Novel stem-loop RNA and drug-bearing DNA hybrid nanostructures specific to LNCaP prostate carcinoma†
Seung Won
Shin‡
a,
Woo Chul
Song‡
a,
A Ra
Kim
a,
Seung-Woo
Cho
b,
Dong-Ik
Kim
c and
Soong Ho
Um
*ad
aSchool of Chemical Engineering, Sungkyunkwan University, Suwon, Gyeonggi-do 440-746, South Korea
bDepartment of Biotechnology, Yonsei University, Seoul, 120-749, South Korea
cDivision of Vascular Surgery, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, 135-710, South Korea
dSKKU Advanced Institute of Nanotechnology (SAINT), Sungkyunkwan University, Suwon, Gyeonggi-do 440-746, South Korea. E-mail: sh.um@skku.edu; Fax: +82 31 290 7272; Tel: +82 31 290 7348
First published on 23rd August 2013
Abstract
Advances in nanotechnology have resulted in the introduction of new materials for therapeutic and diagnostic purposes. In particular, DNA and RNA are viewed as representative and generic nano-blocks because of their physiochemical characteristics of specificity and nanoscopic-level accuracy. In addition, the intrinsic biocompatibility of DNA and RNA and their immune stimulation effects make these molecules ideal candidates for the rational design of novel bio-drug molecules. Recently, we reported novel RNA–DNA hybrid stem-loop structures that target and are endocytosed by LNCaP prostate cancer cells with high specificity. To effectively ligate the DNA and RNA modules in this research, we thoroughly evaluated and optimized several ligation parameters, and observed that we could enhance the ligation efficacy by changing the overhang sequences. A change in sequence information (GCAT) resulted in a 4-fold increase in ligation efficiency in comparison with other ligation factors. To determine the in vitro cellular targeting ability of the nanostructures, RNA–DNA hybrid constructs were complexed with gold nanorods (AuNRs), and the ability of these nanorods to target prostate cancer cells was highest at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 molar ratio of LNCaP cancer-specific looped A10 RNA to stem-DNA. Furthermore, doxorubicin (Dox) as a representative anti-prostate cancer therapeutic was loaded into the DNA–RNA hybrid nanostructures. Our results indicate that RNA–DNA hybrid constructs are effective anti-prostate cancer drug delivery platforms and can be employed for both discovery and delivery.
:10 molar ratio of LNCaP cancer-specific looped A10 RNA to stem-DNA. Furthermore, doxorubicin (Dox) as a representative anti-prostate cancer therapeutic was loaded into the DNA–RNA hybrid nanostructures. Our results indicate that RNA–DNA hybrid constructs are effective anti-prostate cancer drug delivery platforms and can be employed for both discovery and delivery.
1. Introduction
With the advent of nanotechnology, a variety of natural subdivisions or synthetic replicates learning from nature have been built using biological ingredients such as DNA, RNA, peptides, proteins, and even cells as building blocks.1 They have been used to study biological mechanisms and metabolism2 and investigate disease processes.3 In particular, DNA and RNA are among the most promising material resources in terms of their characteristic features4 of flexible rigidity, structural variability, simple topological conformation, intrinsic biocompatibility, precise nanoaccuracy, and easy manipulation via restriction enzymes. Therefore, various nucleic acid nanostructures, including box-,5 tube-,6 tic-tac-toe-,7 honeycomb-,8 network-,9 origami-,10,11 and emoticon-type nanostructures, have been synthesized.12These nanostructures have been utilized in several scientific and engineering studies ranging from basic science studies to medical applications.13,14 For instance, dendritic DNA molecules formed by the assembly of Y-shaped DNA blocks were used for the diagnostic and multiplexed detection of toxic pathogens within a few minutes.15 By using anisotropic sequence design strategies, DNA nanostructures with several modules positioned in different directions have also been designed.16 A three-dimensional DNA-networked gel matrix9 that encapsulated a variety of drug molecules for sustained drug delivery was shown to be effective in in vitro transplantation therapy. By exploiting the genetic code, several functional and therapeutic proteins that could be of use for salutary diagnostic applications have been generated.17 In addition, the formation of nanoparticles through lipid formulations, polymer processing, and immunoengineering18 yielded DNA-based B- or T-cell therapies that effectively destroyed malicious melanomas in a murine model both in vitro and in vivo.19
Recently, our group reported novel stem-loop RNA and drug-bearing DNA hybrid nanostructures conjugated with gold nanorods that target LNCaP cancer cells and that can store anti-cancer therapeutics for potential prostate cancer therapy.20 To achieve the advanced therapeutic system, two characteristics are needed. One is the specific binding affinity for target molecules to avoid the nonspecific delivery phenomenon and another is the efficient capacity to store therapeutic drugs. In the aspect of specific binding affinity, some RNA aptamers could be attractive resources, but they hardly have efficient drug-storage capacity. So the DNAs were adapted to improve the drug-storage capacity. New DNA–RNA hybrid systems could be a rational platform for novel drug delivery systems. In this research, we focus more on some parameters to improve the efficacy of the nanostructure, enhance the ligation yield of the RNA–DNA hybrids and increase the cellular uptake efficiency depending on their surface compositions on gold nanorods of the nanostructure. To construct RNA–DNA hybrids, nucleic acid fragments can be synthesized through various linkage methods including chemical conjugation21 or base hybridization.22 During DNA synthesis, single-stranded DNAs can be conjugated with single-stranded RNA molecules through phosphodiester bond linkages. Alternatively, double-stranded DNAs can be hybridized with double-stranded RNAs via complementary overhangs, eventually forming a hybrid complex, via T4 ligase enzyme activity. The ligation efficacy is a major determinant of whether RNA–DNA hybrid complex formation will be successful or not. Ligation procedures are dominated by several internal or external reaction parameters, including the environmental temperature, time, ion concentration, sequence specificity, and other parameters. Additionally, we conjugated gold nanorods with various mixtures of A10 RNA as well as DNA. The LNCaP specificity of the nanostructures was evaluated by determining the cellular uptake efficiency of the nanostructure complexes. The best cellular uptake occurred when gold nanorods with a 20% A10 RNA surface composition were used.
2. Experimental section
2.1 Double-stranded DNA and RNA fragments
All DNA and RNA molecules were purchased from Integrated DNA Technology. We used a looped RNA aptameric probe with 43 bases, termed A10, which is specific to the prostate-specific membrane antigen (PSMA). Stem DNA scaffolds (designated ds40) comprised 40 base pairs with 4 sticky end bases, and are shown in Fig. 1a–b along with the RNA hairpins. All DNA and A10 sequences are described in Fig. 1c. All fragments were prepared via conventional methods as described previously.23 | ||
| Fig. 1 Depiction of the two nucleic acid segments used for ligation. The A10 RNA aptamer forms a hairpin structure. The DNA segment is composed of two single 40 and 44 base strands. The A10 segment is composed of one single 43 base strand with four overhang bases. Note that the overhang bases of A10 are deoxyribonucleotides. (a) Ligation of the A10 RNA aptamer with 40-bp DNA (ds40). Two A10 overhang sequences, ATGC and TAGC, were used. (b) The most stable folded structure of A10. (c) Detailed information for A10 and ds40; The 5′ end of A10 was modified by phosphorylation and the 5′ end of ds40 with 40 bases was modified by thiolation whereas the 5′ end of complementary ds40 with 44 bases was functionalized by phosphorylation. Red colors in the sequence body represent specific overhang sequences for DNA–RNA hybridization. | ||
2.2 Stem-loop DNA–RNA nanoarchitecture (A10–ds40)
A stem-loop complex was produced by mixing 1.957 μg (74.6 pmol) of ds40, 1.032 μg (74.6 pmol) of A10, T4 DNA ligase, and T4 DNA ligase buffer. The solution was kept at room temperature for over 4 hours, during which time T4 ligase activity was maximal. Different ligation conditions were tested. The final products were loaded on 3% agarose gels and subjected to 100 V for 40 minutes. The gels were post-stained with ethidium bromide (EtBr) solution at a final concentration of 2 μg ml−1, and the bands were observed using a GELDoc-it Imaging system under Launch VisionWorksLS UPV. To evaluate the relative band intensity, Image J software, provided by the NIH, was used.2.3 Incorporation of gold nanorods with mixed surface compositions of either ds40 or A10–ds40 hybrids (A10–ds40–AuNRs)
To amplify the amount of RNA–DNA hybrids, gold nanorods were used as templates because it is easy to strongly link nucleic acids to gold via thiol conjugate chemistry.24 A10–ds40 hybrid nanostructures were bound to gold nanorods. Instead of full coverage of the nanostructures, different amounts of ds40 were mixed with A10. To accurately control the ratios, the A10–ds40 hybrids were quantified by agarose gel electrophoresis analysis. Gel electrophoresis was typically performed using 1× TAE buffer at 80 V for 70 minutes. Different amounts of ds40, namely 0.25, 0.5, 0.75 and 1 μg, were used as calibration standards. Each A10–ds40 ligated sample was loaded onto an agarose gel and its intensity was compared with that of 1 μg of ds40. Gel electrophoresis band intensities were analysed using Image J software. By mixing additional ds40 with the A10–ds40 hybrid ligation stock, a wide variety of mixed solutions, including both A10–ds40 and ds40 solutions, were prepared. We used gold nanorods with a width of 10 nm, a length of 40 nm and an aspect ratio of 4. To cap mixed architectures, the gold nanorods were washed thoroughly with nuclease-free water to remove excessive cetrimonium bromide (CTAB) molecules. After each step, the samples were centrifuged at 13000 rpm for 5 min, the supernatant was discarded, and the gold nanorods were redispersed in nuclease-free water. Lastly, the gold nanorods were mixed together with the prepared mixed RNA–DNA hybrid solutions and a small quantity of tris(2-carboxyethyl)phosphine (TCEP) reducing agent. Gold nanorods were mixed with DNA (ds40) in a 100-fold molar amount. Afterwards, the samples were incubated on a tube rotator at 300 rpm at room temperature.
2.4 Quantitative measurement of A10–ds40–AuNRs in LNCaP cells
LNCaP cells, which express PSMA on their membranes, were seeded in 24-well subculture plates. The cells were incubated at 37 °C under 5% CO2 for 38 hours considering the doubling time of LNCaPs until the confluency reached 90%. Each well plate was washed with 500 μl 1× phosphate buffered saline (PBS) solution. The plates were then incubated for an additional 20 minutes after the addition of 1× binding buffer. After discarding the supernatant, various A10–ds40–AuNR complexes were added to the adherent LNCaP cells followed by 2 hours of incubation in 1× binding buffer. Afterwards, the cells at the bottom of the wells and the cell debris in the well supernatants were measured separately. SpectraMax M5 Multi-mode Microplate Readers (Molecular Devices, USA) were used to measure the absorbance intensity of the gold nanorods at 808 nm.2.5 Preparation of doxorubicin-functionalized A10–ds40–AuNR
Before the A10–ds40 hybrids were reacted with the gold nanorods as described above, the A10–ds40 hybrids were intercalated with doxorubicin.20 The A10–ds40 hybrids were mixed with a 10-fold molar excess of Dox to occupy the intercalating sites of ds40. The mixed solutions were incubated at 25 °C and stirred in a 550 rpm Eppendorf Thermomixer Comfort overnight in the dark. Excess doxorubicin was removed using a Millipore Amicon 3 kDa filter to obtain A10–ds40 hybrids intercalated with doxorubicin (A10–ds40–Dox). Lastly, the A10–ds40–Dox complexes were attached to gold nanorods in the same way as described above to obtain A10–ds40–Dox–AuNRs.2.6 Confocal laser scanning microscopic imaging of A10–ds40–Dox–AuNRs in LNCaP cells
Two cell lines were selected to observe the uptake of A10–ds40–Dox–AuNRs: the PSMA-positive prostate carcinoma cell line LNCaP, and the PSMA-negative prostate carcinoma cell line PC3. Cells from the two cell lines were pre-cultured using conventional cell culture methods. For the cellular uptake experiment, cells were seeded in 4-well CultureSlide slides (BD Falcon) that were filled with Opti-MEM (GIBCO) to give up to 70% confluency. The cells were incubated for 2 hours with a 10 μM solution of free doxorubicin or with a 10 μM solution of the A10–ds40–Dox–AuNR complex in a CO2 incubator. The cells were then rinsed with 1× PBS and successively fixed with 4% formaldehyde for 10 minutes. Finally, they were transferred to mounting media and observed using a VIS confocal laser scanning microscope system equipped with a 10× objective lens.3. Results and discussion
3.1. Evaluation of the ligation efficacy of the A10–ds40 constructs
A looped RNA aptamer with 39 bases specific to a prostate cancer marker and a stem (linear)-shaped DNA fragment of 40 bases were ligated through T4 DNA ligase activity (Fig. 1a–c). The most stable folded structure of the A10 RNA aptamer was predicted by OligoAnalyzer 3.1 software provided by Integrated DNA Technology. Several environmental conditions, including the external variables of incubation time, temperature, reaction volume, and rotation speed, and the internal variables of DNA/RNA molar ratios, reaction nucleotide mass, and 5-prime overhang sequences, were evaluated to determine which conditions lead to the optimum ligation efficacy. To analyze the ligation yields, the intensities of EtBr-stained bands, which correspond to nucleic acid masses,25 were compared via Image J software. Image J software arbitrarily estimates the absolute area intensity of each band in contrast to background signals. Consequently, ligation yields were obtained by comparing the band intensities of A10–ds40 hybrid bands with ds40 calibration standards, and the significance of differences in ligation yields among the various ligation conditions was calculated by either ANOVA or t-tests. The mobility pattern of A10–ds40 hybrid nanostructures synthesized using various conditions are shown in Fig. 2 and 3. Generally, two bands were observed; a bottom band corresponding to A10 or ds40 molecules, and a top band corresponding to stem-loop A10–ds40 hybrid nanostructures. | ||
| Fig. 2 Gel electrophoretic mobility shift assay (GEMSA) images of the A10–ds40 hybrid nanostructures. In each image, lane 1 represents a DNA ladder of 25 base pairs and the last two lanes contain ds40 and A10 as controls. The lanes in the middle represent A10–ds40 hybrids obtained under diverse experimental conditions. They include differences in (a) the total volumes: 12, 24 and 48 μl (lanes 2, 3 and 4), (b) centrifugation speed: 0 and 300 rpm (lanes 2 and 3), (c) total nucleic acid mass: 6 and 12 μg (lanes 2 and 3), (d) 5-prime overhang sequence: GCTA and GCAT (lanes 2 and 3), (e) A10 ratio with a fixed amount of ds40: 1:1, 1:2 and 1:3 (lanes 2, 3 and 4), (f) ds40 ratio with a constant A10 amount: 1:1, 1:2 and 1:3 (lanes 2, 3 and 4), (g) ligation incubation time: 6, 12 and 24 hours (lanes 2, 3 and 4), and (h) incubation temperature: 6, 16 and 24 °C (lanes 2, 3 and 4). The white arrows show different ligation yields for the two overhang sequences. In (d), the left arrow indicates the A10–ds40 hybrids ligated via complementary hybridization of GCTA overhang sequences, while the right arrow indicates A10–ds40 hybrids ligated via hybridization of GCAT overhang sequences. | ||
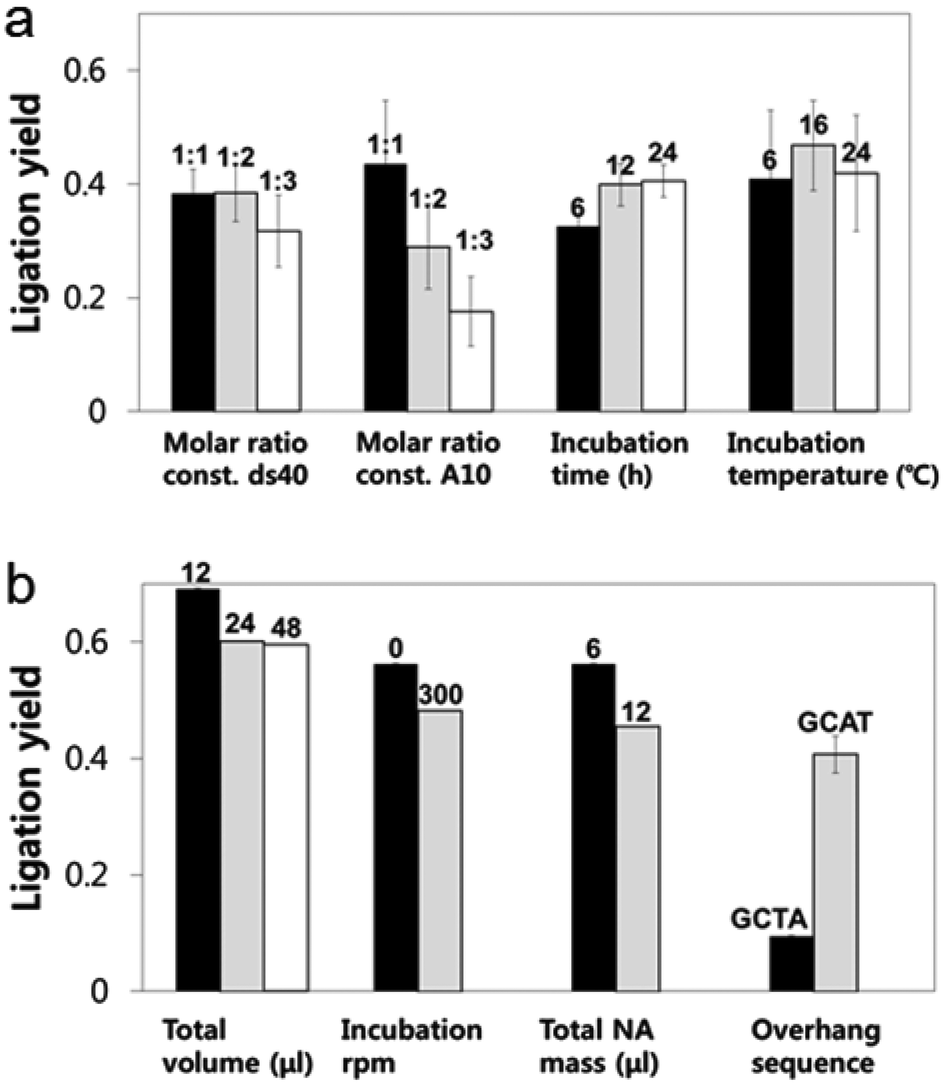 | ||
| Fig. 3 Ligation yield and efficiency variations according to (a) external and (b) internal factors. Specific sequences had enhanced ligation efficiency (p < ∼0.05). Other variables had no significant effect on the ligation efficiency. When the A10 ratio was constant, an increased ds40 amount resulted in a reduction in ligation efficacy, but this was not statistically significant. “NA” refers to nucleic acid. | ||
To evaluate the effects of external factors on the ligation efficiency, the ligation reaction was performed at different temperatures or for different times. A sample with a molar ratio of 1:1 of A10 to ds40 was incubated for 6, 12, or 24 h. The samples were also exposed to temperatures of 4–8, 16, or 24 °C. The three variables had no substantial effect on the ligation yield, as shown in Fig. 2 and 3. In addition, internal factors including the relative molar ratio of A10, the overall size of A10 (Fig. 1b), and the non-palindromic overhang sequence variants were also assessed. To determine the effect of the relative molar ratio of A10, the amount of A10 used in the reaction was varied, and the ds40 amount was then fixed at 1.957 μg. A similar ligation efficiency was observed for all the conditions, except when the complimentary overhang sequences or the head sizes of the secondary RNAs were varied (Fig. S1†). The only factor that affected the ligation efficiency was the complimentary A10 overhang sequence (p = 0.01). An increase in either the A10 or ds40 concentration resulted in a significant decrease of the entire ligation yield (Fig. 2 and 3 and Table S1†). It may be ascribed to the increase in the concentration of total nucleic acids while the concentration of A10, ds40 or ligated products remain constant. Hence, the variation of the molar ratio of A10 or ds40 had no effect on the product yield. We further examined the sequence variables at the A10 and ds40 overhangs. When the overhang sequence of A10 was changed from GCTA to GCAT, a strong increase in the band intensity of the ligated products was observed, and the relative ligation efficacy and yield were 4-fold higher than those of A10 molecules with GCTA overhangs (lane 3 in Fig. 2d and column 8 in Table S1†). We concluded that the T4 ligase activity increases due to the replacement of the last base of A10 with thymine. It should be noted that in RNA interference studies, double-stranded siRNAs select one or two specific bases in the terminal position, allowing double-stranded siRNA to complex easily with the RNA-included silencing complex (RISC) following recognition by DICER.26
To further explore the substantial difference in the ligation efficacy of the GCAT and GCTA systems, two different sizes of RNA were ligated with the same DNAs: the A10 aptamer molecule specific to prostate cancer, and a pancreas cancer-specific aptamer molecule termed “Pan”. A10 is larger than Pan; the molecular weight of A10 is 12121.5 g mol−1 and it is 39 bases long, while that of Pan is 9243.6 g mol−1 and it is 29 bases long. The stems of both RNAs are about 19 bases in length, while the loop parts vary in length. A10 has a larger loop than Pan; 20 bases for A10 (Fig. 1b–c) and 6 bases for Pan (Fig. S2†). The length of A10 is approximately 20.4 nm, while that of Pan is 19.4 nm. The ligation efficacy differed markedly according to the RNA size (note Fig. S1†). The enhanced ligation efficacy observed for Pan may be ascribed to its smaller size, which might result in a weaker steric hindrance. According to a previous study performed at a constant temperature and pressure, the enthalpy of polyethylene oxide is thermodynamically determined by its molecular size.27 This thermodynamic theory could be applied to molecules with analogous structure, such as stem-loop RNA–DNA hybrids. Accordingly, the RNA polymer that we used in this study may have a higher enthalpy than RNAs of lower molecular weight. Therefore, the A10 polymer that we used in this study may have been able to overcome the binding energy of overhang hybridizations.
Accordingly, we conclude that the sequence information and molecular weight of RNA molecules during DNA–RNA hybrid complexation play a significant role in ligation efficacy. The construction of stem-loop A10–ds40 complexes was optimal at a 1:1 molar ratio of A10 and ds40 with GCAT overhangs.
3.2. Estimation of the cellular uptake efficacy of A10–ds40 nanostructures
To date, a number of studies have been aimed at optimizing the rational design of various nanoparticles for the enhancement of their cellular uptake efficiency. Some of them insisted on the existence of several significant issues regarding the particulate's design affecting the cellular permeability. Representatively, they involve ligand density, surface modification, and even target cell (in this case, it should be a cancer cell) heterogeneity.28,29 In addition, the surface charge of the nanoparticle affects its cellular uptake capacity. It is known that negative charges of particles reduce their cellular uptake.30 As live cells' membrane charges are negative, negatively charged particles entering the target cells could be disturbed by their electrostatic repulsion. Therefore, the surface charge of nanoparticles should be considered to improve their cellular uptake capacity.To amplify the targeting signals that nanostructures possess and also to apply the A10–ds40 hybrid nanostructures in vitro, we used gold nanorods (AuNR) as a templating platform. According to our previous studies, the physiochemical properties of AuNRs conjugated with A10–ds40 were substantially altered.20 For instance, A10–ds40–AuNRs had a size of approximately 83.3 ± 20.7 nm and a surface charge of −0.61 mV, which was different from 26.6 ± 6.2 nm and 48.3 mV for bare AuNRs, and over 80% of the initial A10–ds40 was bound to AuNRs. We varied the surface compositions of AuNRs to optimize their uptake efficiency by model cancers, LNCaP cells. ds40 and A10–ds40 amounts were estimated quantitatively and A10–ds40 mixed solutions were prepared along with A10 at percentages of 10, 20, 30 and 40%, according to the mixture ratio presented in Fig. S3 and Table S2.† Mixed solutions with various A10 ratios were conjugated with AuNRs using thiol chemistry. It was proven that the chemical surface compositions on gold nanorods can be controlled by fine-tuning the feeding ratios of the chemical components.31 As mentioned above, 80 mol of terminally thiolated oligonucleotides were conjugated to 1 mol of AuNRs. The A10 density on AuNRs can be obtained by a simple deduction method, in which the molar ratios of ds40 and A10 are simply calculated. In the case where AuNRs were conjugated with A10 at different percentages (10, 20, 30 and 40%), the ratios of A10 to AuNR were 8, 16, 24 and 32, respectively.
It is known that AuNRs have dramatically variable optical and electrical properties that are determined by their size, aspect ratio, and external environment of the gold nanorods.32 For example, a red shift phenomenon in longitudinal plasma resonance occurs when the aspect ratio increases, but the transverse resonance is not affected by an increase in the aspect ratio. The combination of AuNRs with the specific cell targeting capacity of RNA–DNA hybrid nanostructures yields A10–ds40-conjugated AuNRs that can be used for visualization. AuNRs with dimensions of 10 × 40 nm have unique optical properties; they absorb at 808 nm, which is a near-infrared wavelength, and the absorbance is proportional to the amount of AuNRs in free solution as well as in a cell.33 Therefore, the cellular uptake of AuNRs (10 × 40 nm) can be measured by measuring the absorbance at 808 nm. The cellular uptake efficacy of AuNRs with various surface compositions was assessed in plated cells (Fig. 4b) and in cell debris in the supernatant (Fig. S4†). When A10–ds40 was mixed with free ds40, a 20% proportion of A10 resulted in the greatest uptake of A10–ds40–AuNR nanostructures into LNCaP cancer cells. Overall, we conclude that a 2:10 ratio of A10–ds40 and free ds40 was optimal for cellular uptake. These results show that, by finely tuning the surface density of the aptamers, we could determine the specific surface composition of AuNR which enabled us to increase the cellular uptake ability and reduce the electrostatic repulsion simultaneously. Thus, it was reasonable that the cellular uptake tendency could have the highest value at a specific surface composition of nanoparticles. In our case, the surface composition containing 20% A10 had the maximum LNCaP cellular uptake efficiency.
 | ||
| Fig. 4 (a) Schematic drawing of the cellular uptake of A10–ds40–AuNR complexes functionalized with doxorubicin. These nanostructures comprised A10, ds40, and gold nanorods. A10 recognizes the prostate-specific membrane antigen (PSMA), whereas ds40 functions as a storage vehicle for anticancer drugs, Dox. The endocytosis of A10–ds40–AuNR complexes was likely due to specific binding interactions between A10 and PSMA. (b) Evaluation of the prostate cancer cell uptake efficacy of A10–ds40–AuNR complexes with varying surface compositions of gold nanorods. By measuring the AuNR absorbance intensities at 808 nm for different A10 ratios, the cellular uptake efficiency of plated target cells was assessed. | ||
3.3. LNCaP specific targeting of A10–ds40–AuNRs as potential anti-cancer therapeutics
Doxorubicin (Dox) is a widely used anti-cancer drug, and is very effective against various cancers, including prostate cancer.34 Dox can intercalate very well into double-stranded DNA and shows strong fluorescence at 585 nm.35,36 In the past, Dox was selectively bound to prostate specific RNA aptamers with GC-rich regions.37 Based on these characteristic features, we expected that A10–ds40 hybrids containing doxorubicin would have cancer-specific therapeutic effects in LNCaP cells. Our previous research revealed that there were seven or eight Dox molecules per ds40 molecule and 4000–5000 Dox molecules per AuNR.20 The stem-shaped ds40 template provides target sites for Dox intercalations while the loop-structured A10 RNA can bind specifically to the prostate-specific membrane antigen (PSMA).We have evaluated the active targeting therapeutic effects of the RNA–DNA hybrids in vitro by using two prostate cancer strains: LNCaP (PMSA-positive) and PC3 (PSMA-negative). LNCaP and PC3 cells were treated with free doxorubicin or A10–ds40–AuNR structures containing doxorubicin. As expected, A10–ds40–AuNR attached to LNCaP cells through the specific affinity of A10 for PSMA, and the doxorubicin molecules embedded into the DNA template visualize this. The cells were monitored by confocal laser scanning microscopy using doxorubicin's fluorescence emission wavelength. Free doxorubicin was able to penetrate the cellular membranes of both LNCaP and PC3 cells (Fig. 5a–b). Because intracellular doxorubicin intercalates into chromosomes and inhibits replication, it can cause non-specific cell apoptosis.38 In contrast, the A10–ds40–AuNRs with intercalated doxorubicin were selectively taken up by LNCaPs via specific A10–PSMA interactions (Fig. 5c–d), inducing specific cell apoptosis.20
 | ||
| Fig. 5 In vitro cellular uptake images obtained using confocal laser microscopy. The excitation and emission wavelengths of doxorubicin are 480 nm and 580 nm, respectively. The red dots represent doxorubicin in the cytosol of prostate cancer cells. (a) LNCaP and (b) PC3 cells were treated with 10 μM of free doxorubicin. Then, (c) LNCaP and (d) PC3 cells were treated with 10 μM of the A10–ds40–dxo-AuNR complexes. The scale bar is 20 μm. | ||
4. Conclusions
We have optimized a variety of physiochemical parameters to improve the efficiency of the ligation of A10 and ds40 to create novel A10–ds40 hybrid nanostructures. Only the base sequence of the overhang affected the ligation efficacy. When we evaluated the two A10 overhang sequences of GCAT and GCTA, we found that GCAT was superior to GCTA with regard to enhanced ligation. In addition, the ligation efficiency of Pan, which has a smaller head size (or molecular weight) but the same GCTA overhang sequence as A10, to DNA sequences was more efficient. The RNAs that we used were able to interact specifically with PSMA markers on prostate cancer cell surfaces. Moreover, we showed that the 40-base long DNA fragments that we used can incorporate anti-cancer drugs such as doxorubicin, which possesses a phenyl ring that can interact with DNA.35,36 The merger of tumor-specific RNA aptamers and DNA based anti-cancer storage structures yields RNA–DNA hybrid nanostructures that are novel, site-specific drug delivery systems.We coated gold nanorods with the A10–ds40 hybrid nanostructures to enhance the potential anti-cancer effect of the hybrid drug complexes. By varying the surface compositions of the gold nanorods capped with both A10–ds40 hybrids and ds40, we found that the optimal composition of the A10–ds40–AuNRs for cellular uptake was with a 2:10 molar ratio of A10 to ds40. The drug hybrid complex only targeted LNCaP carcinoma. Although we used the gold nanorods in this study simply as a platform for the A10–DNA hybrids, gold nanorods have tremendous possibilities for cancer therapy because of their inherent optical and thermal characteristics. RNA–DNA–AuNR nanostructures could therefore potentially be used in multidisciplinary cancer research fields. We anticipate that the rational design of RNA–DNA–AuNR nanostructures as described in this paper will advance several anti-cancer pharmaceutics in the near future.
Acknowledgements
We thank Changyoon Baek for providing technical assistance with the confocal laser scanning microscope. This work was partially supported by a grant from the Korea Health Technology R&D Project of the Ministry of Health & Welfare of the Republic of Korea (Grant No. A110552) and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (Grant No. 2013R1A1A2016781).References
- M. Sarikaya, C. Tamerler, A. K.-Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed
.
- A. Csiszár, B. Hoffmann and R. Merkel, Langmuir, 2009, 25, 5753–5761 CrossRef PubMed
- J. W. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Nat. Rev. Drug Discovery, 2011, 10, 521–535 CrossRef CAS PubMed
- S. Peng, T. L. Derrien, J. Cui, C. Xu and D. Luo, Mater. Today, 2012, 15, 190–194 CrossRef CAS
- E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. P. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73–76 CrossRef CAS PubMed
- O. I. Willner, R. Orbach, A. Henning, C. Teller, O. Yehezkeli, M. Mertig, D. Harries and I. Willner, Nat. Commun., 2011, 2, 540 CrossRef PubMed
- M. N. Stojanovic and D. A. Stefanovic, Nat. Biotechnol., 2003, 21, 1069–1074 CrossRef CAS PubMed
- H. Sun, W. Li and L. Wu, Langmuir, 2009, 25, 10466–10472 CrossRef CAS PubMed
- S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Nat. Mater., 2006, 5, 797–801 CrossRef CAS PubMed
- Z. Li, M. Liu, L. Wang, J. Nangreave, H. Yan and Y. Liu, J. Am. Chem. Soc., 2010, 132, 13545–13552 CrossRef CAS PubMed
- A. M. Hung, C. M. Micheel, L. D. Bozano, L. W. Osterbur, G. M. Wallraff and J. N. Cha, Nat. Nanotechnol., 2010, 5, 121–126 CrossRef CAS PubMed
- B. Wei, M. Dai and P. Yin, Nature, 2012, 485, 623–627 CrossRef CAS PubMed
- N. C. Seeman, Mol. Biotechnol., 2007, 37(3), 246–257 CrossRef CAS PubMed
- R. F. Service, Science, 2011, 332, 1140–1143 CrossRef CAS PubMed
- S. H. Um, J. B. Lee, S. Y. Kwon, Y. Li and D. Luo, Nat. Protoc., 2006, 1, 995–1000 CrossRef CAS PubMed
- J. B. Lee, Y. H. Roh, S. H. Um, H. Funabashi, W. Cheng, J. J. Cha, P. Kiatwuthinon, D. A. Muller and D. Luo, Nat. Nanotechnol., 2009, 4, 430–436 CrossRef CAS PubMed
- N. Park, S. H. Um, H. Funabashi, J. Xu and D. Luo, Nat. Mater., 2009, 8, 432–437 CrossRef CAS PubMed
- J. J. Moon, H. Suh, A. Bershteyn, M. T. Stephan, H. Liu, B. Huang, M. Sohail, S. Luo, S. H. Um, H. Khant, J. T. Goodwin, J. Ramos, W. Chiu and D. J. Irvine, Nat. Mater., 2011, 10, 243–251 CrossRef CAS PubMed
- M. T. Stephan, J. J. Moon, S. H. Um, A. Bershteyn and D. J. Irvine, Nat. Med., 2010, 16, 1035–1041 CrossRef CAS PubMed
-
A. R. Kim, S. W. Shin, S.-W. Cho, J. Y. Lee, D.-I. Kim and S. H. Um, Adv. Health. Mater. DOI:10.1002/adhm.201200471, in press
- N. G. Dolinnaya, N. I. Sokolova, D. T. Ashirbekova and Z. A. Shabarova, Nucleic Acids Res., 1991, 19, 3067–3072 CrossRef CAS PubMed
- D. R. Horspool, R. J. N. Coope and R. A. Holt, BMC Res. Notes, 2010, 3, 291–299 CrossRef PubMed
- K. Harada and L. E. Orgel, Nucleic Acids Res., 1993, 21, 2287–2291 CrossRef CAS PubMed
- J. Liu and Y. Lu, Nat. Protoc., 2006, 1(1), 246–252 CrossRef CAS PubMed
- V. Bonasera, S. Alberti and A. Sacchetti, BioTechniques, 2007, 43, 173–176 CrossRef CAS PubMed
- D. Castanotto and J. J. Rossi, Nature, 2009, 457, 426–433 CrossRef CAS PubMed
- P. J. Sánchez-Soto, J. M. Ginés, M. J. Arias, Cs. Novák and A. Ruiz-Conde, J. Therm. Anal. Calorim., 2002, 67, 189–197 CrossRef
- F. Gu, L. Zhang, B. A. Teply, N. Mann, A. Wang, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(7), 2586–2591 CrossRef CAS PubMed
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed
- A. S. Mikhail and C. Allen, J. Controlled Release, 2009, 138, 214–223 CrossRef CAS PubMed
- M. Zheng and X. Huang, J. Am. Chem. Soc., 2004, 126, 12047–12054 CrossRef CAS PubMed
- C. Z. Li, K. B. Male, S. Hrapovic and J. H. T. Luong, Chem. Commun., 2005, 3924–3926 RSC
- C. Wang and J. Irudayaraj, Small, 2008, 4, 2204–2208 CrossRef CAS PubMed
- R. B. Weiss, Semin. Oncol., 1992, 19(6), 670–686 CAS
- L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188–200 CrossRef CAS PubMed
- M. P. Schotanus, K. S. Aumann and K. Sinniah, Langmuir, 2002, 18, 5333–5336 CrossRef CAS
- V. Bagalkot, O. C. Farokhzad, R. Langer and S. Jon, Angew. Chem., Int. Ed., 2006, 45(48), 8149–8152 CrossRef CAS PubMed
- F. A. Fornari, J. K. Randolph, J. C. Yalowich, M. K. Ritke and D. A. Gewirtz, Mol. Pharmacol., 1994, 45(4), 649–656 CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60136f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |