
A new focus for quality in chemical measurement
Michael
Thompson
*
School of Biological and Chemical Sciences, Birkbeck University of London, Malet Street, London WC1E 7HX. E-mail: M.Thompson@bbk.ac.uk
Abstract
A sound metrological infrastructure for chemical measurement is a requirement for universal comparability of the results used in international trade. Over the last few decades such a system has been put in place and a corresponding improvement in quality has been evident. But an undue emphasis on traceability and a particular approach to uncertainty have now become counterproductive. Rather than leading to further improvements in the quality of analytical results, these widely-held precepts have distracted analytical chemists from the real problems and progress has stalled. The counter-arguments proposed here are that: (a) broken traceability to the SI is seldom a problem in chemical measurement, both because the metrological infrastructure is very effective and because the applications of analysis seldom demand a relative uncertainty smaller than 1%; (b) the most serious sources of error in analysis usually arise from shortcomings in the chemical preparation of the test solution and matrix mismatch between test solution and calibrators, so cannot meaningfully be attributed to a broken traceability to the SI; (c) the cause-and-effect approach to uncertainty has shortcomings and leads to an ineffectual validation of measurement procedures; (d) properly validated analytical procedures are by definition already fit for purpose, and problems arise only when the analyst deviates from the procedure or when the test materials fall outside its validated scope.
Note on scope and terminology
The following discussion applies to well-found laboratories capable of meeting the requirements of accreditation. To avoid misunderstanding, I have adhered to the VIM3 hierarchy of methodology-related terms.1Measurement principles simply refer to the physical phenomenon observed (e.g., atomic spectrometry). Methods refer to generic descriptions of the operations involved (e.g., flame atomic absorption after acid decomposition). Procedures contain sufficient detail to allow any suitably trained person to perform the measurement in an almost exactly reproducible manner. Here a procedure is taken to include a specification of its ‘scope’, the range of matrices and analyte concentrations covered by the validation. Only procedures are considered in any detail.Introduction
The formation of the European Union brought into sharp focus the need for the mutual recognition of certificates relating to safety, so that the passage of goods over national boundaries would not be hindered by the need for duplicate testing. For this measure to be acceptable, it was necessary for the results of chemical measurement to be reliable and comparable no matter what their origin. This in turn implied that making such measurements should be on a demonstrably sound metrological basis. As an outcome, from the early 1990s there has been an upsurge of interest in the quality of chemical measurement and in clarifying its metrological foundations. This resulted in the rise of proficiency testing, accreditation, national reference laboratories undertaking key comparisons, and the production of GUM2 with its numerous derivative international protocols, standards and guides. While the quality of chemical measurement overall has improved under this regime, there are still shortcomings that we need to eliminate or reduce. Unfortunately an unfounded emphasis on a narrow selection of precepts related to traceability has diverted the attention of analytical chemists away from the real problems.Let me first convince the reader that there are still problems. A few examples suffice, brought to light by proficiency testing or blind quality control.
• GeoPT is an international proficiency testing scheme for rock analysis in which about 70 participants report results for up to 70 elements in one test material per round.3 Criteria for appropriate uncertainty used in calculating z-scores are published before the test. In round 32 (2012), about 7% of the absolute z-scores exceeded 5.0. This was by no means unusual. The proportion was higher in round 33 (2013). (An absolute z-score of 5.0 implies that the deviation of the participant's result from the assigned value was five times greater than the uncertainty defined by the scheme as fit for purpose. The proportion of the absolute z-scores exceeding 5.0 expected in a population of laboratories compliant with the prescribed criterion for uncertainty is about 0.00005%.)
• FAPAS is a large international proficiency testing scheme for foodstuff analysis.4 Criteria for appropriate uncertainty used in calculating z-scores are published before the test. Data were taken from the latest 15 rounds posted on 21st May 2014. These rounds provided tests for 38 combinations of diverse food matrices and analytes. A proportion of 5.1% (78/1529) of the absolute z-scores exceeded 5.0.
• In a recent study from an undisclosed source, a series of blind duplicate samples were sent to a laboratory holding an accreditation that included in its specification a maximum level of uncertainty umax for the analysis of the named analytes and test material. The repeatability standard deviation ![[small sigma, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e111.gif) r estimated from the duplicate results was about 2umax. (It is impossible for the true value σr to exceed the true standard uncertainty υ. Usually in analytical procedures we find σr ≈ υ/2.)
r estimated from the duplicate results was about 2umax. (It is impossible for the true value σr to exceed the true standard uncertainty υ. Usually in analytical procedures we find σr ≈ υ/2.)
A noteworthy feature of the first two examples is that in proficiency tests the participants know that their results will be under third-party scrutiny: they were presumably striving to conform to the schemes' prescribed criteria of uncertainty and so get ‘satisfactory’ z-scores. Further important points emerge. (a) In most instances a properly designed internal quality control system would have flagged up any unacceptable performance before the results were released by the laboratory to the proficiency test scheme or customer. We can infer that the laboratories reporting these very unsatisfactory results were not just conducting the analysis ineptly but also employing an inadequate IQC system. (b) In food analysis, many of the procedures would have been validated by published collaborative (inter-laboratory) study, widely regarded as the most thorough type of validation. We see that using a carefully validated (and in most instances accredited) procedure does not in itself make a laboratory immune from problems. (c) In the third example the laboratory involved was apparently unable to make an adequate estimate of the uncertainty of its measurements results.
Seemingly the huge international efforts put into improving the reliability of chemical measurement has not been completely effective. In my view, this shortcoming persists because certain precepts, as interpreted today, are emphasised beyond their utility and have become counterproductive. These questionable assumptions can be summarised as follows.
• Chemical measurement is unreliable because some existing procedures are not fit for purpose.
• This unfitness for purpose is the outcome of a breakdown of traceability of the result to SI units.
• The problem could be rectified only if measurement uncertainty could be determined by a procedure appropriate for the determination of the physical constants of nature.
These assumptions have diverted analysts' attention away from the real causes of the remaining problems. In contrast I contend that: (a) that most established analytical procedures are fit for purpose—validated procedures are fit for purpose by definition—but problems in the results of chemical measurement are caused by deviation from the validated procedure; (b) that there is no breakdown in traceability to the SI in the overwhelming majority of analytical procedures; and (c) that while the cause-and-effect method is useful to illustrate the uncertainty concept, in chemical measurement it is not appropriate for routine analysis—it is not uniquely valid and, in the majority of instances, gives rise to an underestimate.
Validation and fitness for purpose
Evaluating the uncertainty associated with a procedure is the all-important outcome of validation. But a rational basis for the decision to use a particular analytical procedure demands that we know in advance the level of uncertainty that will best fulfil the customer's needs. We can then compare this fit-for-purpose uncertainty with the uncertainty of any candidate validated procedure. It is difficult, however, to attach an objective value to the uncertainty that represents fitness for purpose. Instead, we rely nearly always on professional judgement and an agreement between analyst and customer. Such judgements are implicitly financial. An uncertainty that is too great will tend to result in inept decisions with possibly-severe financial penalties: one that is too small will be unduly expensive to procure. These principles can be made quantitative although there are several practical difficulties that have yet to be resolved in detail.5Despite these difficulties surrounding fitness for purpose, evolutionary tendencies come to our aid without any high-level intervention or financial modelling. In an established application sector, such as food analysis, the suite of procedures in use evolves over time by a process of adaption. Procedures that are unnecessarily accurate (and therefore needlessly expensive) tend to be set aside in favour of cheaper and less accurate ones. Procedures that give rise to too great a proportion of inept decisions tend to be replaced by more accurate (and more expensive) ones. The eventual outcome is a suite of procedures giving results with appropriate (fit-for-purpose) uncertainties over the whole range of relevant concentrations. This process is thought to be the origin of the empirically-observed Horwitz function that describes the trend of reproducibility standard deviation as a function of mass fraction in the analysis of foodstuffs.6,7 Because of this tendency to adaptation, it is a cogent supposition that most procedures used in established fields are already fit for purpose. It is therefore only departure from such a procedure that causes serious analytical errors.
Many, perhaps most, of such departures can be attributed to human error.8,9 Assuming the existence of a well-found laboratory with traceable standards, analytical results are likely to be inaccurate when either of the following occurs: (a) deviation from a well validated procedure; or (b) the use of a procedure outside the scope of its validation. It is clear from this that validation plays an axial role in obtaining accurate results, so it essential that critical aspects of validation are well understood and executed. However, two aspects of validation—the definition of the applicable scope of a procedure and the estimation of the uncertainty associated with its results—commonly fall short of requirements.
What exactly is ‘traceability’?
The need in chemical measurement for traceability to Le Système international d'unités (SI) through an ‘unbroken chain of calibrations’ is fundamental and as such hardly needs emphasising. In recent metrologically-slanted literature, however, numerous papers strongly draw attention to this principle, as if a breakdown in traceability were a common failing in analytical laboratories. The reality is quite different. Routine analytical results seldom need a relative uncertainty smaller than 1% to be fit for purpose. Only when analysis supports such industries as precious metal production is a smaller relative uncertainty required. But by virtue of a high-quality international measurement infrastructure, the SI standards of mass, amount and volume can be easily translated to the analyst's bench, under routine conditions, with a relative uncertainty better than 0.1%. Shortcomings in chemical measurement therefore cannot reasonably be attributed to a breakdown in the traceability chain. Undue attention to traceability to the SI, therefore, will seldom reduce true (as opposed to estimated) uncertainty but often distract the analyst's attention away from the real causes of problems. Moreover, even if it resulted, a reduction of uncertainty below the level that defines fitness for purpose would be pointless scientifically and wasteful economically.A schematic diagram of a typical chemical measurement procedure (Fig 1) helps to pinpoint the major sources of uncertainty. The actions requiring traceability to the SI are clearly marked but other, almost always greater, causes of error cannot be referred back to the SI in any meaningful way. Setting aside uncertainty from sampling as a separate issue, the major problems encountered in analysis concern (a) the efficacy of the chemical preparation of the test solution from the test portion, (b) mistakes in the preparation of the calibrators, and (c) the comparison between the test solution and the calibrators via the selected measurement principle. The test solution can deviate from the correct concentration by virtue of loss or gain of analyte, loss through incomplete chemical treatment of the test portion or gain by contamination from the laboratory environment. Then the comparison between the treated test solution and calibrators can suffer from loss or gain of the net analytical signal brought about by matrix mismatch. These almost ubiquitous features of chemical analysis can seldom be predicted from theory and, in any event, cannot be meaningfully referred to the SI. On those grounds we can reject the idea that shortcomings in chemical measurement are related to incomplete traceability to the SI.
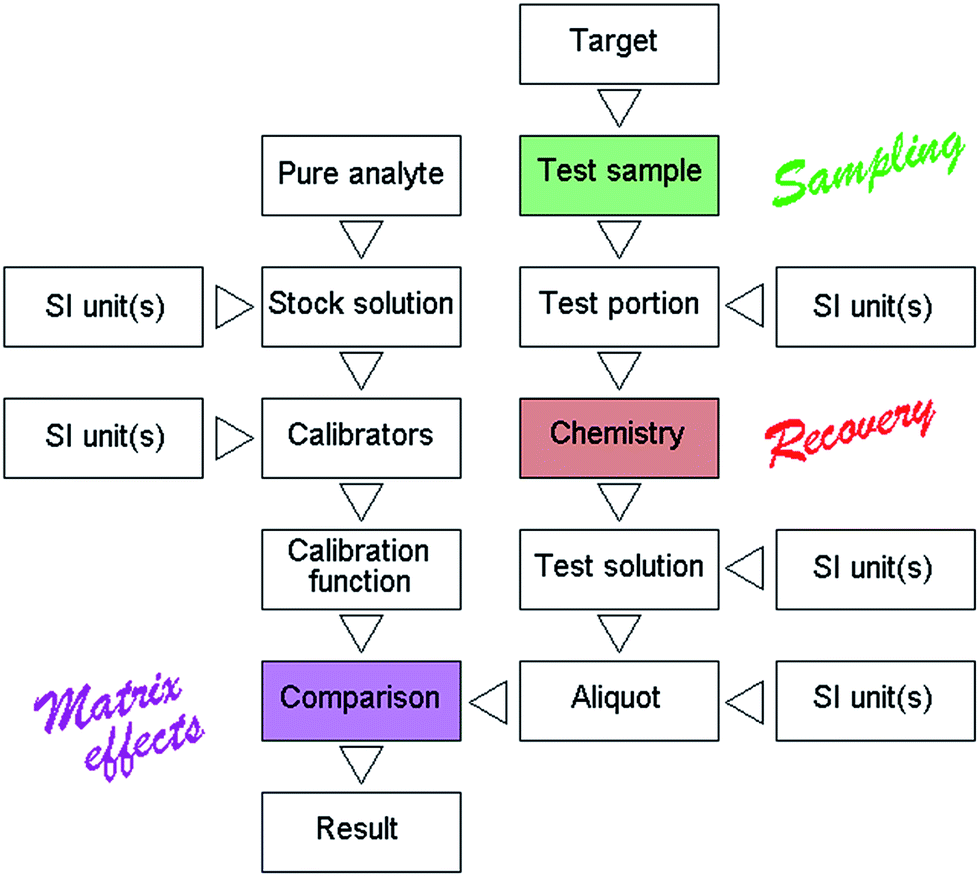 | ||
| Fig. 1 Schematic diagram of a chemical measurement, showing actions requiring traceability to the SI, and features that act as major sources of uncertainty (colour). | ||
Estimating uncertainty
This undue emphasis on traceability is related to the estimation of uncertainty, which demands ‘an unbroken chain of calibrations’ traceable to the SI.1 In the corresponding estimation procedure, the scientist creates a statistical model of the measurement procedure by iteratively breaking down the analytical procedure into sub-procedures until they cannot be further so divided. Then a variance is attributed to each of these irreducible sub-procedures and the variances are combined according to the mathematical laws of error propagation to give the square of the standard uncertainty.2,10 This is sometimes referred to as the ‘cause-and-effect’ method or, less formally, the ‘bottom-up’ or ‘splitter’ approach. The method is held by some of its advocates to be uniquely valid, even an invariant truth rather than an estimate.11 This latter assertion is plainly incorrect when all of the component variances to be combined are themselves estimates based on finite (often very small) numbers of observations, on conventional values, on manufacturers' specifications, or even on judgement alone.There are two types of problem inherent in the splitter approach that potentially affect all measurements but beset chemical measurements in particular, namely (a) parametric, and (b) structural, shortcomings in the statistical model of the procedure. A parametric problem occurs when the variance attributed to a sub-procedure is incorrect. This could be rectified in principle by substituting the correct value into the model. But how would the analyst detect such a problem? It could only be done by comparison of the outcome with an independent estimate of uncertainty, a sort of reference value, to show that the splitter estimate was incorrect. No such reference value exists. The more serious problem occurs when the structure of the model itself is incomplete. Such an occurrence is commonplace to some degree in analytical procedures: there are usually numerous factors, both inherent and extraneous, of which the level could vary and thus conceivably affect the net analytical signal: there is also a potentially much larger number of their possible interactions. There is no way of rectifying this problem because few of these factors or interactions can be modelled adequately and most of them are neither detected nor even suspected. This problem gives rise to ‘dark uncertainty’.12,13 On this basis alone there are grounds for suspecting that the ‘splitter’ method will tend to underestimate the uncertainty associated with chemical measurement. As we shall see there is much evidence supporting this contention.
The extreme alternative method is the ‘top-down’ or ‘lumper’ approach, in which the standard uncertainty is equated with the reproducibility standard deviation sR of results of a procedure based on an inter-laboratory study such as a collaborative trial. The essential idea here is that the variable factors and their interactions that affect an analytical result will be close-to-randomly sampled in a sufficiently large study. Many practitioners object to this idea on the grounds that the complete scope for variation among the factors will be insufficiently sampled in an interlaboratory study. On that basis alone ‘splitters’ consider that sR will be tend to underestimate standard uncertainty. They also object to sR on the specious grounds that the estimate does not take procedure bias into account, and (equivalently) that we can say nothing about its traceability. As we have seen above, there is no substantive issue with traceability to the SI in most chemical measurement, because we can safely assume that all of the participants in a collaborative trial will have well-found laboratories with traceably calibrated equipment. Moreover, any candidate procedure will have been subjected to a very careful consideration in respect of bias before the very costly interlaboratory study is undertaken.
We thus see two schools of thought each claiming (possibly correctly) that the rival method of estimating uncertainty will tend to produce an underestimate. The inference can only be that the method tending to give the greater estimate will be closer to correct, but may still be somewhat too small. In several instances where such comparisons have been made, we see that sR tends to exceed the splitter estimate by a factor typically approaching two.12
Additional evidence can be gleaned from interlaboratory studies in which participants report their results with associated uncertainty estimates. In such studies, the reported uncertainties usually fail to account for the variation among the results.12 Furthermore, it is an almost invariable observation that in collaborative trials of a procedure, sR exceeds the repeatability standard deviation sr by a typical factor of two.14 Were there no dark uncertainty, the two standard deviations would be tend to be equal, that is, the repeatability standard deviation would account for all of the variation between laboratories. A large number of studies have thus demonstrated a tendency for analysts to underestimate their uncertainties. This seems to be an outcome of inter alia an incomplete validation.
(Note: in practice among analytical chemists there is a continuum of approaches to estimating uncertainty that fall between the extremes of ‘splitting’ and ‘lumping’.)
What is going wrong, and how can we cure it?
What then accounts for the remaining problems in chemical measurement if not a broken traceability chain? A central issue seems to be a failure to estimate a realistic uncertainty during validation. Some of the physical factors contributing to this problem can be readily identified. One such is the tendency of laboratories to base their uncertainty on repeatability standard deviation sr, when it is clear that sR is a more appropriate starting point.8 Moreover, the method used to estimate sr is itself flawed when it is based on rapidly repeated analyses of a single certified reference material (CRM). This method is deficient in several different ways.15 Firstly, CRMs are usually more finely divided and closer to homogeneity than the routine test samples. This tends to make the variance smaller than apposite for routine analysis. Secondly, the replicated test solutions are likely to be analysed in an unbroken sequence in the shortest possible time, which is again atypical of routine operation and in itself leads to underestimation. Thirdly, focussing all of the effort on a single reference material precludes any exploration of the defined scope of both matrix variation and analyte concentration, and thereby excludes these further sources of variability. (The more serious mistake of attempting to estimate sr by repeated measurements of a single test solution or calibrator hopefully does not need to be considered here.) There are almost certainly in addition other features of chemical analysis that contribute to the overall ‘dark uncertainty’ that leads to the ubiquitous but unexplained variation among laboratories.Finally there are psychological factors involved as well as physical: obtaining a small uncertainty tends to be seen as a measure of an analyst's skill, a laboratory's competence, or a method developer's ingenuity, a situation that tends to generate an unconscious data selection bias. If sr looks disappointingly large, it is tempting simply to repeat the experiment and, should a smaller sr emerge, to use that value.
Where a procedure has been validated, that is with a good estimate of the uncertainty, any problems in the accuracy of the results mostly seem to stem from human error,8 a deviation by the analyst from the validated procedure. This deviation can take two forms, a departure from the documented details of the procedure, or an application of the procedure to test materials outside the scope of the validation. Moreover, either of these problems can affect a whole run of analysis or be sporadic—restricted to just a few of the test materials that comprise the run. (A ‘run’ is the period during which repeatability conditions are regarded as prevailing.) Another reported major cause of problems is undetected instrument failure. When any of these circumstances occur, it renders the true uncertainty completely unknown (not simply underestimated), sometimes to an extent that has practical consequences.
Benefits and limitations of internal quality control
Internal quality control (IQC), if properly devised and executed,16 can go a long way towards detecting such occasional problems before the analytical results in a run are released and, moreover, can trigger an investigation of the problem. Logically, unless every run of analysis is subjected to IQC, we cannot be confident with a high probability that an uncertainty properly determined at validation is maintained during the long-term use of a procedure.However, IQC (as currently understood16) cannot guard against problems arising from the use of a procedure outside the scope of its validation. Analysts must be at all times aware of the possibility of encountering test materials with compositions outside the validated scope. In validating a procedure, the defined range of matrix types among the test materials must be reasonably limited. Operational categories such as ‘food’ or ‘soil’ may in some cases be far too inclusive. Soil for example could consist largely of silica, or clay minerals, or chalk, or peat, or, in tropical countries, laterite (largely an Fe2O3–Al2O3 mixture), or any of their various mixtures. Such diverse matrices could have a marked influence on the efficacy of a chemical decomposition and, just as importantly, on matrix effects encountered during the subsequent comparison of the test solution and calibrators. The scope must also include a specification of the concentration range covered by the validation process. This is because uncertainty tends to vary widely with the concentration of the analyte.
The surrogate test material used in IQC, however, with its invariant matrix and fixed analyte concentration, would show no untoward effect in a run that included out-of-scope test materials. Sporadic (transient) problems affecting a small part of a run are also unlikely to be detected by internal quality control. Several methods are available for detecting sporadic blunders and these measures should where possible be deployed as routine alongside IQC. Methods for detecting an out-of-scope matrix are not as yet well documented.17
Conclusions
The foregoing discussion demonstrates that poor analytical performance can seldom be reasonably attributed to a breakdown in traceability to the SI. In contrast, poor performance is clearly linked to a broad tendency for analysts to underestimate their uncertainty. This in turn stems from inadequate models of the measurement procedure, which can take no account of the ‘dark uncertainty’ that is ubiquitous in the results of interlaboratory studies (and presumably generally). However, a major proportion of poor results could be eliminated by the deployment of a well-designed internal quality control system, backed by regular participation in proficiency test schemes and the use of certified reference materials. A further proportion of poor results could be eliminated by the development of measures to detect the inadvertent use of a procedure on test materials outside its validated scope.The reproducibility standard deviation in an unbiased procedure, estimated by interlaboratory study, is usually a reasonable estimate of uncertainty. However, collaborative trials are very costly to carry out and, apart from in the food sector, seldom attempted. Sometimes, when a subset of the participants use very similar procedures, a value of σR can be estimated from results of proficiency tests. An alternative value, which might at least serve as a benchmark for comparison, could be taken as double the repeatability standard deviation, which can be estimated in a single-laboratory validation. An independent estimate that was substantially less than such a benchmark should be regarded as prima facie suspect. For this purpose, however, the repeatability value must be estimated under conditions of measurement that are all but impossible to simulate in a one-off validation exercise—the estimation must rest on real-life conditions, that is, when the procedure is ‘bedded down’ in routine use on routine test materials.16 The estimation protocol outlined in Appendix A is one such.
I must acknowledge that there exists a so-far unresolved problem in using realistic estimates of uncertainty in a commercial context. A laboratory offering an analytical service with a realistic uncertainty is likely to lose custom to a competitor claiming an unrealistically lower uncertainty at the same price. This commercial pressure encourages the use of approaches to estimation that bias the apparent uncertainty downwards. This tendency might at first seem impossible to forestall in an open market, but there are countermeasures that could be taken at all levels of the sociology of chemical measurement.
• Analytical laboratory managers could educate their customers in the commercial benefits of having fit-for-purpose results with realistically estimated uncertainties.
• Quality managers in laboratories could ensure that an adequate IQC programme is in place, and that some attempt is made to detect sporadic errors and to identify out-of-scope test materials.
• Customers from the outset could make clear to laboratories that they intend to apply blind quality control to ensure that laboratories fulfil contractual uncertainty specifications.18
• Funding agencies could ring-fence adequate support for studies relating to uncertainty and quality in chemical measurement.
• Accreditation agencies could pay special attention to the way in which uncertainties are estimated by a candidate laboratory, and to ensure that any uncertainties advertised or claimed were consistent with the laboratory's proficiency test scores and with the records of an adequate internal quality control system.
• Universities and colleges providing higher education in chemical measurement could make better provision for the coverage of quality issues in chemical analysis supporting industry, trade and public service.
• Professional bodies could set standards for such training in analytical quality and uncertainty that have to be fulfilled for the educational institution to receive endorsement of its courses, or for an analytical chemist to receive chartered status. They could further disseminate impartial information about uncertainty and quality to the wider community.
Appendix A
The following protocol gives rise to an estimate of repeatability standard deviation, where appropriate as a function of the analyte concentration, that takes full account of variations in conditions (a) within runs, and (b) among allowed variations in the matrix of the test materials and the concentrations of the analytes.• Adhere always to the validated procedure and its defined scope, under routine conditions of measurement.
• Within each run of analysis, make the measurement on duplicate test portions of all (or a random selection) of the routine test samples. Place the duplicate test portions at individually randomised positions within the sequence of test portions in the run.
• Repeat the above in a number of separate runs.
• Estimate the repeatability standard deviation, where appropriate as a function of concentration, by considering the median of the absolute differences between corresponding pairs of results.18
Notes and references
- JCGM 200, International vocabulary of basic and general terms in metrology (VIM), 3rd edn, 2008, http://www.bipm.org/vim Search PubMed.
- JCGM100, Guide to the expression of uncertainty in measurement (GUM), Bureau International des Poids et Mesures, Sèvres, France, 2008, http://www.bipm.org/en/publications/guides/gum.html Search PubMed.
- GeoPT, International Association of Geoanalysts, (Sec) Ms Jennifer Cook, British Geological Survey, Keyworth, NG12 5GG.
- FAPAS, Food and Environment Research Agency, Central Science Laboratory, Sand Hutton, York, YO41 1LZ.
- T. Fearn, S. Fisher, M. Thompson and S. R. L. Ellison, Analyst, 2002, 127, 818–824 RSC.
- W. Horwitz, L. R. Kamps and K. W. Boyer, J. - Assoc. Off. Anal. Chem., 1980, 63, 1344–1354 CAS.
- M. Thompson, Analyst, 1999, 124, 991 RSC.
- S. R. L. Ellison and W. A. Hardcastle, Accredit. Qual. Assur., 2012, 17, 453–464 CrossRef PubMed.
- Analytical Methods Committee, AMCTB no. 56, Anal. Methods, 2013, 5, 2914–2915 RSC.
- Quantifying uncertainty in analytical measurement, Eurachem/CITAC Guide, ed. A. Williams, S. L. R. Ellison and M. Roesslein, 2nd edn, 2000, http://www.eurachem.com/ Search PubMed.
- Anonymous referees.
- M. Thompson and S. R. L. Ellison, Accredit. Qual. Assur., 2011, 16, 483–487 CrossRef.
- Analytical Methods Committee, AMCTB no. 53, Anal. Methods, 2012, 4, 2609–2612 RSC.
- M. Thompson and P. J. Lowthian, J. AOAC Int., 1997, 80, 676–679 CAS.
- M. Thompson, Anal. Methods, 2012, 4, 1598–1611 RSC.
- M. Thompson and B. Magnusson, Accredit. Qual. Assur., 2013, 18, 271–278 CrossRef.
- Analytical Methods Committee, AMCTB no. 49, www.rsc.org/amc.
- Analytical Methods Committee, AMCTB no. 54, Anal. Methods, 2012, 4, 3521–3523 RSC.
| This journal is © The Royal Society of Chemistry 2014 |