
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA direct enzyme-linked immunosorbent assay (ELISA) for the quantitative evaluation of Janus Kinase 3 (JAK3) inhibitors
Silke M.
Bauer
,
Matthias
Gehringer
and
Stefan A.
Laufer
*
Department of Pharmaceutical and Medicinal Chemistry, Institute of Pharmaceutical Sciences, Eberhard Karls University of Tuebingen, D-72076 Tuebingen, Germany. E-mail: stefan.laufer@uni-tuebingen.de; Fax: +49-7071-295037; Tel: +49-7071-2978788
First published on 5th August 2014
Abstract
The availability of a robust, precise and cost-efficient test system is the prerequisite for the performance of any medicinal chemistry program, especially in an academic environment. For the identification of novel Janus Kinase 3 inhibitors and determination of structure–activity relationships, a direct enzyme-linked immunosorbent assay was developed. Being applicable in a standard laboratory, the main advantage over existing methods is the basic equipment requirement as this assay does not need special handling originating from the use of cell cultures or radiometric readout. In contrast to earlier methods relying on two-step procedures, our assay uses a single peroxidase conjugated antibody to quantify and detect substrate phosphorylation. This optimized assay features a limit of detection of 0.032 μg mL−1, a wide dynamic range of 0.638 OD units and an appropriate Z′ factor of 0.66. The determined IC50 value for Tofacitinib was 3.5 ± 0.6 nM (n = 18). A detailed assay protocol custom-tailored for the JAK3 kinase domain and a step-by-step manual for optimizing specific assay conditions are presented. This ELISA primarily addresses researchers within the academic setting facing the task to identify lead structures and explore structure–activity relationships of JAK3 inhibitors at reasonable costs.
Introduction
Janus kinases (JAKs) are a group of non-receptor tyrosine kinases playing a fundamental role in the immune system, hematopoiesis and interferone signaling. Consequently, the four JAK family members JAK 1,2,3 and Tyrosine Kinase 2 are highly attractive targets for treatment of inflammatory and oncological disorders.1–3Several inhibitors of JAK1 and/or JAK2 as well as pan-JAK inhibitors are currently under clinical investigation for treatment of inflammatory disorders like rheumatoid arthritis (RA) and autoimmune diseases such as Crohn's disease and psoriasis. Recently, the pan-JAK inhibitor Tofacitinib has been approved by the US Food and Drug Administration (FDA) for its application in patients with severe RA inadequately responding to methotrexate.4 However, the European Medicines Agency (EMA) refused the marketing authorization of Tofacitinib in 2013 due to an increased risk for the occurrence of serious infections among other severe side effects.5
In contrast to the other JAK family members, the expression of JAK3 is restricted to the hematopoietic tissue where it exclusively associates with cytokine receptors bearing the common gamma-chain (γc) subunit, enabling JAK3 to specifically operate in immune cells. Consequently, JAK3 represents an attractive target for immunosuppression with limited side effects. Nevertheless, no selective JAK3 inhibitor is available for therapy so far and there is a lack of sufficiently characterized compounds with a distinct selectivity profile. Despite its poor selectivity within the JAK family Knapp et al. suggested the pan-JAK inhibitor Tofacitinib as a high-quality chemical probe for JAK3.6 To evaluate the JAK3 activity of our Tofacitinib-derived compound library,7 we modified, adapted and optimized a general enzyme-linked immunosorbent assay (ELISA) for the detection of protein tyrosine kinase activity previously established by Lázaro et al.8 Earlier published non-cellular JAK3 assays were based on radiometric formats or depended on multi-step procedures using expensive reagents. In contrast, our protocol is operationally simple, robust, and performable at a low cost making it a useful tool for routine screening of JAK3 inhibitors, especially in an academic environment. The ELISA reported herein allows the determination of inhibitory potencies in the low nanomolar range and the IC50 values obtained were subsequently confirmed by commercial radiometric and cellular assays. As a result of our research in this area, we recently described a new class of highly potent tricyclic JAK inhibitors with superior selectivity compared to Tofacitinib.9
In the general assay procedure reported by Lázaro et al., an artificial synthetic polypeptide consisting of L-glutamic acid and L-tyrosine in random distribution with a molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of glutamic acid to tyrosine is adsorbed to the wells of the assay plate. In the presence of tyrosine kinase and ATP, the synthetic polypeptide is phosphorylated on its tyrosine residues while ADP is generated. The amount of the phosphorylated kinase substrate directly correlates with kinase activity. The comparison of the maximum phosphorylation level with the phosphorylation degree achieved when the JAK3 kinase reaction is carried out in the presence of different inhibitor concentrations allows the determination of IC50 values.
:1 of glutamic acid to tyrosine is adsorbed to the wells of the assay plate. In the presence of tyrosine kinase and ATP, the synthetic polypeptide is phosphorylated on its tyrosine residues while ADP is generated. The amount of the phosphorylated kinase substrate directly correlates with kinase activity. The comparison of the maximum phosphorylation level with the phosphorylation degree achieved when the JAK3 kinase reaction is carried out in the presence of different inhibitor concentrations allows the determination of IC50 values.
For detection, Lázaro's protocol applies a two-step procedure. As a primary antibody, a monoclonal mouse anti-phosphotyrosine antibody is used while a goat anti-mouse immunoglobulin G (IgG) antibody labeled with peroxidase serves as a secondary antibody. This method requires two incubation steps, one for each antibody reaction. In contrast, our novel protocol uses a single monoclonal anti-phosphotyrosine antibody conjugated with peroxidase for directly detecting the degree of phosphorylation of the synthetic polypeptide, as illustrated in Fig. 1.
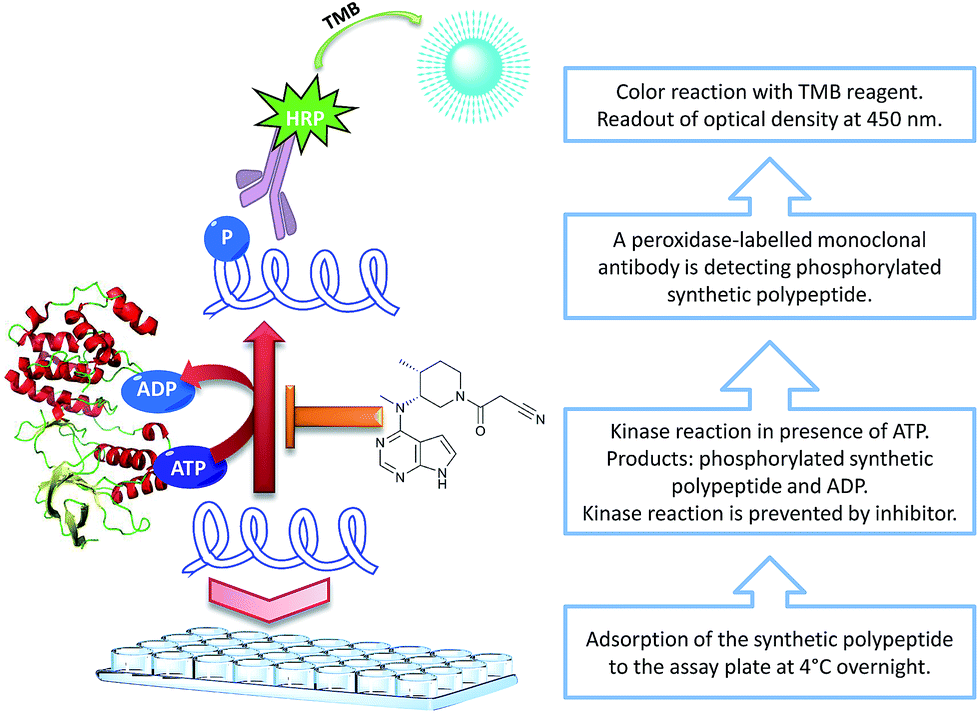 | ||
| Fig. 1 Schematic workflow of the established direct JAK3 ELISA. | ||
Experimental
Materials and methods
Determination of the Z′ factor and dynamic range
The Z′ factor was calculated according to the general equation
The dynamic range is expressed by the term [μp − μn].10
The variables of the equation refer to the following abbreviations: p: OD450 of positive control, n: OD450 of negative control, σ: standard deviation of OD450, and μ: mean of OD450.
Determination of the limit of detection (LOD)
A calibration curve of the phosphorylated polypeptide was established and detection was carried out according to the conditions described in the general assay procedure. The LOD was assessed by linear regression of the calibration curve applying the following equation:11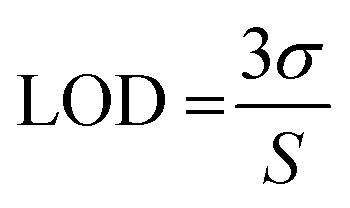
The following abbreviations are used for the variables of the equation: σ: standard deviation of the response and S: slope of the calibration curve.
Calculation of the percentage of inhibition
The percentage of inhibition is calculated according to the following equation:OD450STIM: the optical density at 450 nm for the positive control (STIM); OD450Sample: the optical density at 450 nm for the kinase reaction in the presence of a test compound in the respective dilution; OD450NSB: the optical density at 450 nm for nonspecific binding (NSB).

Adaption and optimization of the assay settings
To determine the optimal amount of the kinase substrate, a concentration row was prepared by dissolving the synthetic polypeptide in PBS. 100 μL of coating solution containing concentrations from 100 μg mL−1 to 0.625 μg mL−1 of the synthetic polypeptide were added to the wells of the assay plate. The plate was covered with a lid and coated overnight at 4 °C. After the coating step, a kinase buffer (KB) with an ATP concentration of 100 μM was prepared. After one hour of incubation with pure KB and KB containing 60 ng mL−1 JAK3 kinase and the subsequent antibody incubation step (dilution factor 1:60000), optical densities (OD) of the nonspecific binding (NSB) signal and the maximum signal were detected at 450 nm (OD450). For titration of the enzyme concentration, increasing amounts of JAK3 kinase (15 ng mL−1 to 3840 ng mL−1) were used while the ATP concentration of the KB was kept constant at 100 μM. The subsequent procedure was performed as described above for the determination of the kinase substrate concentration. For evaluating the dilution factor of the monoclonal peroxidase-labelled anti-phosphotyrosine antibody, the wells of the assay plate were coated with a 5 μg mL−1 kinase substrate overnight at 4 °C. Pure KB with an ATP concentration of 1.4 μM was incubated as the negative control for the detection of the NSB signal. For determination of the positive control indicating the maximum signal, a KB containing 1.4 μM ATP and 60 ng mL−1 of JAK3 kinase was employed. A dilution row with increasing amounts of the peroxidase-labelled anti-phosphotyrosine antibody was prepared in blocking buffer (BB: 0.1% Tween 20 and 0.25% bovine serum albumin in PBS) which had been adjusted to pH 6.5. 100 μL of the respective antibody dilution were added to the respective wells for both, positive and negative control, and incubated for one hour. The Km value (Michaelis constant) for ATP was measured after coating the assay plate using 100 μg mL−1 of the polypeptidic kinase substrate. The kinase reaction was carried out at a kinase concentration of 60 ng mL−1 while varying the ATP concentrations from 0.1 μM to 1000 μM, maintaining the assay procedure as described above.
Results and discussion
General assay procedure
Prior to performing the assay, 10 mM stock solutions of samples and the reference compound Tofacitinib were prepared in dimethyl sulfoxide (DMSO). The polypeptidic kinase substrate was dissolved in PBS to obtain a final concentration of 5 μg mL−1. 100 μL of this solution were added into all wells of the assay plate with an Eppendorf Multipette® plus equipped with a 5 mL Eppendorf Combitip plus®. The plate was closed with a lid and stored overnight at 4 °C for adsorption of the polypeptidic kinase substrate to its surface.A KB containing 100 mM HEPES, 10 mM MgCl2, 4 mM dithiothreitol, 0.1 mM Na3VO4, and 1.4 μM ATP was prepared in ultra-pure water and the pH was adjusted to 6.8–6.9 by dropwise addition of 1 M HCl. This KB solution served as a medium for both, preparation of the JAK3 enzyme solution and dilution of test compounds/reference. The enzyme solution contained a final concentration of 60 ng mL−1 of JAK3. This enzyme solution was apportioned into Eppendorf reaction tubes in an ice-cooled rack. Dilution of the compounds was carried out in a two-step procedure. First, the 10 mM stock solutions of samples/reference were serially diluted in KB to a 100-fold excess of the final assay concentration via 10-fold dilution steps. As a second step of the dilution procedure, the respective volume of the 100-fold pre-dilutions was added to the JAK3 enzyme solution in Eppendorf reaction tubes. Thereby, final assay concentrations of samples and Tofacitinib were obtained.
It must be pointed out that vortexing of solutions containing JAK3 should be performed gently and only for few seconds as it significantly reduces the kinase activity leading to very shallow and non-sigmoid shaped inhibition curves.
Subsequently, all wells of the assay plate were rinsed three times with a PBS-containing wash bottle. Prior to the kinase reaction step, the remaining free binding sites were blocked by treatment with BB for 12 minutes at room temperature. After completion of the blocking step, the BB was discarded and the plate was rinsed three times with PBS containing 0.1% Tween-20 (polyoxyethylene (20) sorbitan monolaurate) (PBST) using a wash bottle. For draining, the plate was left upside down for 5 minutes on an absorptive surface. Potentially existing air bubbles originating from the washing step were removed by patting the assay plate carefully on a soft and absorptive surface. For the kinase reaction, 50 μL of the diluted samples and Tofacitinib were transferred from the Eppendorf reaction tubes to the corresponding wells of the assay plate. For positive control (STIM), indicating maximum phosphorylation, 50 μL of JAK3 solution were pipetted into the respective wells. For determination of NSB, 50 μL of plain KB were added into the respective wells. The pipetting of the kinase containing testing solutions to the assay plate was performed carefully but as quickly as possible (max. 5 minutes) since the degree of phosphorylation and therefore the detected kinase activity is time-dependent. During the kinase reaction, the plate was covered with a lid and incubated for one hour at 37 °C, gently shaking at 150 rounds per minute in the incubator. After the kinase reaction, the content of the plate was discarded. The plate was washed three times with PBST, drained and patted following the same procedure as described before.
While the plate was draining, the monoclonal anti-phosphotyrosine–peroxidase conjugated antibody was diluted 1:60000 in BB which has been adjusted to pH 6.5. 100 μL of the antibody dilution were pipetted into all wells using an Eppendorf Multipette® plus equipped with a 5 mL Eppendorf Combitip plus®. For incubation of the antibody reaction, the plate was again covered with a lid and incubated as described for the kinase reaction step.
After the last mentioned incubation, the antibody solution was discarded and the assay plate was rinsed with PBST to remove any unbound antibody. The plate was again drained upside down on an absorptive surface and patted as described before.
Finally, 50 μL of the TMB substrate reagent were added into all wells using an Eppendorf Multipette® plus equipped with a 2.5 mL Eppendorf Combitip plus® and left for 5 minutes in the dark while a blue color developed. The color reaction was stopped by pipetting 25 μL of 1 M sulphuric acid into each well using the afore-mentioned pipette leading to a color change from blue to yellow. The OD was read out in an ELISA microplate reader at 450 nm.
Adaption and optimization of the assay settings
The general protocol published by Lázaro et al. for the detection of protein tyrosine kinase activity was adapted to the specific requirements of the JAK3 kinase domain. As illustrated in the schematic ELISA procedure (Fig. 1), two kinase substrates, namely ATP and the synthetic polypeptide, are involved in the kinase reaction of this ELISA. Copeland et al. defined this kind of mechanism as a bisubstrate enzyme reaction.12 We could confirm the above-mentioned polypeptide to be a suitable substrate for this kind of kinase activity assay as it is recognized and sufficiently phosphorylated by JAK3. When defining conditions for kinase activity assays based on a bisubstrate reaction, the apparent Km value for one substrate needs to be determined at the saturation concentration of the other substrate.12 As the assay will be carried out at the Km value of ATP, the amount of polypeptidic kinase substrate adsorbed to the assay plate must be kept at the saturation concentration to avoid a limitation of potential phosphorylation sites. Consequently, a decreased signal magnitude will only be caused by a reduced kinase activity as a result of kinase inhibition by the kinase inhibitor. In contrast, the ATP concentration was kept at 100 mM for the determination of the required synthetic polypeptide concentration, to guarantee saturation of this substrate component. This time, the concentration of the polypeptide adsorbed to the assay plate was varied from 0.63 μg mL−1 to 100 μg mL−1. The signal magnitude measured using a 5 μg mL−1 polypeptidic kinase substrate was comparable to signal responses obtained with 10 μg mL−1 or 100 μg mL−1 of the afore-mentioned substrate (Fig. 2A). Thus, a concentration of 5 μg mL−1 of the synthetic polypeptide was considered as sufficient. In order to ascertain both, the optimal kinase concentration and the sensitivity of this ELISA method, the linearity of the relationship between signal and kinase concentration was determined. Concentrations between 15 ng mL−1 and 3840 ng mL−1 of JAK3 were incubated at an ATP concentration of 100 μM, to rule out that the kinase activity and the obtained signal magnitude were limited by ATP-depletion. In Fig. 2C, the OD450 is plotted as a function of the kinase concentration. For JAK3 enzyme concentrations up to 60 ng mL−1, the measured OD450 values are directly proportional to the applied kinase concentrations. Within this linear range, a decrease of kinase activity directly results in a proportional loss of the signal response. A further enhancement of JAK3 concentration does not result in a further proportional increase of analytical signal. Moreover, the employment of excessive kinase concentrations can diminish the sensitivity of such an assay system and thus disables the sufficient discrimination of small differences in inhibitory activity. A decrease of kinase activity by different inhibitor concentrations will not lead to a proportional reduction of the phosphorylation degree when applying excessive amounts of kinase. A linear regression of the optical densities corresponding to the respective kinase concentrations of the linear part of Fig. 2C was performed. The slope of the resulting regression line indicates the assay's sensitivity as a function of the kinase concentration. We determined a slope of 0.013 OD450 units per ng JAK3 * mL and a coefficient of determination (R2) of 0.982 proving the correlation between assay sensitivity and the kinase concentration. Since a concentration of 60 ng mL−1 JAK3 combines both, signal linearity and high signal magnitude, this concentration was considered as the optimal kinase concentration for this assay system. | ||
| Fig. 2 (A) Saturation curve of the synthetic polypeptidic kinase substrate consisting of L-glutamic acid and L-tyrosine in a molar ratio of 4:1 after an overnight coating step at 4 °C. Different concentrations of the polypeptidic kinase substrate are plotted against the OD450. The maximum signal represents the maximum signal achieved after incubation with JAK3 kinase and ATP. The NSB signal indicates the background noise in the absence of kinase. (B) Saturation curve of the monoclonal anti-phosphotyrosine peroxidase-conjugated antibody. Coating and kinase reaction for positive and negative controls were performed as described in the Experimental section. After the kinase reaction, increasing dilutions of the antibody were added to both wells for negative and positive controls for determination of maximum- and NSB signals followed by one hour of incubation. Antibody dilutions are plotted against the obtained OD450 for maximum- and NSB signals. (C) Determination of the optimal kinase concentration and the relationship between kinase concentration and resulting signal response. The assay procedure was performed as described in the Experimental section with an ATP concentration of 100 μM. (D) Hyperbolic relationship for the degree of substrate phosphorylation as a function of the ATP concentration. For determination of the apparent Km value of this bisubstrate reaction, the polypeptidic kinase substrate was used in a concentration of 100 μg mL−1 to ensure saturation. Different ATP concentrations were applied while all other parameters remained constant. Each data point is the arithmetic mean of at least three separate experiments ± standard deviation. | ||
The precision of this ELISA method employing a JAK3 concentration of 60 ng mL−1 was verified by a low standard deviation of 0.03 OD units.
In a subsequent step, the appropriate working dilution for the peroxidase-labelled monoclonal anti-phosphotyrosine antibody was investigated (Fig. 2B). Therefore, antibody dilutions were added to the wells with positive and negative controls to determine the respective maximum- and the NSB signal magnitude. NSB is caused by binding of the antibody to the non-phosphorylated polypeptidic kinase substrate or other interferents. An OD450 below 0.1 was considered appropriate. The best signal-to-noise ratio was achieved with an antibody dilution of 1:60000.
The Km value for ATP was determined by titrating ATP concentrations between 0.1 μM and 1000 μM while keeping all other assay parameters constant (Fig. 2D). The rate of reaction in the presence of a large excess of ATP (1000 μM) was considered maximal and consequently defined as the maximum degree of phosphorylation (100%). According to Copeland et al., special care needs to be taken when setting substrate concentrations for enzymatic reactions involving more than one substrate in order to develop a balanced assay system.12 As the assay is based on a bisubstrate enzyme reaction, the assay plate was coated using 100 μg mL−1 of the synthetic polypeptide ensuring saturation conditions for determination of the apparent Km value for ATP, which was found to be 0.7 μM. As reported in previous work of Goettert et al., an ATP concentration of maximum twice the Km value is acceptable and should not be exceeded.13 Applying 1.4 μM ATP (twice the Km value) provided a good assay signal magnitude and assay stability on the one hand, and the necessary sensitivity for identification of ATP-competitive kinase inhibitors on the other hand.
The Z′ factor is a common tool to evaluate the quality of an assay. According to Zhang et al., an assay with a Z′ factor between 0.5 and 1 is considered an excellent assay.10 A Z′ factor of 0.66 confirmed this ELISA with its optimized settings as suitable for routine inhibitor screening. For further evaluation of the developed ELISA method, the LOD was determined according to the recommendations of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) by linear regression of a calibration curve (R2 0.997).11 The analytical performance of the optimized assay settings was confirmed with an LOD of 0.032 μg mL−1 phosphorylated polypeptidic kinase substrate and a dynamic range of 0.638 OD units.
Within the structural class of N-substituted 7H-pyrrolo[2,3-d]pyrimidines, Tofacitinib and two less potent inhibitors (1 and 2 in Table 1) described by Flanagan et al. were tested applying the optimized assay parameters. The resulting inhibition curves were steep and sigmoid-shaped. For the reference Tofacitinib and compound 1/2, IC50 values of 3.5 ± 0.6 nM (n = 18), 154 ± 0.04 nM (n = 3) and 311 ± 0.03 nM (n = 3) were obtained. These IC50 values were compared with the IC50 values previously reported by Flanagan et al., even though it must be emphasized that direct comparison of IC50 values determined in different assays is precarious.14 However, IC50 values determined with our optimized ELISA correlate reasonably with those reported by Flanagan et al. (Table 1).


In summary, the ELISA protocol established by Lázaro et al. was transformed from an indirect to a direct format, as a second incubation step with a secondary detection antibody becomes redundant when the primary antibody is directly labelled with the detection enzyme. Moreover, potential cross reactivity arising from a secondary detection antibody that curtails the specificity of the ELISA response and therefore the accuracy of the readout data is avoided. Application of a single monoclonal detection antibody is less likely to cause interference with other than the target epitope. Apart from inter- and intra-assay signal consistency, non-specific signals are evaded and therefore the assay sensitivity and precision is improved.
Due to its sensitivity, this ELISA allows the investigation of SARs even within a series of structurally similar inhibitors.9
For exploration of selectivity, the extension to other members of the JAK family is conceivable in the same format. Furthermore, this assay protocol is suitable to be transferred to high-throughput screening campaigns using automated robotic instrumentation, making it applicable for the measurement of large numbers of samples at a time. Thus, it might also attract the attention of industrial researchers.
Conclusions
In the current report, we describe an optimized protocol for a direct JAK3 ELISA. Previously published non-cellular JAK3 assays apply radiometric formats or depend on multi-step procedures using expensive reagents or require special laboratory equipment. The advantages over existing protocols include easy performance, robustness, cost-efficiency and avoidance of radioisotopes. The assay is readily discriminating the effects of minor changes in the inhibitor structure on kinase inhibition and is thus suited for lead optimization. Essential instructions for successful establishment and handling of this ELISA are reported in detail. Unlike the general method reported by Lázaro et al., only one monoclonal anti-phosphotyrosine peroxidase conjugated antibody is required for detection, avoiding a second incubation time, cross reactivity and additional costs.Abbreviations
| ATP | Adenosine-5′-triphosphate |
| BB | Blocking buffer |
| BSA | Bovine serum albumin |
| DMSO | Dimethyl sulfoxide |
| EMA | European medicines agency |
| ELISA | Enzyme-linked immunosorbent assay |
| FDA | Food and drug administration |
| HEPES | (2-[4-(2-Hydroxyethyl)piperazin-1-yl]ethanesulfonic acid) |
| ICH | International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use |
| IgG | Immunoglobulin G |
| JAK | Janus kinase |
| JAK3 | Janus kinase 3 |
| KB | Kinase buffer |
| K m | Michaelis constant |
| LOD | Limit of detection |
| NSB | Nonspecific binding |
| OD | Optical density |
| OD450 | Optical density at 450 nm |
| PBS | Phosphate-buffered saline |
| PBST | PBS containing 0.1% Tween-20 |
| R 2 | Coefficient of determination |
| RA | Rheumatoid arthritis |
| SARs | Structure–activity relationships |
| TMB | 3,3′,5,5′-Tetra-methylbenzidine |
| Tween-20 | Polyoxyethylene (20) sorbitan monolaurate |
| γ c | Common gamma chain interleukin receptor (IL-2RG) |
Acknowledgements
The authors would like to thank L. Fischer and K. Bauer for successful laboratory co-work during the establishment of this assay. Special thanks to Jun.-Prof. Dr Pierre Koch and Peter Keck for their help, support and advice.References
- B. H. Kim, S. R. Oh, C. H. Yin, S. Lee, E. A. Kim, M. S. Kim, C. Sandoval, S. Jayabose, E. A. Bach, H. K. Lee and G. H. Baeg, Br. J. Haematol., 2010, 148, 132–143 CrossRef CAS PubMed.
- C. Haan, C. Rolvering, F. Raulf, M. Kapp, P. Druckes, G. Thoma, I. Behrmann and H. G. Zerwes, Chem. Biol., 2011, 18, 314–323 CrossRef CAS PubMed.
- M. Pesu, F. Candotti, M. Husa, S. R. Hofmann, L. D. Notarangelo and J. J. O'Shea, Immunol. Rev., 2005, 203, 127–142 CrossRef CAS PubMed.
- J. J. O'Shea, A. Kontzias, K. Yamaoka, Y. Tanaka and A. Laurence, Ann. Rheum. Dis., 2013, 72, 111–115 CrossRef PubMed.
- EMA, Refusal of the marketing authorisation for Xeljanz (tofacitinib), http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/002542/WC500146629.pdf.
- S. Knapp, P. Arruda, J. Blagg, S. Burley, D. H. Drewry, A. Edwards, D. Fabbro, P. Gillespie, N. S. Gray, B. Kuster, K. E. Lackey, P. Mazzafera, N. C. O. Tomkinson, T. M. Willson, P. Workman and W. J. Zuercher, Nat. Chem. Biol., 2013, 9, 3–7 CrossRef CAS PubMed.
- M. Gehringer, M. Forster, E. Pfaffenrot, S. M. Bauer and S. A. Laufer, ChemMedChem, 2014 DOI:10.1002/cmdc.201402252.
- I. Lazaro, M. Gonzalez, G. Roy, L. M. Villar and P. Gonzalez-Porque, Anal. Biochem., 1991, 192, 257–261 CrossRef CAS.
- M. Gehringer, E. Pfaffenrot, S. Bauer and S. A. Laufer, ChemMedChem, 2014, 9, 277–281 CrossRef CAS PubMed.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, in Current Step 4 version, Parent Guideline dated 27 October 1994 (Complementary Guideline on Methodology dated 6 November 1996 incorporated in November 2005), 2005.
- R. A. Copeland, Anal. Biochem., 2003, 320, 1–12 CrossRef CAS.
- M. Goettert, S. Luik, R. Graeser and S. A. Laufer, J. Pharm. Biomed. Anal., 2011, 55, 236–240 CrossRef CAS PubMed.
- M. E. Flanagan, T. A. Blumenkopf, W. H. Brissette, M. F. Brown, J. M. Casavant, C. Shang-Poa, J. L. Doty, E. A. Elliott, M. B. Fisher, M. Hines, C. Kent, E. M. Kudlacz, B. M. Lillie, K. S. Magnuson, S. P. McCurdy, M. J. Munchhof, B. D. Perry, P. S. Sawyer, T. J. Strelevitz, C. Subramanyam, J. M. Sun, D. A. Whipple and P. S. Changelian, J. Med. Chem., 2010, 53, 8468–8484 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |