO and pure GO system after 2 days of culture (Fig. 9b). This is consistent with immunofluorescence studies and points toward the improved biological response associated with biomineralized HAP in the CS–GO system.
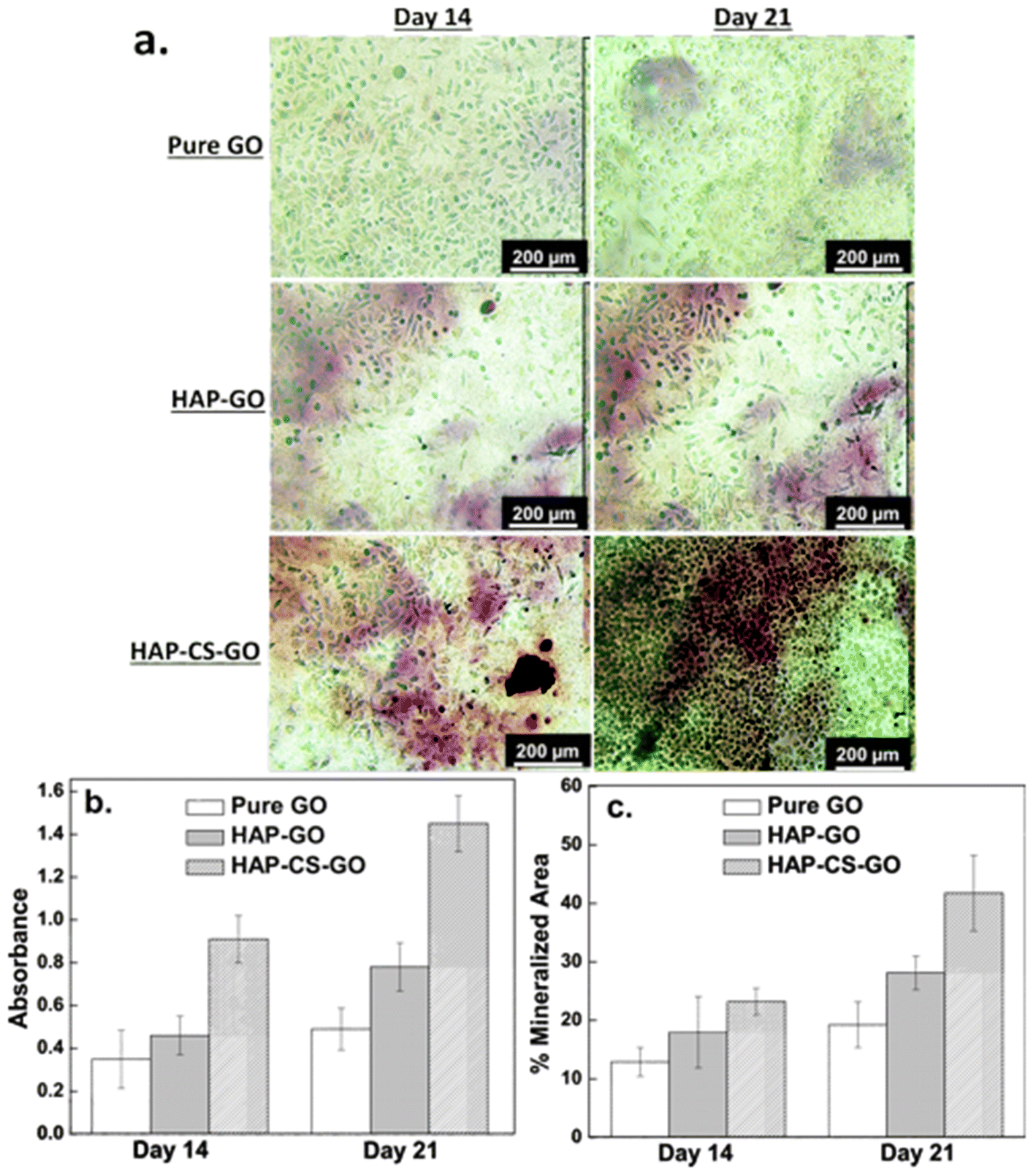 | ||
| Fig. 10 (a) Alizarin red (AR) staining of pre-osteoblasts cells cultured on pure GO, HAP–GO, HAP–CS–GO surfaces for 14 and 21 days. (b) Quantitative estimation of AR dye by, and (c) mineralized area (%) illustrating enhanced mineralization on HAP–CS–GO. | ||
On day 14, the number of red pixels followed the sequence: HAP–CS–GO > HAP–GO > GO. A marked increase in the number of the red pixels was observed in all the three systems on day 21. Compared to pure GO and HAP–GO, the cells cultured in the HAP–CS–GO system were observed to be in intimate contact with the deposited mineral. As shown in Fig. 10b, ARS staining was significantly more prominent in the HAP–CS–GO system compared to the HAP–GO system on days 14 and 21 (p < 0.005). This is most likely to be related to the presence of CS, which enhances calcium binding and matrix deposition.
A similar trend was observed when the mineralized area was quantified (Fig. 10c). The mineralized area increased from day 14 to day 21 for all the three systems, but was statistically more significant for the HAP–CS–GO system (p < 0.005). Mineralization is considered to be the strongest indicator of osteoblast differentiation and osteogenesis. HAP–CS–GO promoted the formation of nodules displaying a higher degree of mineralization than HAP–GO. Promotion of mineralization is considered the “gold standard” in assays and confirms that HAP–CS–GO provides an environment conducive to osteogenesis. Here again, the osteogenic properties of CS are responsible for the highly mineralized nodules that form in the HAP–CS–GO composite material system.
FTIR spectroscopy after 21 days of culture revealed that osteoblasts cultured in HAP mineralized systems exhibited distinct, bone mineral specific peaks (phosphate ν1 and ν3 peaks at 900–1200 cm−1, arrows in Fig. 11a). Osteoblasts displayed amide I peaks corresponding to the collagen matrix at 1590–1700 cm−1. After 21 days of culture, the mineral-to-matrix ratio, quantified as the area ratio of phosphate to amide peaks, was significantly greater for the HAP–CS–GO system relative to cells in the HAP–GO system. Both the quantity of mineralization (e.g., mineralized area) and quality of mineralization (e.g., mineral-to-matrix ratio) are important in evaluating the bone cell differentiation of osteoblastic cells.42,43 For instance, it was demonstrated that staining techniques alone are sometimes not adequate to identify bone minerals.42 The study with MC3T3 cells showed positive von Kossa staining but bone-like minerals were not detected with FTIR.42 The fetal rat calvarial cells cultured in vitro displayed a mineral-to-matrix ratio between 1.9 and 3.7, depending on the concentration of ascorbic acid and β-glycerophosphate. These ratios were lower than the control mineral-to-matrix ratio of 5.04 obtained for 16-day-old rat calvaria from intact thin bone sections. Moreover, these ratios were lower than the control ratio of 4.35 obtained for normal intact bovine cortical bone.42 The mineral-to-matrix ratios obtained in our study were 1.76 (HAP–CS–GO), 1.43 (HAP–GO), and 0.68 (pure GO), and are lower compared to reported control ratios (5.04 or 4.35). If we consider our data and the data reported in the literature, we conclude that in vitro culture of osteoblast-like cells may result in mineral-to-matrix ratios lower than that of the intact bone (in vivo). This suggests an important consideration in skeletal tissue engineering; i.e., an optimal cell culture environment is important to induce bone forming cells to produce strong biomimetic bone-like minerals exhibiting a mineral-to-matrix ratio similar to the actual bone.
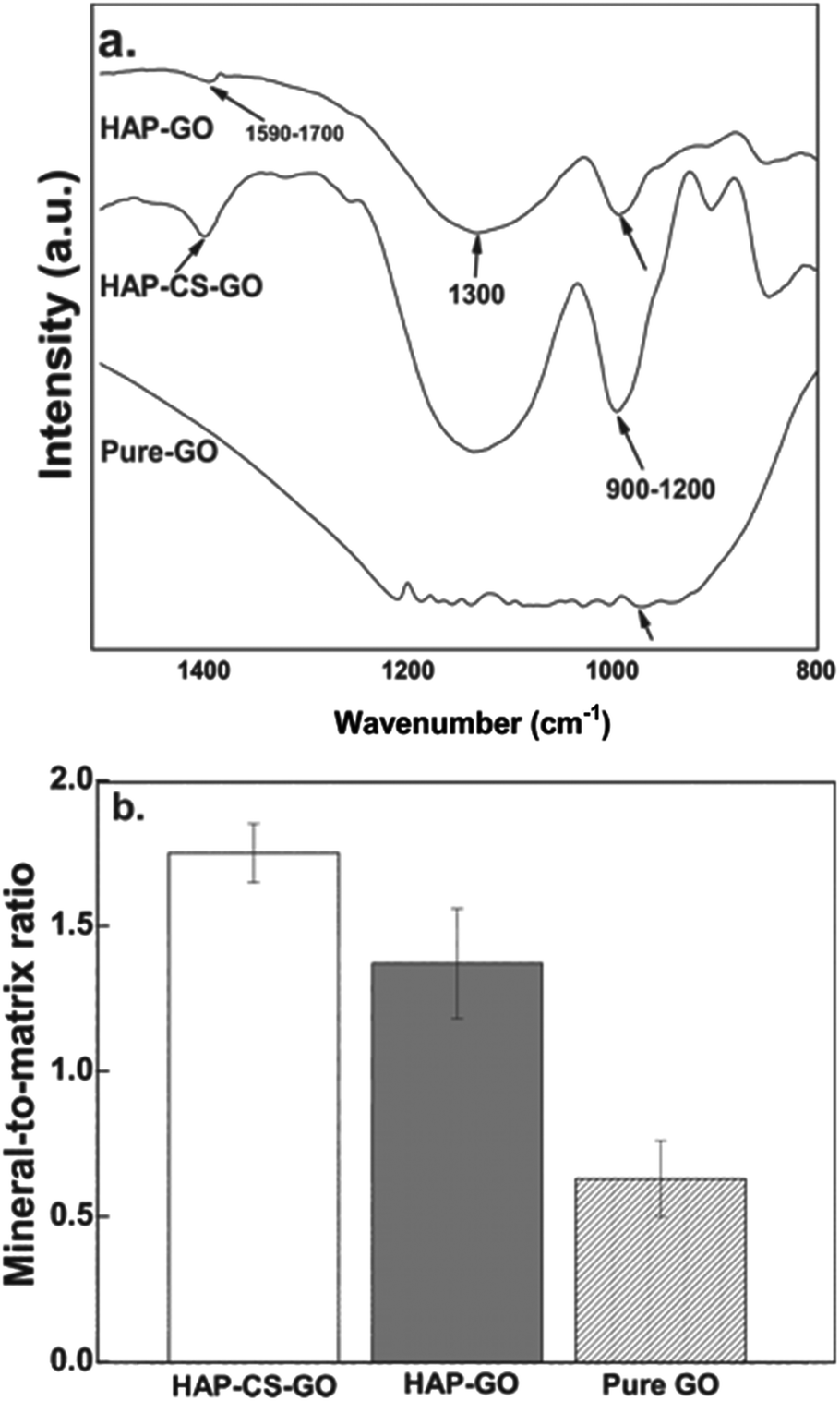 | ||
| Fig. 11 (a) Representative FTIR spectra of osteoblasts cultured for 21 days on pure GO, HAP–GO, and HAP–CS–GO surfaces. Arrows at 900–1200 cm−1 represent phosphate and mineralization peaks, while arrowheads at 1590–1700 cm−1 represent collagen amide i peaks. (b) Mineral-to-matrix-ratio of osteoblast cells after 21 days of culture calculated by the area ratio of phosphate and amide peaks. | ||
To the best of our understanding, this is the first study that explores the in vitro behavior of pre-osteoblasts in the biomineralized CS–GO system with advantages over other multicomponent based composites reported in the literature because of superior biocompatibility of CS. This study also strengthens our understanding of HAP. Given that the mineralization of HAP on GO and CS–GO is simple, feasible, and occurs under mild conditions without employing any extraneous toxic crosslinking agents (such as glyoxal, glutaraldehyde, and diepoxy compounds), we believe that such a system has the potential for application in wound management, drug delivery, and tissue engineering.
4. General conclusions
The study outlines the successful mineralization of the chitosan–graphene oxide (CS–GO) conjugate system in simulated body fluid (SBF). Fourier transform infrared spectroscopy indicated extensive biomineralization of the CS–GO system as compared to GO and is attributed to stronger electrostatic interactions between the functional groups present in the CS–GO system and calcium ions. The combination of the chitosan–graphene oxide conjugate and biomineralization is beneficial in favorably modulating cellular activity (osteoblast functions: cell attachment, proliferation, actin, vinculin and fibronectin expression). Overall, the mineralization of the CS–GO system in the synthesis of a hybrid composite exhibits superior osteoblasts compatibility compared to pure GO and HAP–GO systems.References
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Biomimetic systems for hydroxyapatite mineralization inspired by bone and enamel, Chem. Rev., 2008, 108(11), 4754–4783 CrossRef CAS PubMed.
- S. Dasgupta, S. S. Banerjee, A. Bandyopadhyay and S. Bose, Zn and Mg doped hydroxyapatite nanoparticles for controlled release of protein, Langmuir, 2010, 26, 4958–4964 CrossRef CAS PubMed.
- W. Bonfield, M. D. Grynpas, A. E. Tully, J. Bowman and J. Abram, Hydroxyapatite reinforced polyethylene- a mechanically compatible implant material for bone replacement, Biomaterials, 1981, 2, 185–186 CrossRef CAS.
- W. Suchanek and M. Yoshimura, Processing and properties of hydroxyapatite-based biomaterials for use as hard tissue replacement implants, J. Mater. Res., 1998, 13, 94–117 CrossRef CAS.
- X. Zheng, M. Huang and C. Ding, Bone strength of plasma-sprayed hydroxyapatite/Ti composite coatings, Biomaterials, 2000, 21, 841–849 CrossRef CAS.
- R. Olivares, S. E. Rodil and H. Azarte, Osteoinduction properties of graphite-like amorphous carbon films evaluated in-vitro, Diamond Related Mater., 2007, 16, 1858–1867 CrossRef CAS PubMed.
- T. Kulia, S. Bhadra, D. Yao and N. H. Kim, Recent advances in graphene based polymer composites, Prog. Polym. Sci., 2010, 35(11), 1350–1375 CrossRef PubMed.
- D. Depan, B. Girase, S. H. Shah and R. D. K. Misra, Structure-process-property relationship of the polar graphene-oxide-mediated cellular response and stimulated growth of osteoblasts on hybrid chitosan network structure nanocomposite scaffolds, Acta Biomater., 2011, 7(9), 3432–3445 CrossRef CAS PubMed.
- X. Zhang, J. Yin, C. Peng, W. Hu, Z. Zhu, W. Li, C. Fan and Q. Huang, Distribution and biocompatibility studies of graphene oxide in mice after intravenous administration, Carbon, 2011, 49, 986–995 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. Suk, J. R. Potts and R. S. Ruoff, Graphene and graphene oxide: synthesis, properties, and applicationsAdv. Mater. 2010, 2235, 3906–3924 Search PubMed.
- H. Fan, L. Wang, K. Zhao, N. Li, Z. Shi, Z. Ge and Z. Jin, Fabrication, mechanical properties, and biocompatibility of graphene-reinforced chitosan composite, Biomacromolecules, 2010, 11, 2345–2351 CrossRef CAS PubMed.
- S. Agarwal, X. Zhou, F. Ye, Q. He, G. C. K. Chen, J. Soo, F. Boey, H. Zhang and P. Chen, Interfacing live cells with nanocarbon substrate, Langmuir, 2010, 26, 2244–2247 CrossRef CAS PubMed.
- J. T. Zhu, H. M. Wong, K. W. K. Yeung and S. C. Tjong, Spark plasma sintered hydroxyapatite/graphite nanosheet and hydroxyapatite/multiwalled carbon nanotube composites: mechanical and in vitro cellular properties, Adv. Eng. Mater., 2010, 13, 336–341 CrossRef.
- N. Huebsch and D. J. Mooney, Inspiration and applications in the evolution of biomaterials, Nature, 2009, 462, 426–432 CrossRef CAS PubMed.
- S. Kim, Y. Kim, T. Yoon, S. Park, I. Cho and E. Kim, The characteristics of a hydroxyapatite–chitosan–PMMA bone cement, Biomaterials, 2004, 25(26), 5715–5723 CrossRef CAS PubMed.
- J. Li, Y. Chen, Y. Yin, F. Yao and K. Yao, Modulation of nano-hydroxyapatite size via formation on chitosan–gelatin network film in situ, Biomaterials, 2007, 28(5), 781–790 CrossRef CAS PubMed.
- L. Zhang, P. Tang, W. Zhang, M. Xu and Y. Wang, Effect of chitosan as a dispersant on collagen-hydroxyapatite composite matrices, Tissue Eng. Part C Methods, 2010, 16(1), 71–79 CrossRef CAS PubMed.
- K. Katti, D. Katti and R. Dash, Synthesis and characterization of a novel chitosan/montmorillonite/hydroxyapatite nanocomposite for bone tissue engineering, Biomed. Mater., 2008, 3(3), 4122 CrossRef PubMed.
- (a) Y.-H. Nien and C. Huang, The mechanical study of acrylic bone cement reinforced with carbon nanotube, Mater. Sci. Eng., B, 2010, 169(1–3), 134–137 CrossRef CAS PubMed; (b) J. Li, N. Ren, J. Qiu, X. Mou and H. Liu, Graphene oxide-reinforced biodegradable genipin-cross-linked chitosan fluorescent biocomposite film and its cytocompatibility, Int. J. Nanomed., 2013, 8, 3415–3426 CrossRef CAS PubMed.
- R. D. K. Misra, D. Depan and J. S. Shah, Structure-process-functional property relationship of nanostructured carbon mediated cellular response for soft-tissue reconstruction and replacement, Acta Biomater., 2012, 8(5), 1908–1917 CrossRef CAS PubMed.
- L. F. Bonewald, S. E. Harris, J. Rosser, M. R. Dallas, S. L. Dallas and N. P. Camacho, Von Kossa staining alone is not sufficient to confirm that mineralization in vitro represents bone formation, Calcif Tissue Int., 2003, 72, 537–547 CrossRef CAS PubMed.
- E. A. Nauman, D. M. Ebenstein, K. F. Hughes, L. Pruitt, B. P. Halloran and D. D. Bikle, Mechanical and chemical characteristics of mineral produced by basic fibroblast growth factor-treated bone marrow stromal cells in vitro, Tissue Eng., 2002, 8, 931–939 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, How useful in SBF in predicting in vivo bone bioactivity, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- A. W. Xu, Y. Ma and H. Colfen, Biomimetic mineralization, J. Mater. Chem., 2007, 17, 415–449 RSC.
- D. K. James and G. Antonios, Review: mineralization of synthetic polymer scaffold for bone tissue engineering, Tissue Eng., 2007, 13, 927–938 CrossRef PubMed.
- M. Tanahashi and T. Matsuda, Surface functional group dependence on apatite formation on self-assembled monolayers in a simulated body fluid, J. Biomed. Mater. Res., 1997, 34, 305–315 CrossRef CAS.
- H. Cao, L. Zhang, H. Zheng and Z. Wang, Hydroxyapatite nanocrystals for biomedical applications, J. Phys. Chem. C, 2010, 114, 18352–7 CAS.
- H. Wang and C. A. Blitterswijk, The role of three-dimensional polymeric scaffold configuration on the uniformity of connective tissue formation by adipose stromal cells, Biomaterials, 2010, 31(15), 4322–4329 CrossRef CAS PubMed.
- Q. Chen, M. S. Kinch, T. H. Lin, K. Burridge and R. L. Juliano, Integrin-mediated cell adhesion activates mitogen-activated protein kinase, J. Biol. Chem., 1994, 269(43), 26602–5 CAS.
- H. S. Cheung, M. T. Story and D. J. McCarty, Mitogenic effects of HA and calcium pyrophosphate dihydrate crystals on cultured mammalian cells, Arthritis Rheum., 1984, 27(6), 665–674 CrossRef.
- H. S. Cheung and D. J. McCarty, Mitogenesis induced by calcium containing crystal: role of intracellular dissolution, Exp. Cell Res., 1985, 157, 63–70 CrossRef CAS.
- C. Knabe, R. Gildenhaar, G. Berger, W. Ostapowicwz, R. Fitzer, R. J. Radlanski and U. Gross, Morphological evaluation of osteoblasts cultured on different calcium phosphate ceramics, Biomaterials, 1997, 18(20), 1339–1347 CrossRef CAS.
- A. M. Moursi, C. H. Damsky, J. Lull, D. Zimmerman, S. B. Doty, S. Aota and R. K. Globus, Fibronectin regulates calvarial osteoblast differentiation, J. Cell Sci., 1996, 109, 1369–1380 CAS.
- A. M. Moursi, A. V. Winnard, P. L. Winnard, J. J. Lannutti and R. R. Seghi, Enhanced osteoblast response to a polymethylmethacrylate-hydroxyapatite composite, Biomaterials, 2002, 23, 133–144 CrossRef CAS.
- G. Daculsi, P. Pliet, M. Cottrel and G. Guicheux, Role of fibronectin during biological apatite crystal nucleation: ultrastructural characterization, J. Biomed. Mater. Res., 1997, 47, 228–233 CrossRef.
- H. Wang and C. A. Blitterswijk, The role of three-dimensional polymeric scaffold configuration on the uniformity of connective tissue formation by adipose stromal cells, Biomaterials, 2010, 31, 4322–4329 CrossRef CAS PubMed.
- M. C. Siebers, P. J. Brugge, X. F. walboomers and J. A. Jansen, Integrins as linker proteins between osteoblasts and bone replacing materials. A critical review, Biomaterials, 2005, 26, 137–146 CrossRef CAS PubMed.
- F. Denizot and R. Lang, Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability, J. Immunol. Methods, 1986, 89, 271–277 CrossRef CAS.
- T. A. Owen, M. Aronow, V. Shalhoub, L. M. Barone, L. Wilming, M. S. Tassinari, M. B. Kennedy, S. Pockwinse, J. B. Lian and G. S. Stein, Progressive development of the rate osteoblast phenotype in vitro: reciprocal relationships in expression of genes associated with osteoblast proliferation and differentiation during formation of the bone extracellular matrix, J. Cell Physiol., 1990, 143, 420–430 CrossRef CAS PubMed.
- P. V. Hauschka, J. B. Lian, D. E. C. Cole and C. M. Gundberg, Osteocalcin and matrix Gla protein: vitamin K dependent proteins in bone, Physiol. Rev., 1989, 69, 990–1047 CAS.
- J. Y. Choi, B. H. Lee, K. B. Song, R. W. Park, I. S. Kim, K. Y. Sohn, J. S. Jo and H. M. Ryoo, Expression patterns of bone-related proteins during osteoblastic differentiation in Mc3T3-E1 cells, J. Celzl. Biochem., 1996, 61, 609–618 CrossRef CAS.
- L. F. Bonewald, S. E. Harris, J. Rosser, M. R. Dallas, S. L. Dallas and N. P. Camacho, von Kossa staining alone is not sufficient to confirm that mineralization in vitro represents bone formation, Calcif Tissue Int., 2003, 72, 537–547 CrossRef CAS PubMed.
- E. A. Nauman, D. M. Ebenstein, K. F. Hughes, L. Pruitt, B. P. Halloran and D. D. Bikle, Mechanical and chemical characteristics of mineral produced by basic fibroblast growth factor-treated bone marrow stromal cells in vitro, Tissue Eng., 2002, 8, 931–939 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |