 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sensitive electrochemical determination of yohimbine in primary bark of natural aphrodisiacs using boron-doped diamond electrode†
Ľ.
Švorc
*a,
D. M.
Stanković
b,
E.
Mehmeti
c and
K.
Kalcher
c
aInstitute of Analytical Chemistry, Faculty of Chemical and Food Technology, Slovak University of Technology in Bratislava, Radlinského 9, Bratislava, SK-812 37, Slovak Republic. E-mail: lubomir.svorc@stuba.sk
bDepartment of Analytical Chemistry, Innovation Center of the Faculty of Chemistry, University of Belgrade, Studentskitrg 12-16, Belgrade, 11000, Serbia
cInstitute of Chemistry – Analytical Chemistry, Karl-Franzens University, Universitätsplatz 1, Graz, A-8010, Austria
First published on 16th April 2014
Abstract
For the first time, a simple and sensitive analytical method for the direct determination of yohimbine is presented using differential pulse voltammetry with a boron-doped diamond electrode. Two irreversible oxidation peaks, a distinct one at +0.80 and a second poorly-defined one at +1.65 V, were observed when cyclic voltammetry was carried out in Britton–Robinson buffer solution at pH 7 (vs. Ag/AgCl). With optimized differential pulse voltammetric parameters (pulse amplitude 100 mV, pulse time 25 ms, step potential 5 mV and scan rate 10 mV s−1), the current response of yohimbine at +0.80 V was linearly proportional to the concentration in the range from 0.25 to 90.9 μmol L−1 with a low detection limit of 0.13 μmol L−1 (0.046 mg L−1) and a good repeatability (relative standard deviation of 2.5% at 18.4 μmol L−1 for n = 6). The practical applicability of the developed method was demonstrated by the assessment of the total content of yohimbine in extracts of the primary bark of natural aphrodisiacs such as Pausinystalia yohimbe and Rauvolfia serpentina with recoveries in the range of 92–97%. The proposed electrochemical procedure represents an inexpensive and effective analytical alternative for the quality control analysis of products containing yohimbine and other biologically and structurally related alkaloids used as natural dietary supplements.
1. Introduction
Yohimbine (17α-hydroxy-yohimban-16α-carboxylic acid methyl ester, YOH) is an important naturally occurring indole alkaloide. It primarily acts as a stimulant inhibiting the function of monoamine oxidase.1 As an antagonist of α2-receptors it can increase brain noradrenaline cell firing and release.2 Some of its side effects are also manifested as anxiety, headaches and increased urinary output.3Pausinystalia yohimbe (Corynanthe yohimbe) and Rauvolfia serpentina are medicinal trees native to Africa, and South and East Asia. Their bark contains YOH as the main species used both clinically and traditionally to treat cardiac diseases and sexual dysfunction.4 Considering the consumption of YOH with dietary supplements or therapeutic pharmaceuticals, and the important impact of YOH on the human system, the development of novel, simple and sensitive procedures for the determination of YOH is significant.Various analytical methods for the detection and quantification of YOH and other structurally related alkaloids in different matrices (plant, bark, pharmaceuticals, urine etc.) have been described in previous scientific papers. Most of them involve spectrometry5,6 and separation methods such as capillary electrophoresis,7 high-performance liquid chromatography with electrochemical detection,8,9 UV spectrophotometry,10 mass spectrometry11,12 and gas chromatography combined with mass spectrometry.13–15 These methods usually offer very useful analytical information in terms of the identification and quantification of YOH and its degradation products and/or metabolites. However, they require highly sophisticated and expensive instrumentation and often require a long analysis time and laborious sample pretreatment processes (derivatization, purification, sorbent extraction steps). Furthermore, the demands for highly skilled workers often constrain their use in routine analytical practice. In this sense, the development of novel, simple, cheap and sensitive analytical methods for the detection and quantification of YOH and other alkaloids in various matrices is still of great interest.
Electroanalytical techniques have been confirmed to be excellent alternatives for the determination of many biologically relevant electroactive compounds, as they are simple and inexpensive and require a relatively short analysis time. Moreover, they yield useful information on the kinetics and charge transfer mechanisms involved in a particular electrode reaction.16 Concerning the electrochemistry of YOH, however, there is only one short report available on its basic voltammetric characterization using platinum and gold rotating disc electrodes.17 An oxidation peak at a potential of about +1.0 V vs. SCE in 0.1 M sulphuric acid was observed for high concentrations of YOH. At low concentrations limiting current plateaus were obtained due to the adsorption of oxidation products on the electrode surface. Similarly, when traditional carbonaceous electrodes were used difficulties arose from electrode passivation and the poor electrochemical activity of YOH. Therefore only a very limited number of papers have been published on the electrochemical determination of YOH. Furthermore, it is also interesting that there has been no report on the use of modified electrodes for the determination of YOH.
Nowadays, boron-doped diamond (BDD) has attracted a great amount of attention as a unique electrode material due to its superior electrochemical properties such as its wide potential window, low background current, high sensitivity and long-term stability as well as the negligible adsorption of neutral and polar species due to the presence of sp3 hybridized diamond carbon atoms.18,19 However, the analytical performance of BDD electrodes is strongly affected by the quantity of doping agent, morphologic factors, the presence of impurities and crystallographic orientation.20 Besides these aspects, the physical and chemical properties of BDD electrodes are also influenced by controllable (hydrogen or oxygen) surface termination achieved either by exploitation of oxygen and hydrogen plasmas or by electrochemical pretreatment with highly anodic and cathodic potentials.21 Our research group has extensively applied BDD electrodes as effective alternatives to traditional electrode materials for the sensitive determination of various biologically active compounds important in the area of environmental,22–25 food26 and drug analysis.27–32 Likewise, various applications of BDD electrodes in the electroanalysis of organic compounds have been recently reported by different groups under the leadership of Şentürk,33–35 Fatibello-Filho36–38 and Ozkan.39,40
In this paper, we describe the development of a novel, simple and sensitive analytical method for the direct electrochemical determination of YOH as a cost-effective and less time-consuming alternative to separation techniques. A study of the voltammetric behavior of YOH on BDD electrodes is also presented. Moreover, to our knowledge, no attempt related to the electrochemical determination of YOH using a BDD electrode has been carried out up to now. The practical applicability of the developed method is demonstrated by determining the total content of this alkaloid in the extracts of the bark of natural aphrodisiacs such as Pausinystalia yohimbe and Rauvolfia serpentina.
2. Experimental
2.1. Chemicals
Yohimbine hydrochloride (purity ≥ 98%, YOH) was purchased from Sigma-Aldrich (Austria) and used without any further purification. Britton–Robinson (BR) buffer solution was prepared by mixing phosphoric acid, acetic acid and boric acid (with all components at 40 mmol L−1 concentration) and adjusting the pH of the buffer with sodium hydroxide (0.2 mol L−1). A stock standard solution of YOH (1 mmol L−1) was prepared by dissolving 39.1 mg of its solid hydrochloride standard in 100 mL water, the solution was then stored in a refrigerator at 4–6 °C prior to use. Working solutions of lower concentrations of YOH were freshly prepared on the day of the experiment by appropriate dilution with the supporting electrolyte. All other chemicals were of analytical reagent grade. Deionized water with a resistivity not less than 18 MΩ cm (Millipore Milli-Q system) was used for the preparation of all the solutions.2.2. Apparatus
Voltammetric measurements were performed with an AUTOLAB PGSTAT 302N (Metrohm Autolab B.V., The Netherlands) potentiostat/galvanostat controlled by NOVA 1.9 electrochemical software. The three electrode system consisted of Ag/AgCl/3 M KCl and platinum wire as the reference and counter electrodes, respectively. A BDD electrode (Windsor Scientific Ltd, Slough, Berkshire, United Kingdom) embedded in a polyether ether ketone (PEEK) body with an inner diameter of 3 mm, a resistivity of 0.075 Ω cm and a boron doping level of 1000 ppm (as declared by the supplier) was used as the working electrode. All of the pH values were measured using a pH meter (Orion, model 1230) with a combined electrode (glass-reference electrodes), which was calibrated weekly with standard buffer solutions. Each potential reported in this paper is given against the Ag/AgCl/3 M KCl reference electrode at a laboratory temperature of 25 ± 1 °C.2.3. Measurement procedures
A known volume of stock solution of YOH was made up to 40 mL with supporting electrolyte and subsequently transferred into the voltammetric cell. Prior to its first use every day, the BDD electrode was rinsed with deionized water and rubbed very gently with a piece of damp silk cloth until the surface of the electrode had a mirror-like appearance. Subsequently, it was anodically pretreated by applying +2 V for 180 s in 1 M H2SO4 solution in order to clean the electrode surface (get rid of any impurities) followed by cathodic pretreatment at −2 V for 180 s to attain a predominantly hydrogen-terminated surface. In this sense, the hydrogen-terminated surface of the BDD electrode is explicitly the most commonly used in electrochemical measurements in our laboratory. The nonpolar hydrogen-terminated surface gives this electrode a hydrophobic nature, thus its ability to adsorb polar substances (YOH) in the analyzed solution is very low. The hydrogen-terminated BDD electrodes are also highly stable and sensitive for the analysis of a number of biological species. On the other hand, short oxygen plasma treatment leads to oxygen-containing functional groups on the BDD electrode making its surface hydrophilic and thus more prone to the adsorption of polar substances. This is the explicit reason for the use of the hydrogen-terminated BDD electrode, thus we haven't tried to perform the cathodic pretreatment (hydrogen termination) as the first step followed by anodic pretreatment (oxygen termination) in order to attain a more hydrophobic surface which is resistant to fouling by the analyte or products of the electrode reaction. The peak currents in cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were evaluated without background correction. Every point of the calibration curve represents the corresponding average of three successive measurements of the standard solution of YOH. Linear least-square regression (OriginPro 8.0, OriginLab Corporation, USA) was used for the evaluation of the calibration curve; the relevant results (slope and intercept) are reported with a confidence interval of 95% probability. The detection limit was calculated as three times the standard deviation for the blank solution divided by the slope of the calibration curve. The recovery analysis and analysis of real samples was performed by the standard addition method.2.4. Sample preparation
Commercial products (natural aphrodisiacs) of primary bark from Pausinystalia yohimbe and Rauvolfia serpentina were purchased in a local shop in Vienna. One gram of dried and powdered bark was macerated with 20 mL of methanol, filtered and the filtrate evaporated to dryness at 70 °C. The residues were dissolved in 30 mL of HCl (2%, m m−1) and filtered. The filtrate was neutralized to pH 7 with 0.1 M NaOH and extracted three times with 20 mL of chloroform. The chloroform layers were combined and evaporated to dryness; the residues were subsequently dissolved in 20 mL of methanol and filtered.41 The extract was filled with deionized water to 50 mL in a volumetric flask. An aliquot amount of the prepared sample (200 μL) was added to 40 mL of BR buffer solution at pH 7 in the electrochemical cell and analyzed by the standard addition method.3. Results and discussion
3.1. Electrochemical behavior of YOH on the BDD electrode
CV measurements were conducted to investigate the electrochemical behavior of YOH on a BDD electrode employing various supporting electrolytes, such as BR (pH 2–12) and phosphate (pH 2–8) buffer solutions as well as acetic acid (0.1 M), sulfuric acid (0.1 M) and nitric acid (0.1 M). It was found that all of the used acids were not convenient as there was no measurable or detectable current response for YOH. The analyte gave a signal in BR and phosphate buffers, and the electrochemical behavior in both electrolytes was similar. Nevertheless, in BR buffer solution (pH 7), the analytical signal was characterized by a higher magnitude, a lower background current and better repeatability when compared to that observed in phosphate buffer (pH 7). Hence, the former supporting electrolyte was chosen for further studies.Fig. 1 displays typical cyclic voltammograms in the absence (curve a) and presence of 0.1 mmol L−1 YOH (curve b) in BR buffer solution at pH 7 over the whole investigated working potential range. In the anodic scan a well-defined oxidation peak appears at +0.82 V, whereas a second one at +1.65 V is only poorly established with a small magnitude. In the reverse scan no reduction peak could be observed demonstrating that the charge transfer during the redox reaction of YOH on the BDD electrode is totally irreversible. Furthermore, in the absence of YOH no voltammetric peak was recorded and the background current appears to be low.
 | ||
| Fig. 1 Cyclic voltammograms of (a) 0 mol L−1 and (b) 0.1 mmol L−1 YOH in BR buffer solution at pH 7 on a BDD electrode at a a scan rate of 50 mV s−1. The effect of pH on the peak potential Ep (■) and peak current Ip (▲) of the first peak of YOH on the BDD electrode is shown in the inset. | ||
3.2. Effect of pH
The effect of pH on the peak potential of the first oxidation peak (Ep) with the corresponding peak current (Ip) of 0.1 mmol L−1 YOH was systematically investigated by CV in the pH range of 2–12 using BR buffer solution. This study revealed that Ep underwent a shift towards more negative values with an increase of the pH of the supporting electrolyte as depicted in the inset of Fig. 1. This behavior confirms the participation of protons in the oxidation reaction of YOH on a BDD electrode. The dependence is linear over the whole studied pH range and may be expressed by the following equation:| Ep (V) = 1.197 (V) − 0.049 × pH, R2 = 0.998 |
The calculated slope indicates the involvement of the same number of protons and electrons in the electrode reaction of YOH on the BDD electrode. To our knowledge, there is only one short report on the basic voltammetric characterization of YOH using platinum and gold rotating disc electrodes suggesting a mechanism for the electrochemical oxidation of YOH, which served as a basis for our interpretation.17 Although a closer clarification of the oxidation mechanism on the BDD electrode has been beyond the aim of this study, the proposed overall oxidation mechanism of YOH is believed to involve four electrons and protons. In this sense, the first oxidation peak at around +0.82 V may be assigned to the oxidation of the hydroxyl group of YOH including the loss of two electrons and two protons to give a ring ketone. Subsequently, the second and very poorly-defined peak at about +1.65 V is probably associated with the oxidative deprotonization of the carbon at position 6. Moreover, YOH can offer a number of sites for deprotonization up to 12 electrons in pairs.17 The proposed mechanism is shown in Scheme 1.
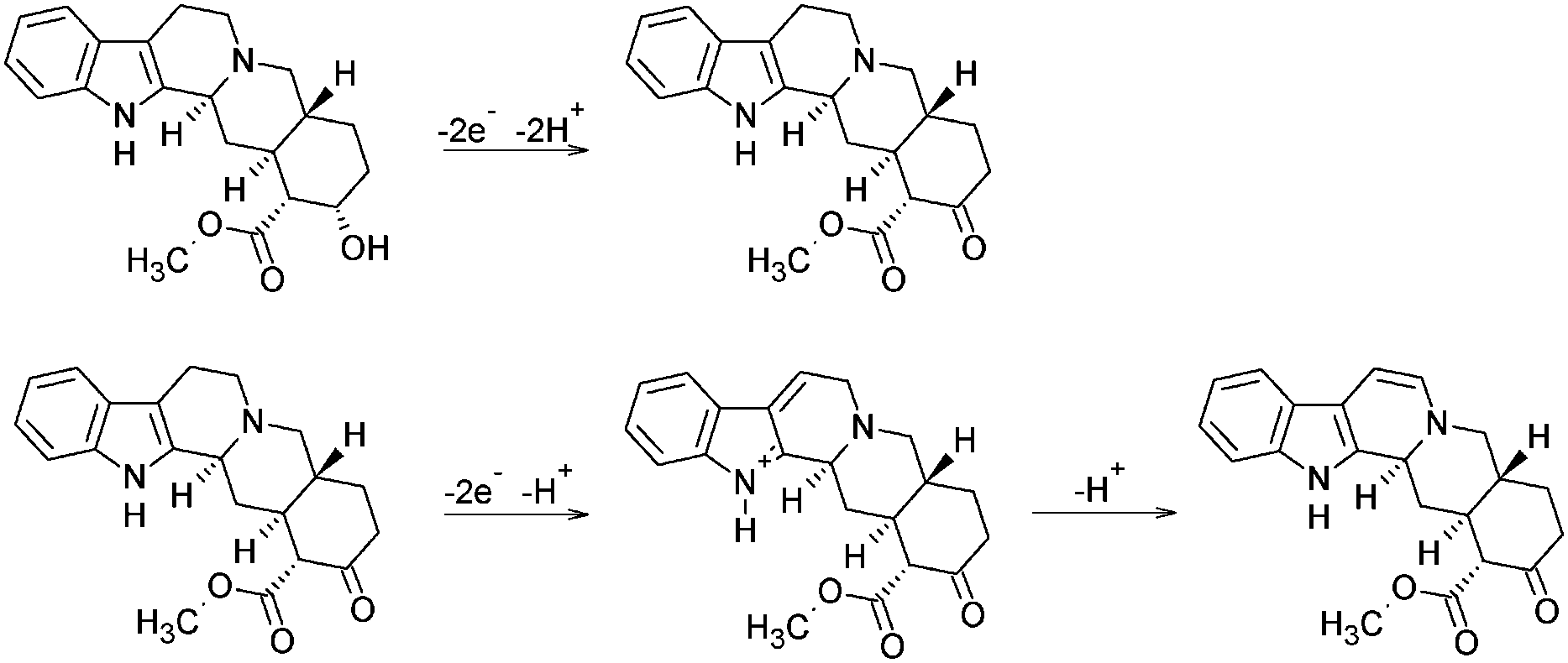 | ||
| Scheme 1 Mechanism of electrochemical oxidation of YOH. | ||
The effect of the pH on the peak currents (Ip) of YOH was also studied in the pH range of 2–12 using BR buffer solution. The achieved results demonstrated that the peak current increased up to pH 7, after which it declined sharply (inset of Fig. 1). Therefore, pH 7 represented the most appropriate pH value of the BR buffer solution for further measurements.
3.3. Effect of scan rate
The effect of the scan rate (v) on the peak current (Ip) was tested by CV measurements for characterization of the nature of the mass transport of YOH during its electrode reaction in BR buffer solution at pH 7 on the BDD electrode (rate-determining step). Considering the first oxidation peak of YOH its current increased linearly with the square root of the scan rate within the range of 20–130 mV s−1, indicating that the electrode process is controlled by diffusion (Fig. 2).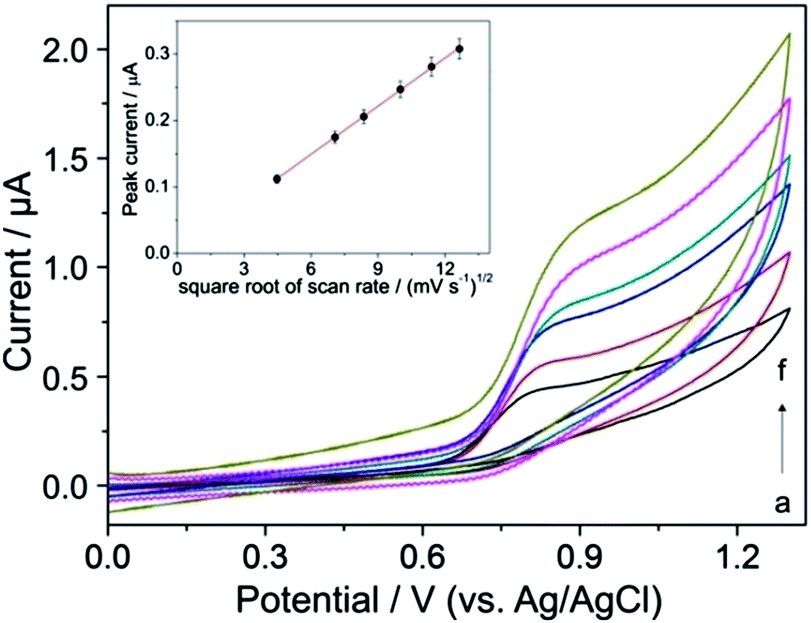 | ||
| Fig. 2 Cyclic voltammograms of 0.1 mmol L−1 YOH at various scan rates (v): (a) 20, (b) 50, (c) 70, (d) 100, (e) 130 and (f) 160 mV s−1 in BR buffer solution at pH 7 on the BDD electrode. The peak current Ip as a function of v1/2 for the first oxidation peak of YOH is shown in the inset. | ||
The linear dependence (inset of Fig. 2) can be expressed by the following equation:
| Ip (μA) = −0.004 (μA) + 0.024 × v1/2 (mV s−1), R2 = 0.999 |
The peak potential of YOH slightly shifted towards more positive values as the scan rate increased which is typical for irreversible electrochemical reactions.26–29
3.4. Optimization of DPV parameters
DPV was chosen for the determination of YOH as a sensitive pulse voltammetric technique with good discrimination against the background current. This powerful technique has been applied in the trace determination of numerous compounds.23–27,29 Optimization of DPV parameters was performed in order to obtain current responses for the oxidation of YOH (first oxidation peak) with highest magnitude. The pulse amplitude (a) and the pulse time (t) with a step potential of 5 mV and scan rate of 10 mV s−1 were considered as parameters to assess the optimum experimental set-up. Variation of a in the range of 10–200 mV (with a pulse time fixed at 25 ms) showed that the current response of the oxidation peak of YOH increased, but this effect was associated with a broadening of the peak at the same time as depicted in Fig. 3. However, at values above 100 mV, the oxidation signal of YOH became much wider and the background current became significantly higher in addition to which a slight shift of the peak towards less positive values was observed.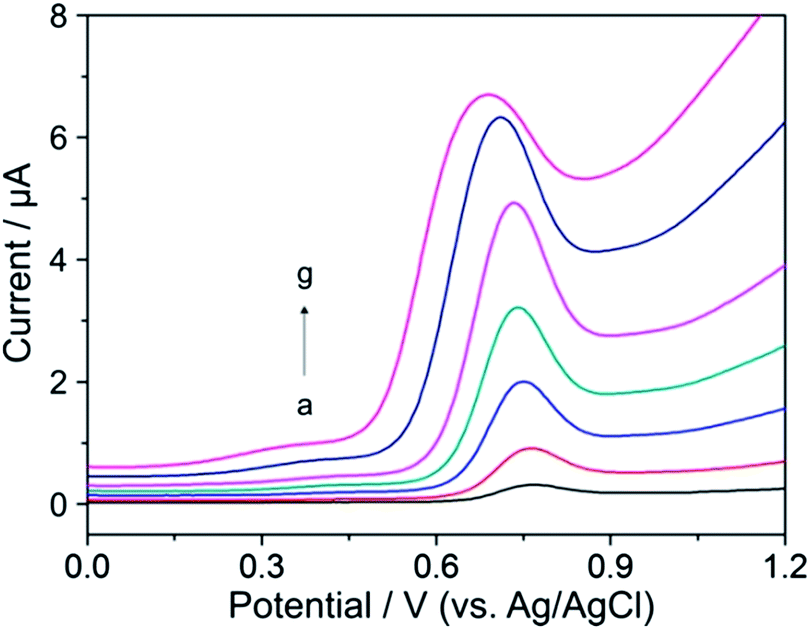 | ||
| Fig. 3 Differential pulse voltammograms of 0.1 mmol L−1 YOH with various pulse amplitudes: (a) 10, (b) 25, (c) 50, (d) 75, (e) 100, (f) 150 and (g) 200 mV at a pulse time of 25 ms, step potential of 5 mV and scan rate of 10 mV s−1 in BR buffer solution at pH 7 on the BDD electrode. | ||
In the case of t, the peak current of YOH decreased with its increase in the range of 5–100 ms. Fig. 4 shows that below a t value of 25 ms, the magnitude of the peak current was higher than the background current. Above this value, the current response of YOH was not so sensitive. A peak current with satisfactory magnitude, high repeatability and lower background was observed at 25 ms with a pulse amplitude of 100 mV. Overall, a pulse amplitude of 100 mV, pulse time of 25 ms, step potential of 5 mV and scan rate of 10 mV s−1 represent the most suitable values for the determination of YOH using the BDD electrode.
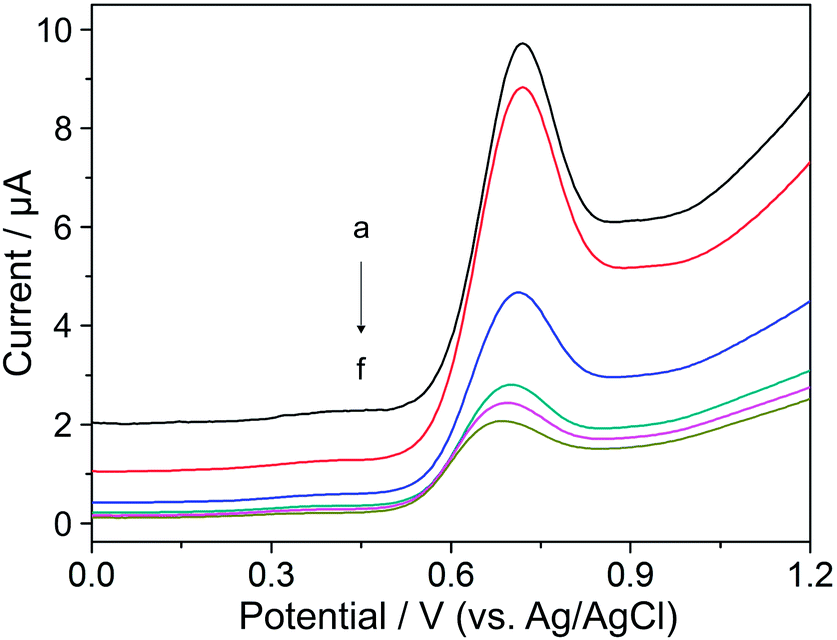 | ||
| Fig. 4 Differential pulse voltammograms of 0.1 mmol L−1 YOH for different pulse times: (a) 5, (b) 10, (c) 25, (d) 50, (e) 75 and (f) 100 ms at a pulse amplitude of 100 mV, step potential of 5 mV and scan rate of 10 mV s−1 in BR buffer solution at pH 7 on the BDD electrode. | ||
3.5. Analytical performance
Once the most suitable experimental conditions for the quantification of YOH were established, DP voltammograms at different concentrations of YOH were recorded to examine the analytical performance of the method using an oxidation peak at around +0.80 V. Fig. 5 shows DP voltammograms recorded after the addition of aliquots of a standard solution of YOH to BR buffer solution at pH 7. The respective calibration curve with the obtained linear relationship between the peak current and the concentration of YOH is presented in the inset of Fig. 5. The statistical assessment of the calibration curve and the analytical characteristics for the developed method are summarized in Table 1. | ||
| Fig. 5 Differential pulse voltammograms of different concentrations of YOH: (a) 0, (b) 0.25, (c) 0.99, (d) 2.00, (e) 2.99, (f) 4.98, (g) 7.44, (h) 9.90, (i) 12.3, (j) 18.4, (k) 24.4, (l) 36.1, (m) 47.6, (n) 69.8 and (o) 90.9 μmol L−1 in BR buffer solution at pH 7 on the BDD electrode at the optimized DPV parameters: pulse amplitude of 100 mV, pulse time of 25 ms, step potential of 5 mV and scan rate of 10 mV s−1. Inset: peak current Ip as a function of the concentration of YOH, standard deviations from three measurements. | ||
| Analytical parameter | Value |
|---|---|
| a Calculated as 3 × SDintercept/slope. b RSD calculated for 6 replicate DPV measurements at 18.4 μM YOH (n = 6). | |
| Peak potential (V vs. Ag/AgCl) | +0.80 |
| Intercept (nA) | 14.1 |
| Standard deviation of intercept (nA) | 1.5 |
| Slope (nA L μmol−1) | 34.2 |
| Standard deviation of slope (nA L μmol−1) | 0.7 |
| Linear concentration range (μmol L−1) | 0.25–90.9 |
| Coefficient of determination (R2) | 0.997 |
| Detection limita (μmol L−1) | 0.13 |
| Repeatabilityb (%) | 2.5 |
The low detection limit of 0.13 μmol L−1 was obtained as a consequence of the high S/N ratio achieved owing to the low and stable background current and the low adsorption propensity of the BDD electrode surface without any chemical surface modification. The repeatability of the developed procedure was tested by carrying out six replicate DPV measurements with 18.4 μmol L−1 YOH under the same operating conditions over a short time interval. The relative standard deviation (RSD) achieved was 2.5% revealing the good repeatability of the method and confirming that minimal adsorption of the components present in the analyzed solution had occurred on the BDD electrode. These results demonstrate the suitability of the BDD electrode for the sensitive and precise determination of YOH.
3.6. Interference study
To evaluate the selectivity of the proposed method, the effect of various possibly interfering compounds was examined using DPV under the optimized experimental conditions with a mixed solution approach containing a fixed concentration of 18.4 μmol L−1 YOH in BR buffer solution at pH 7. The tolerance limit was defined as the maximum concentration of the interfering substance that caused an error of less than ±5% for the determination of YOH. It was found that 150-fold excess of common ions such as Na+, K+, Mg2+, Zn2+, Fe3+, Cu2+, Al3+, Cl−, NO3−, PO43− and SO42− showed only minor effects on the oxidation signal of YOH. Insignificant interferences were also recorded for 100-fold excesses of sugars such as glucose, fructose and sucrose.The accompanying and structurally related indole alkaloids reserpine and ajmalicine were considered to be possibly interfering agents in plant samples (e.g. present together with YOH in Rauvolfia serpentina). When using BR buffer solution at pH 7, the results showed that a 50-fold excess of reserpine and ajmalicine had a negligible effect on the peak current of the first oxidation peak of YOH (see S1 in ESI†). Moreover, a more detailed electrochemical study of these alkaloids with BDD electrodes revealed that both reserpine and ajmalicine are electrochemically inactive at neutral pH in BR buffer solution at cathodic and anodic potentials. However, in preliminary studies, when using acidic and weakly acidic media (pH 1.5–6) and BDD electrodes, both compounds provided small oxidation peaks in the range of potentials of 0.9–1.1. These alkaloids will also be the subject matter of our investigations in further studies. It can be concluded that the proposed method provides good selectivity for the electrochemical determination of YOH.
3.7. Determination of YOH in the bark of natural aphrodisiacs
The extracts from natural aphrodisiacs (primary bark of trees Pausinystalia yohimbe and Rauvolfia serpentina) were analyzed in order to evaluate the validity and the practical applicability of the herein developed methodology. The results obtained using the standard addition method with DPV for n = 3 were as follows: (0.69 ± 0.03)% (m m−1) for Pausinystalia yohimbe and (0.77 ± 0.05)% (m m−1) for Rauvolfia serpentina. Due to the absence of certified values for the amount of YOH in the studied products, the results were compared with literature values, which report approximately 6% indole alkaloids in the bark, of which 10–15% (i.e. 0.6–0.9%) is YOH.4Recovery analyses were performed in order to estimate the accuracy of the proposed analytical procedure for samples of Pausinystalia yohimbe and Rauvolfia serpentina. The results are summarized in Table 2. The recovery values ranged from 92 to 95% for Pausinystalia yohimbe and from 96 to 97% for Rauvolfia serpentina, respectively. These values indicate that there are no significant matrix interferences from the analyzed samples. Thus, YOH can be quantitatively determined by the proposed method, thus being a guarantee of the accuracy and suitability of the voltammetric determination of YOH in samples of this kind.
Typical DP voltammograms of YOH in the extract of bark of Pausinystalia yohimbe are illustrated in Fig. 6. To verify the observed oxidation peak at +0.72 V as YOH (curve b), the sample was spiked with aliquots of the standard solution of YOH and the corresponding DP voltammograms were recorded. As can be seen (curves c–e), the peak increased after each standard addition demonstrating that it could be assigned to the oxidation of YOH. Furthermore, the small oval voltammetric peak at about +1.0 V could be attributed to the oxidation of reserpine and/or ajmalicine which represent the minor alkaloids occurring in the bark of Pausinystalia yohimbe.
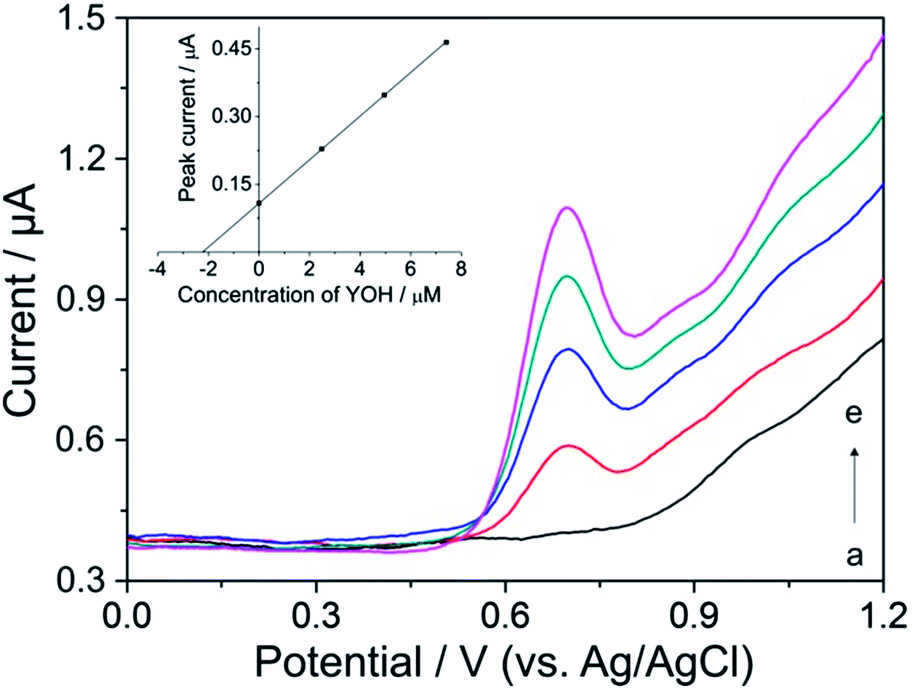 | ||
| Fig. 6 Differential pulse voltammograms for the determination of YOH in the extract of the bark of Pausinystalia yohimbe: (a) blank, (b) after addition of 200 μL of extract and after spiking of (c) 2.5, (d) 5.0 and (e) 7.4 μmol L−1 of standard solution of YOH in BR buffer solution at pH 7 on the BDD electrode. Analysis by the standard addition method is depicted in the inset. The experimental conditions were the same as those in previous measurements. | ||
4. Conclusions
A fully validated DPV procedure using BDD as the electrode material is described for the first time for the simple, sensitive and precise electrochemical determination of YOH. A low detection limit (0.13 μmol L−1) was achieved without any surface modification clearly demonstrating the superiority of this electrode material. Moreover, the developed methodology can be considered to be the first voltammetric analytical method for the determination of YOH, because until now there there have been no published reports which have described the use of bare and/or modified electrodes for the determination of YOH, except for a short report dealing with the application of gold and platinum electrodes without any reference to analytical characteristics.17The proposed procedure is considerably more inexpensive than separation methods for the determination of YOH, especially HPLC and GC techniques. The practical applicability of the method was successfully demonstrated by assessment of the total YOH content in extracts of the primary bark of natural aphrodisiacs with good recoveries. Considering the results achieved in this study, the BDD electrode is a good candidate for possible applications as a sensitive electrochemical sensor and a cost-effective alternative for future use in the quality control analysis of dietary supplements containing various alkaloids.
Acknowledgements
This work has been supported by the Grant Agency of the Slovak Republic (grant no. 1/0051/13), the Slovak Research and Development Agency under the contract no. APVV-0797-11 and the Ministry of Education and Science of the Republic of Serbia (project no. OI 172030). Support from the bilateral program Action: Austria-Slovakia is gratefully acknowledged.References
- J. Kim, A. Cowan, R. Lisek, N. Raymondi, A. Rosenthal, D. D. Hirsch and S. M. Rawls, Brain Res., 2011, 1384, 110–117 CrossRef CAS PubMed.
- J.-X. Zhu, F.-Y. Xu, W.-J. Xu, Y. Zhao, C.-L. Qu, J.-S. Tang, D. M. Barry, J.-Q. Du and F.-Q. Huo, Exp. Neurol., 2013, 248, 381–386 CrossRef CAS PubMed.
- M. Yeung, L. Lu, A. M. Hughes, D. Treit and C. T. Dickson, Neuropharmacology, 2013, 75, 47–52 CrossRef CAS PubMed.
- Z. Tchoundjeu, M.-L. Mpeck, E. Asaah and A. Amougou, For. Ecol. Manage., 2004, 188, 175–183 CrossRef PubMed.
- D. K. Singh, B. Srivastava and A. Sahu, Anal. Sci., 2004, 20, 571–573 CrossRef CAS.
- B. D. Joshi, P. Tandon and S. Jain, BIBECHANA, 2012, 8, 73–80 CrossRef.
- Q. Chen, P. Li, H. Yang, B. Li, J. Zhu and L. Peng, Anal. Bioanal. Chem., 2010, 398, 937–942 CrossRef CAS PubMed.
- M. R. Goldberg, L. Speier and D. Robertson, J. Liq. Chromatogr., 1984, 7, 1003–1012 CrossRef CAS.
- M. Hariharan, S. Guthrie, E. K. Kindt, T. V. Noord and L. J. Grunhaus, J. Liq. Chromatogr., 1991, 14, 351–364 CrossRef CAS.
- M. Farouk, L. Abd El-Aziz, A. E. El-Gindy and E. Shokry, Bulletin of Faculty of Pharmacy, Cairo University, 2011, vol. 49, pp. 67–79 Search PubMed.
- J. Sun and P. Chen, J. Pharm. Biomed. Anal., 2012, 61, 142–149 CrossRef CAS PubMed.
- S. Gupta, K. Shanker and S. K. Srivastava, J. Pharm. Biomed. Anal., 2012, 66, 33–39 CrossRef CAS PubMed.
- Q. Chen, P. Li, Z. Zhang, K. Li, J. Liu and Q. Li, J. Sep. Sci., 2008, 31, 2211–2218 CrossRef CAS PubMed.
- A. Pelander, I. Ojanperä, J. Sistonen, I. Rasanenand and E. Vuori, J. Anal. Toxicol., 2003, 27, 226–232 CrossRef CAS PubMed.
- V. Dumestre-Toulet, V. Cirimele, S. Gromb, T. Belooussoff, D. Lavault, B. Ludes and P. Kintz, Forensic Sci. Int., 2002, 126, 71–76 CrossRef CAS.
- R. Sokolová, Š. Ramešová, I. Degano, M. Hromadová, M. Gál and J. Žabka, Chem. Commun., 2012, 48, 3433–3435 RSC.
- E. Bishop and W. Hussein, Analyst, 1984, 109, 965–966 RSC.
- H. Sun, L. Dong, H. Yu and M. Huo, Russ. J. Electrochem., 2013, 49, 883–887 CrossRef CAS.
- H. Dejmkova, J. Barek and J. Zima, Int. J. Electrochem. Sci., 2011, 6, 3550–3563 CAS.
- L. C. Melo, D. D. Souza, P. de Lima-Neto and A. N. Correia, Electroanalysis, 2010, 22, 2502–2510 CrossRef CAS.
- D. A. Tryk, H. Tachibana, H. Inoue and A. Fujishima, Diamond Relat. Mater., 2007, 16, 881–887 CrossRef CAS PubMed.
- Ľ. Švorc, M. Rievaj and D. Bustin, Sens. Actuators, B, 2013, 181, 294–300 CrossRef PubMed.
- L. Bandžuchová, Ľ. Švorc, J. Sochr, J. Svítková and J. Chýlková, Electrochim. Acta, 2013, 111, 242–249 CrossRef PubMed.
- E. Culková, Ľ. Švorc, P. Tomčík, J. Durdiak, M. Rievaj, D. Bustin, R. Brescher and J. Lokaj, Pol. J. Environ. Stud., 2013, 22, 1317–1323 Search PubMed.
- Ľ. Švorc, D. M. Stanković and K. Kalcher, Diamond Relat. Mater., 2014, 42, 1–7 CrossRef PubMed.
- Ľ. Švorc, P. Tomčík, J. Svítková, M. Rievaj and D. Bustin, Food Chem., 2012, 135, 1198–1204 CrossRef PubMed.
- Ľ. Švorc, J. Sochr, M. Rievaj, P. Tomčík and D. Bustin, Bioelectrochemistry, 2012, 88, 36–41 CrossRef PubMed.
- Ľ. Švorc, J. Sochr, P. Tomčík, M. Rievaj and D. Bustin, Electrochim. Acta, 2012, 68, 227–234 CrossRef PubMed.
- Ľ. Švorc, J. Sochr, J. Svítková, M. Rievaj and D. Bustin, Electrochim. Acta, 2013, 87, 503–510 CrossRef PubMed.
- Ľ. Švorc and K. Kalcher, Sens. Actuators, B, 2014, 194, 332–342 CrossRef PubMed.
- J. Sochr, Ľ. Švorc, M. Rievaj and D. Bustin, Diamond Relat. Mater., 2014, 43, 5–11 CrossRef CAS PubMed.
- Ľ. Švorc, M. Vojs, P. Michniak, M. Marton, M. Rievaj and D. Bustin, J. Electroanal. Chem., 2014, 717–718, 34–40 CrossRef PubMed.
- Y. Yardım, M. Gülcan and Z. Şentürk, Food Chem., 2013, 141, 1821–1827 CrossRef PubMed.
- P. T. Pınar, Y. Yardım and Z. Şentürk, Cent. Eur. J. Chem., 2013, 11, 1674–1681 CrossRef PubMed.
- Y. Yardım, E. Keskin and Z. Şentürk, Talanta, 2013, 116, 1010–1017 CrossRef PubMed.
- P. B. Deroco, F. C. Vicentini, G. G. Oliveira, R. C. Rocha-Filho and O. Fatibello-Filho, J. Electroanal. Chem., 2014, 719, 19–23 CrossRef CAS PubMed.
- B. C. Lourencao, M. Baccarin, R. A. Medeiros, R. C. Rocha-Filho and O. Fatibello-Filho, J. Electroanal. Chem., 2013, 707, 15–19 CrossRef CAS PubMed.
- J. A. Ardila, E. R. Sartori, R. C. Rocha-Filho and O. Fatibello-Filho, Talanta, 2013, 103, 201–206 CrossRef CAS PubMed.
- F. Agin, N. Karadas, B. Uslu and S. A. Ozkan, Curr. Pharm. Anal., 2013, 9, 299–309 CrossRef CAS.
- N. Karadas, S. Sanli, M. Gumustas and S. A. Ozkan, J. Pharm. Biomed. Anal., 2012, 66, 116–125 CrossRef CAS PubMed.
- S. Gupta, K. Shanker and S. K. Srivastava, J. Pharm. Biomed. Anal., 2012, 66, 33–39 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ay00704b |
| This journal is © The Royal Society of Chemistry 2014 |