Determination of iodinated X-ray contrast agents in pharmaceutical formulations and artificial urine samples by differential pulse voltammetry
Piotr
Markowski
*a,
Katarzyna
Piwowar
b and
Irena
Baranowska
a
aDepartment of Inorganic, Analytical Chemistry and Electrochemistry, Faculty of Chemistry, Silesian University of Technology, 6 B. Krzywoustego Str., 44-100 Gliwice, Poland. E-mail: piotr.markowski@polsl.pl; Fax: +48 32 237 1205; Tel: +48 32 237 2804
bDepartment of Physical Chemistry and Technology of Polymers, Faculty of Chemistry, Silesian University of Technology, 9 M. Strzody Str., 44-100 Gliwice, Poland
First published on 7th April 2014
Abstract
A new differential pulse voltammetry (DPV) technique was developed and validated for the determination of iopromide (IOP), iodixanol (ION) and amidotrizoic acid (DTZA), which belong to the iodinated X-ray contrast agent group in pharmaceutical formulations and artificial urine samples. All measurements were performed in the three-electrode configuration with a glassy carbon electrode (GCE) as a working electrode, Ag|AgCl|KCl(sat.) as a reference and platinum wire as an auxiliary electrode. The supporting electrolytes for determination of selected contrast agents were mixtures of methanol and Britton–Robinson buffers with different pH values. Quantification was performed by means of calibration curve and standard addition methods. The calibration curves for ION, IOP and DTZA were linear over a concentration range of 0.032–0.258, 0.039–0.394 and 0.041–0.326 mM, respectively. Good linear behaviour over the investigated concentration ranges was observed with values of r2 higher than 0.994 for all the iodinated contrast agents (ICA). The limits of detection (LOD) and limits of quantification (LOQ) for all analysed contrast agents were calculated and recovery studies were also performed. The percentage recoveries varied from 94.44 to 101.05% (after SPE procedure). Analytical methods for the preparation of urine samples were worked out and optimized before its voltammetry measurements (solid phase extraction – SPE). The differential pulse voltammetry method described in this work is the first procedure allowing determination of three iodinated X-ray contrast agents (IOP, ION and DTZA) in pharmaceutical formulations and artificial urine samples.
1 Introduction
Iodinated contrast agents (ICA) are commonly used as diagnostic compounds with computer tomography (CT) and X-ray imaging (XRI) to enhance soft tissue contrast, to evaluate blood flow abnormalities and to characterize lesions. The chemical structure of ICA is based on a benzene ring, three or six iodine atoms (responsible for enhancing X-ray) and hydrophilic functional groups (responsible for ensuring water solubility of ICA). Over the last thirty years, the ICA has been shown to be a suitable marker in the determination of the glomerular filtration rate (GFR), because they do not bind to plasma proteins, do not metabolize and they are 100% filtered through the kidneys. GFR has been determined with ICA in humans and in animals such as dogs and cats.1–13The ICA with six iodine atoms is iodixanol (ION), whose systematic (IUPAC) name is 5-{N-[3-(N-{3,5-bis[(2,3-dihydroxypropyl)carbamoyl]-2,4,6-triiodophenyl}acetamido)-2-hydroxypropyl]acetamido}-1-N,3-N-bis(2,3-dihydroxypropyl)-2,4,6-triiodobenzene-1,3-dicarboxamide. Its trade name is VISIPAQUE™. ION is a nonionic, iso-osmolar, dimeric ICA. It was introduced into clinical practice in 1996.14 It is used as a contrast agent during coronary angiography and CT imaging of the brain and body.15,16 Determination of ION in human and animal plasma, urine and serum has been reported using high-performance liquid chromatography (HPLC) with ultraviolet detection (UV)3,17–20 and tandem mass spectrometry (MS/MS) used as detection techniques.21 The colorimetric method for determination of iodixanol in biological fluids was also developed.13,22 The other published methods used the spectrophotometric determination of ION in mammalian cells22 and HPLC method with inductively coupled plasma mass spectrometry as the technique used to detect ION in radiopaque solution for injection (RSI).23
Iopromide (IOP) and amidotrizoic acid (DTZA) are ICA that have three iodine atoms. IOP, whose systematic name is 1-N,3-N-bis(2,3-dihydroxypropyl)-2,4,6-triiodo-5-(2-methoxyacetamido)-1-N-methylbenzene-1,3-dicarboxamide, is a nonionic, low-osmolar contrast agent. It is commonly known as ULTRAVIST™. It was introduced in 1985, and since then has been used in brain and abdominal CT and angiography.24 On the other hand, DTZA, or 3,5-bis(acetylamino)-2,4,6-triiodobenzoic acid, is an ionic, monomeric ICA known as HYPAQUE™ or UROGRAFIN™, and is commonly used as a contrast agent in CT imaging of digestive and urinary systems.25 IOP and DTZA are both determined in water samples using HPLC-MS/MS,26–30 gas chromatography (GC) with MS/MS detection29,30 and measurement of organic iodine by ion chromatography (IC).31 DTZA was also determined by RSI using capillary electrophoresis (CE).32
Samples have been prepared by deproteinization with various agents such as perchloric acid,2,9,33 trichloroacetic acid (TCA),2 trifluoroacetic acid (TFA),11,12,34 acetone,2 dichloromethane,2 hydrochloric acid and methanol,35 zinc sulfate and methanol,36 and acetonitrile.6
In addition, samples have been pretreated using extraction procedures such as liquid–liquid extraction (LLE)37 and solid phase extraction (SPE) with various columns such as LiChrolut® EN,21,27,38 Sep-Pak C18,18 Isolut ENV+ (ref. 25 and 30) and Oasis HLB.29 The extraction procedure developed by Agasøster uses aqueous two-phase partitioning sample preparation.39 The procedure developed by Jacobsen20 requires an automated online dialysis system for sample preparation, and the procedure developed by Denis requires ultrafiltration.21 Some of the published procedures for preparation of biological and water samples used direct dilution.6,23,40
The aim of the present work is to develop a simple and efficient analytical method for quantification of ICA in pharmaceutical formulations and artificial urine by differential pulse voltammetry. Secondly, a validation of the procedure has been carried out, which provides accuracy and reliability.
Voltammetric methods for the determination of iopromide (IOP), iodixanol (ION) and amidotrizoic acid (DTZA) have not yet been described in the literature. To our knowledge, this is the first electroanalytical method that allows determination of IOP, ION and DTZA in pharmaceutical formulations and artificial urine. This method may be considered as a suitable alternative to the existing chromatographic methods.
2 Experimental
2.1 Chemicals, reagents and solutions
Iopromide (∼96% purity) and iodixanol (∼95% purity) were purchased from Toronto Research Chemicals Inc. (Toronto, Canada). Amidotrizoic acid, uric acid (∼99% purity) and hippuric acid were purchased from Sigma-Aldrich (Schnelldorf, Germany). The structures and IUPAC names of analysed X-ray contrast agents are presented in Table 1.| Contrast agents | Molecular structure | IUPAC name |
|---|---|---|
| Iopromide (IOP) |

|
1-N,3-N-Bis(2,3-dihydroxypropyl)-2,4,6-triiodo-5-(2-methoxyacetamido)-1-N-methylbenzene-1,3-dicarboxamide |
| MW = 791.11 | ||
| Iodixanol (ION) |
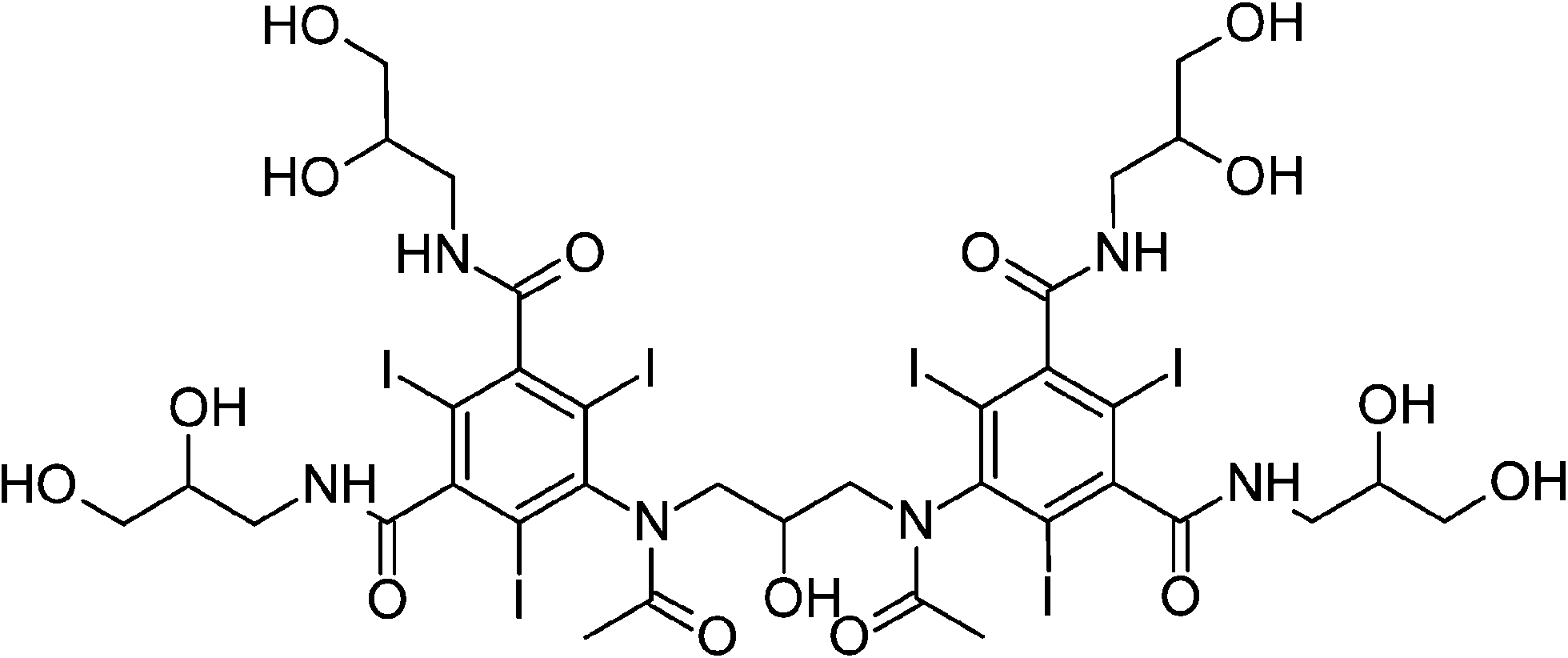
|
5-{N-[3-(N-{3,5-Bis[(2,3-dihydroxypropyl)carbamoyl]-2,4,6-triiodophenyl}acetamido)-2-hydroxypropyl]acetamido}-1-N,3-N-bis(2,3-dihydroxypropyl)-2,4,6-triiodobenzene-1,3-dicarboxamide |
| MW = 1550.18 | ||
| Amidotrizoic acid (DTZA) |
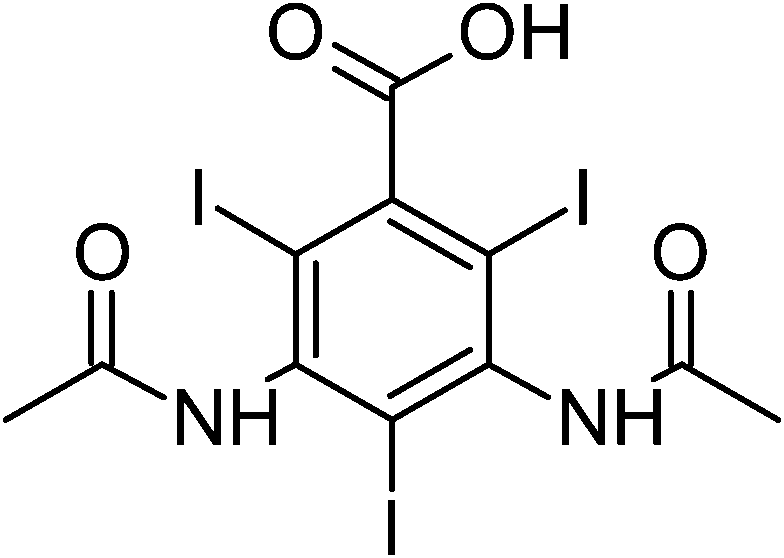
|
3,5-Bis(acetylamino)-2,4,6-triiodobenzoic acid |
| MW = 613.91 |
Urea and creatinine (≥99% purity) were bought from Merck (Darmstadt, Germany). Potassium phosphate monobasic (KH2PO4), sodium phosphate dibasic (Na2HPO4), o-boric acid (H3BO3), acetic acid (CH3COOH), o-phosphoric acid (H3PO4), sodium hydroxide (NaOH) and methanol, all of analytical grade, were purchased from POCH S.A. (Gliwice, Poland). All other chemicals and reagents (acetonitrile and ethyl octane) used were of good commercial quality and were available and obtained from POCH S.A. (Gliwice, Poland). Aqueous solutions were prepared with double-distilled water. Britton–Robinson (BR) buffer solutions were prepared employing standard laboratory procedures.
2.2 Buffer preparation
Britton–Robinson (BR) buffer solutions at different pH values were prepared by mixing appropriate amounts of 200 μM NaOH with 25 mL of a mixed acid that contains 40 μM of each of o-boric, o-phosphoric and acetic acids (H3BO3–H3COOH–H3PO4). Phosphate buffer at pH 6.81 was prepared by mixing 25 mL of 67 μM KH2PO4 with 25 mL of 67 μM Na2HPO4. 67 μM KH2PO4 and Na2HPO4 solutions were prepared by dissolving 0.9 g of each compound in 100 mL water.All of these chemicals were of analytical grade and used without further purification. The pH of the solutions was adjusted by mixing buffer components and was verified before each measurement.
2.3 Stock solutions
Separate stock solutions of analysed iodinated X-ray contrast agents at different concentrations were prepared in 10 mL volumetric flasks by dissolving the appropriate amount of reference substance in a mixture of methanol and water (1/1, v/v). Stock solutions were prepared at the beginning of the study and were stored at 4 °C. Solutions of lower concentrations were prepared by dilution of stock solution with water.2.4 Artificial urine samples
Normal urine is an aqueous mixture of organic and inorganic substances. Most of the constituents are either waste products of cellular metabolism or products derived directly from certain foods. The most important organic substances are urea, uric acid and creatinine.The artificial urine that we used includes between about 55–900 mg L−1 of urea; the concentration of creatinine, when present, is preferably above about 50 mg L−1, and more preferably, between about 350 and 3000 mg L−1. This artificial urine includes an appropriate amount of individual components of human urine such that the artificial sample gives the appearance of being genuine. Furthermore, the artificial urine uses laboratory grade chemicals and is hence safe for handling because there is no risk of disease. These urine samples were stored at 4 °C before analysis.
2.5 Instrumentation
All voltammetric measurements were carried out using a μAUTOLAB Type III potentiostat (Eco-Chemie, The Netherlands) with a glassy carbon electrode (GCE) as a working electrode (1.5 mm diameter). This electrode was polished with 0.03 μm alumina (Buehler), then ultrasonicated in distilled water and finally rinsed with methanol. A platinum rod was used as a counter electrode, and an Ag|AgCl|KCl(sat.) electrode was used as a reference electrode. (All electrodes were purchased from Cypress Systems, Lawrence, USA). Voltammetric measurements were carried out in a 3 mL glass electrochemical cell. All measurements were automated and controlled through the programming capacity of the apparatus.Examined samples were carried through solid-phase extraction (SPE) on a J. T. Baker system (Deventer, The Netherlands) using Waters HLB® cation-exchange (N-vinylpyrrolidone–m-divinylbenzene copolymer) SPE columns (500 mg, 6 mL) (Milford, U.S.A.). The 0.45 μm disposable SPE Nylon Hydrophilic Membrane Filters were purchased from J. T. Baker (Deventer, The Netherlands). All pH measurements were made with an ELMETRON (Zabrze, Poland) Model CP-401 pH meter using a combined glass electrode and calibrated with standard buffers. A HERMILE Z 323 K (Gosheim, Germany) centrifuge was used. Argon was used for the removal of dissolved oxygen from the measured solutions.
2.6 Analytical procedure for ICA determination
All of the electrochemical experiments were carried out at ambient laboratory temperature (22 ± 3 °C). All measurements were carried out in (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) mixtures of BR buffers (at different pH values) and methanol.
:1, v/v) mixtures of BR buffers (at different pH values) and methanol.
Each measurement was repeated six times using fresh sample solution to ensure reproducibility of the results. Each 2 mL mixture of BR buffer and methanol (9/1, v/v) (at different pH values) as a supporting electrolyte was transferred into the 3 mL glass voltammetric cell. With the aim of removing oxygen, the solution was purged with pure argon for 10 min for 30 s before each measurement. After measurement of the electrolyte, the appropriate amount of the relevant compounds was added and voltammograms were recorded for different concentrations of standard solutions.
Before measurements, the GCE was polished manually to a mirror finish using an alumina (1.0, 0.3 and 0.05 μm particle size) paste and thoroughly rinsed with purified water and methanol. Each measurement was repeated six times using fresh sample solutions to ensure reproducibility of the results. Between experiments, the cell was treated with concentrated nitric acid and then washed with water. Parameters for the cyclic and differential pulse voltammetry are presented in Tables 2 and 3.
| Contrast agents | Initial potential [V] | First vertex potential [V] | Second vertex potential [V] | Condition potential [V] | Conditional time [s] | Scan rate [V s−1] | Step potential [V] |
|---|---|---|---|---|---|---|---|
| ION | 0.25 | +1.60 | −0.05 | −1.20 | 30.00 | 0.80 | 0.015 |
| IOP | 0.00 | +1.75 | −0.10 | −1.50 | 30.00 | 0.60 | 0.015 |
| DTZA | 0.20 | +1.70 | +0.10 | −1.10 | 45.00 | 0.80 | 0.015 |
| Contrast agents | Initial potential [V] | Start potential [V] | End potential [V] | Condition potential [V] | Conditional time [s] | Scan rate [V s−1] |
|---|---|---|---|---|---|---|
| ION | 0.25 | +1.60 | −0.10 | −1.20 | 75.00 | 0.80 |
| IOP | 0.00 | +1.75 | −1.00 | −1.50 | 30.00 | 0.60 |
| DTZA | 0.20 | +1.80 | +0.10 | −1.10 | 60.00 | 0.80 |
2.7 Calibration curves
The calibration curves for all examined X-ray contrast agents were determined by the least squares linear regression method, and hence fit to the equation y = ax + b, where “y” indicates the intensity of the current (A), “a” and “b” are constants, and “x” denotes the concentration of the analysed compound. The peak current vs. concentration dependence was recorded in the concentration range of 0.032–0.394 mM. The regression line was used to calculate concentrations of all compounds in the standard solutions based on the peak area ratio. The calibration curves were measured six times.2.8 Sample preparation
Artificial urine sample preparation and extraction methods are described below step by step. A urine sample (2.5 mL) was placed into clean centrifuge tube (10 mL), and then suitable amounts of IOP, ION or DTZA solutions were added to each tube. The solution was mixed with 3 mL phosphate buffer at pH 6.81 and 3 mL methanol. After shaking for 1 min, the obtained mixture was centrifuged for another 10 min at 5000 rpm at room temperature (ca. 22 °C).Then, the sample was transferred to a volumetric flask (10 mL), and water was added to the mark. The obtained sample was filtered through a Bakerbond nylon syringe filter (0.45 μm), and the clear supernatant was then transferred into an Oasis® HLB (500 mg, 6 mL) column. (Earlier this column was conditioned by washing with 4 mL of methanol and 4 mL of water.) X-ray contrast agents were eluted with 5 mL of acetonitrile. The sample was evaporated to dryness under a nitrogen stream at room temperature. The residues were dissolved in 3 mL of BR buffer at pH = 1.81 and 0.5 mL of this solution was transferred to the voltammetric cell. The same procedure was repeated for urine without addition of analytes in order to register a blank test. An overview of the sample preparation procedure for determination of IOP, ION and DTZA in artificial urine is presented in Fig. 1.
 | ||
| Fig. 1 Procedure of artificial urine samples preparation for determination of the IOP, ION and DTZA. | ||
3 Results and discussion
DPV was used for voltammetric measurements due to its good sensitivity and resolving power. The peak current depends on the pH of the medium, concentration and chemical composition of the buffer solution and instrumental parameters. We have studied optimization of the proposed procedure and examined conditions that could affect the results. The current was measured and recorded for the sample solution.As a working electrode for IOP, ION and DTZA determination, a glassy carbon electrode was used, where analysed iodinated X-ray contrast agents were oxidized. For each compound three oxidation peaks were obtained. The analytical peaks selected were: 1.46, 1.40 and 1.32 (vs. Ag|AgCl|KCl(sat.)) for IOP, ION and DTZA, respectively. BR buffer solutions at different pH values were used as supporting electrolytes. When BR buffer at pH 1.81 was used, the peaks of IOP, ION and DTZA were all well defined.
3.1 Development of CV and DPV methods
The aim of the present study is to evaluate and optimize parameters of an analytical method for determination of four selected X-ray contrast agents in pharmaceutical formulations and artificial urine samples. The current was measured and recorded for the sample solution.The first step in the investigation of IOP, ION and DTZA was to check examined compounds for electroactivity in studied conditions (GCE as a working electrode, BR buffer as a supporting electrolyte) by voltammetric methods.
The cyclic voltammetry technique was applied as a diagnostic tool to obtain information about electrochemical oxidation of IOP, ION and DTZA at a glassy carbon electrode in the BR universal buffer between pH values of 1.81 and 11.20.
Fig. 2 shows representative cyclic voltammograms of solutions of IOP, ION and DTZA in mixtures of BR buffer at pH 1.81 and methanol (9/1, v/v), where three well-distinguished peaks proportionally increasing with concentration are observed. For each analyte there are only three peaks appearing in the entire potential range between 0.0 V and 1.75 V, resulting from oxidation processes.
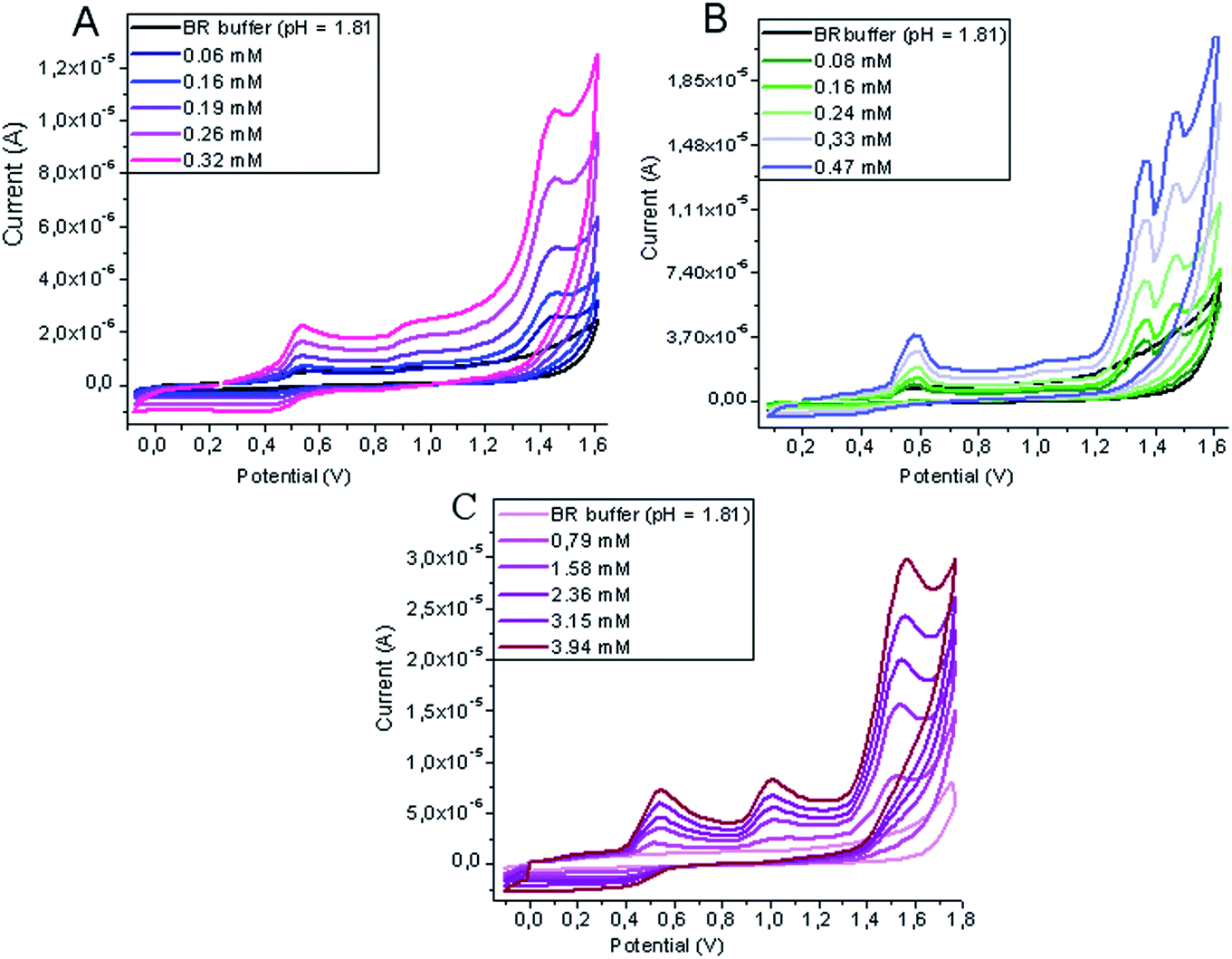 | ||
| Fig. 2 Cyclic voltammograms recorded for determination of ION (A), DTZA (B) and IOP (C) in mixture of BR buffer at pH 1.81–methanol (9/1, v/v) at the GCE (vs. Ag|AgCl||KCl(sat.)). | ||
IOP, ION and DTZA are electroactive compounds, which give three well-defined oxidation peaks at the GCE in the working potential range from −1.0 to 1.5 V in acidic media by DPV (Fig. 3).
 | ||
| Fig. 3 DPV voltammograms recorded for determination of ION (A), IOP (B) and DTZA (C) in mixture of BR buffer at pH 1.81–methanol (9/1, v/v) at the GCE (vs. Ag|AgCl||KCl(sat.)). | ||
The mechanisms of oxidation were connected with the presence of imino and hydroxyl groups in the IOP, ION and DTZA molecules. No peaks were observed in the cathodic scan, pointing to the irreversible nature of the oxidation process.
3.2 Calibration curves and linearity
The calibration curves were measured and evaluated by the least squares linear regression method. The calibrations were linear for IOP, ION and DTZA in the studied concentration ranges. The correlation coefficients of the all calibration curves were high, with values between 0.995 and 0.999. The calibration curves showed linear responses over the whole range of concentrations used in the assay. The equations associated with the calibration are summarized in Table 4.| Contrast agents | Linear range [mM] | a | b | r 2 | LODd [mM] | LOQe [mM] |
|---|---|---|---|---|---|---|
| a Slope. b Intercept. c Correlation coefficient. d Limit of detection. e Limit of quantification. | ||||||
| ION | 0.032–0.258 | 6.420 × 10−7 | −5.505 × 10−9 | 0.998 | 0.010 | 0.029 |
| IOP | 0.039–0.394 | 4.067 × 10−7 | 4.120 × 10−8 | 0.999 | 0.011 | 0.032 |
| DTZA | 0.041–0.326 | 5.906 × 10−7 | 9.464 × 10−9 | 0.995 | 0.013 | 0.040 |
3.3 Effect of pH
The peak current depends on pH of the medium, concentration and chemical composition of the buffer solution, and instrumental parameters. The electrooxidation experiments of analysed X-ray contrast agents were performed in the BR buffers at different pH values. The pH effect of the electrolyte was examined between pH values of 1.81 and 11.20 for IOP and ION, and between 1.81 and 7.24 for DTZA.The position of the peak current obtained by reduction of compounds was strongly pH dependent. The effect of pH for IOP, ION and DTZA can be seen in Fig. 4. The peaks potentials of analysed drugs moved into more positive directions, with increasing acidity of the supporting electrolyte. In acidic media, the peak of the reduction process was narrow and well defined. Therefore, pH 1.81 was chosen as the best pH for analytical applications. The highest peaks in the voltammograms were observed for the lowest value of pH.
 | ||
| Fig. 4 Plot of oxidative peak current response versus pH of BR buffer containing 3.9 × 10−5 M IOP (A), 6.0 × 10−6 M of ION (B), 4.0 × 10−6 M of DTZA (C) at the GCE (vs. Ag|AgCl||KCl(sat.)). | ||
3.4 Effect of scan rate
The influence of the potential scan rate on the peak current of 50 × 10−6 M contrast agent in pH 1.81 BR buffer was investigated at the GCE in the range of 0.2–1.0 V s−1. Oxidative peak currents of IOP (Fig. 5A), ION (Fig. 5B) and DTZA (Fig. 5C) showed linear dependency on the potential scan rate in the 0.2–1.0 V s−1 range with a quite good correlation coefficient, indicating that the oxidative reaction is surface-diffusion controlled. | ||
| Fig. 5 Plot of oxidative peak current versus potential scan rate of 1.18 × 10−4 M of IOP (A), 0.6 × 10−4 M of ION (B) and 0.8 × 10−5 M of DTZA (C) in BR buffer (pH = 1.81) at the GCE (vs. Ag|AgCl||KCl(sat.)). | ||
3.5 Effect of conditioning potential and time
The effect of conditioning potential and time on the oxidative peak current of 20 × 10−5 M IOP, ION and DTZA was investigated in BR buffer pH = 1.81 in the potential range −1.1 to −2.0 V and in the time range 30–120 s at the GCE. Fig. 6 shows the dependence of conditioning potential on the peak current of IOP (A), ION (B) and DTZA (C).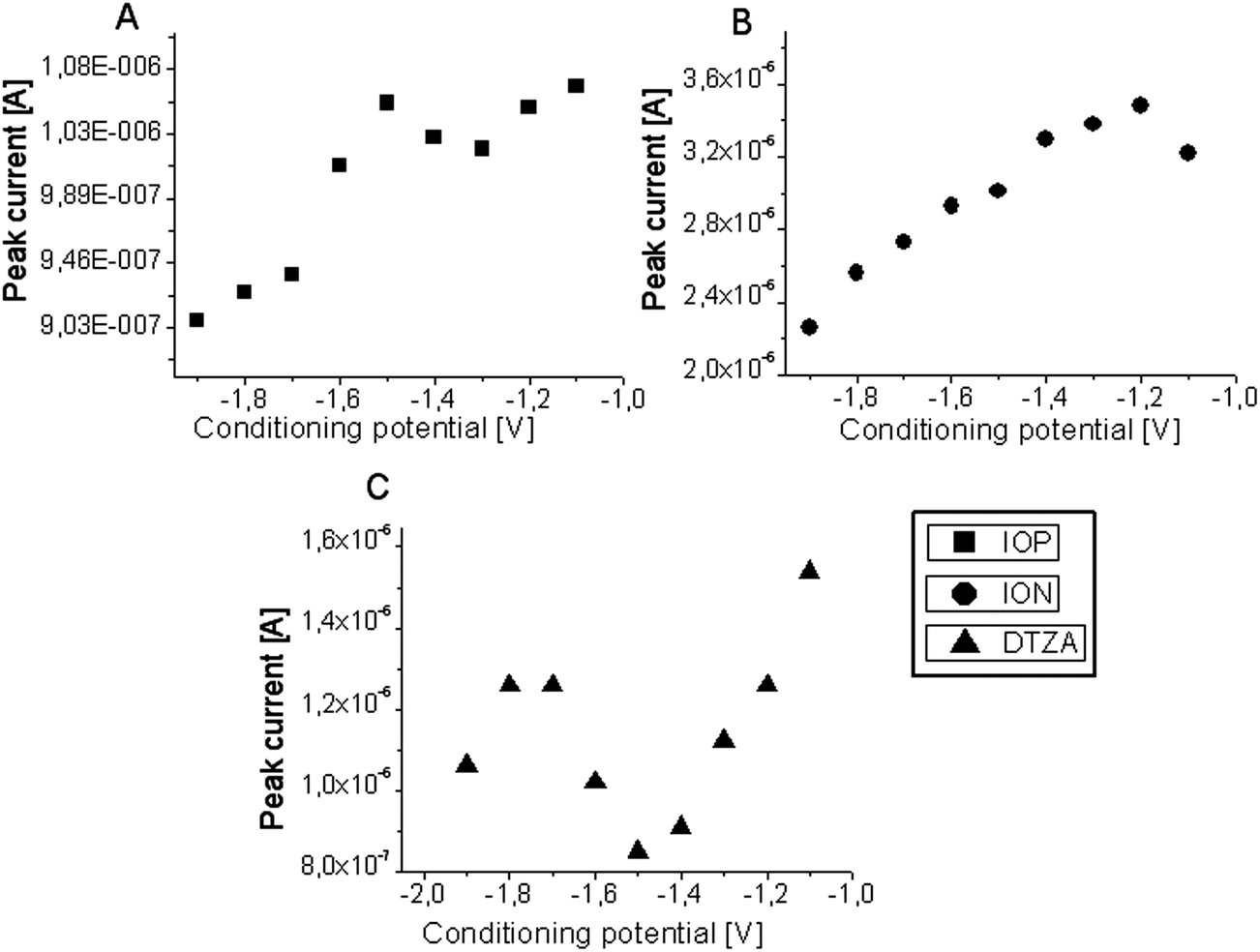 | ||
| Fig. 6 Plot of oxidative peak current response versus conditioning potential of 3.9 × 10−5 M IOP (A), 6.0 × 10−6 M of ION (B), 4.0 × 10−6 M of DTZA (C) at the GCE (vs. Ag|AgCl||KCl(sat.)). | ||
As shown (Fig. 6A), the response of the GCE for IOP decreases with increasing negative conditioning potential to −1.3 V, then the peak current increases to a potential of −1.5 V. The response of the working electrode decreases upon increasing the negative potential. In the case of ION (Fig. 6B) the peak current decreases from −1.2 V with applied higher negative potential. The peak current of DTZA (Fig. 6C) decreases with the increasing negative potential up to −1.5 V, then it increases during the conditioning with the potentials in the range of −1.6 V ÷ −1.8 V, and again decreases with applied potential of −1.9 V.
As shown on Fig. 7A, the peak current of IOP decreases with increasing conditioning time. The dependence of the peak current of ION is different (Fig. 7B)—the peak potential increases with increasing conditioning time to reach a maximum at 75 s, and then decreases with conditioning time. A similar dependence of peak current was observed for DTZA (Fig. 7C) for which the peak current increases with the increasing conditioning potential up to 60 s and then increasing conditioning potential causes a decrease in the peak current.
 | ||
| Fig. 7 Plot of oxidative peak current response versus conditioning time of 3.9 × 10−5 M IOP (A), 6.0 × 10−6 M of ION (B), 4.0 × 10−6 M of DTZA (C) in BR buffer (pH = 1.81) at the GCE (vs. Ag|AgCl||KCl(sat.)). | ||
Hence, −1.5 V and 30 s; −1.2 V and 75 s; and −1.1 V and 60 s were taken as the optimized conditioning potentials and times for the determination of IOP, ION and DTZA, respectively.
3.6 Analyte recoveries
Recoveries of X-ray contrast agents during their separation from urine were analysed. Extraction experiments were first performed using standard solutions, and then the procedure was checked with artificial urine samples. The determination of the recovery rates was carried out from spiked artificial urine samples.The mean recovery of analytes ranged from 94.4 to 101.1%. Recoveries for the studied analytes (IOP, ION and DTZA) in the other protocols ranged from about 69 to 109%.
The coefficient of variation (C.V.) for three successive determinations of ION at a concentration of 0.097 mM is 2.17%; for 0.108 mM IOP, the C.V. is 4.09%; and for 0.122 mM DTZA, it is 2.46%.
The coefficients of variation for the studied X-ray contrast agents (IOP, ION and DTZA) in the other protocols described in the literature ranged from 0.8 to 9.8%.26–40 Obtained recovery results of spiked urine samples are given in Table 5.
| Contrast agents | Concentration added (mM) | Concentration found (mM) | S.D.a (mM) | C.V.b (%) | L (mM) | Recovery (%) | U | |
|---|---|---|---|---|---|---|---|---|
| (mM) | (%) | |||||||
| a Standard deviation of concentrations found. b Coefficient of variation of concentrations found. c Confidence interval (α = 0.05, t = 4.303). d Expanded uncertainty (for confidence level 95%, coverage factor k = 2).41 | ||||||||
| ION | 0.097 | 0.092 | 0.002 | 2.17 | 0.092 ± 0.005 | 94.84 | 6.13 × 10−3 | 6.67 |
| 0.194 | 0.185 | 0.003 | 1.62 | 0.185 ± 0.007 | 95.13 | 10.28 × 10−3 | 5.56 | |
| IOP | 0.108 | 0.102 | 0.005 | 4.90 | 0.102 ± 0.012 | 94.44 | 7.28 × 10−3 | 7.14 |
| 0.275 | 0.273 | 0.004 | 1.46 | 0.273 ± 0.010 | 99.27 | 17.06 × 10−3 | 6.25 | |
| DTZA | 0.122 | 0.123 | 0.003 | 2.46 | 0.123 ± 0.007 | 100.82 | 7.23 × 10−3 | 5.89 |
| 0.285 | 0.288 | 0.006 | 2.10 | 0.288 ± 0.015 | 101.05 | 13.71 × 10−3 | 4.76 | |
3.7 Limits of detection and quantification
The limits of detection (LOD) and limits of quantification (LOQ) for determination of analysed drugs in model solutions were calculated on the peak current using the following equations: LOD = 3 S.D./a and LOQ = 10 S.D./a, where “S.D.” is the standard deviation of the peak currents and “a” is the slope of the related calibration equation.The limit of detection (LOD) was between 0.010 mM and 0.013 mM for the analysed compounds. LODs reported for IOP, ION and DTZA in the literature ranged from 0.02 to 1.43 μg L−1.17–23,26–40
The LOD and LOQ values are summarized in Table 4. Both LOD and LOQ values confirmed the sensitivity of the proposed methods.
3.8 Application of the new DPV method to urine samples and pharmaceutical formulations
Methods of preparation of urine samples for analysis n order to remove matrix effects were elaborated upon and optimized. Preparation procedures of urine samples containing IOP, ION and DTZA by SPE gave good results: the recoveries of these analytes from urine were 94.44–99.27, 94.84–95.13 and 100.82–101.05%, respectively. Received results are averages of three measurements of samples prepared in parallel. The results of these analyses (recoveries, standard deviations, coefficients of variation and confidence intervals) are summarized in Table 5. Analyte identification was performed according to peak potential by comparison with the standard solution, and by the standard addition method.The developed DPV methods for IOP, ION and DTZA determination were applied to pharmaceutical formulations (ULTRAVIST™, VISIPAQUE™ and UROGRAFIN™). Conditions of IOP, ION and DTZA determination in pharmaceutical formulations were elaborated above. Methods to prepare pharmaceutical formulations for analysis in order to remove matrix effect were elaborated.
The data proved the suitability of only diluting as a procedure for the determination of investigated compounds from pharmaceutical formulations.
3.9 Specificity
Sometimes, voltammetric techniques can pose difficulties in the analysis of biological fluids, which contain reducing or oxidizing substances. Methods of urine samples for analysis preparation in order to remove matrix effect were elaborated and optimized.The specificity of the method for the analysis of artificial urine samples was evaluated by the determination of selected iodinated X-ray contrast agents in spiked artificial urine with satisfactory results. The main components of real urine—urea, hippuric acid, uric acid and creatinine—were added to the artificial urine samples. It has been shown that urea, creatinine and hippuric acid are nonelectroactive over a range of potentials in the data measurement conditions.
Under given measurement conditions only the uric acid underwent oxidation. However, this peak does not affect the analyte determination because it has a different potential peak.
The presence of the main components of urine does not interfere in the analysis of ION, IOP and DTZA. No interfering peaks were observed near the peak potentials of examined compounds in artificial urine samples after SPE (Fig. 8).
 | ||
| Fig. 8 DPV voltammograms of ION (A), IOP (B) and DTZA (C) in BR buffer (pH = 1.81) of artificial urine samples after SPE procedure at the GCE (vs. Ag|AgCl||KCl(sat.)). | ||
4 Conclusions
We have developed and validated a new and reliable DPV method for determination of IOP, ION and DTZA and have applied the method both to standard solutions and to artificial urine including spiked analysed contrast agents. DPV was used in the voltammetric measurement owing to its good sensitivity and resolving power. It is well known that DPV is suitable for the analysis of electrochemically active substances. Well-defined oxidation peaks were observed for every studied X-ray contrast. The effect of pH on peak potential and peak current permitted elaborating the best conditions for determination of the compound. The data proved the suitability of the SPE procedure for the extraction of investigated compounds from urine samples.The developed methods showed good recoveries (from 94.4 to 101.1%) for analysed X-ray contrast agents compared with chromatographic methods. It is necessary to underline the fact that it is the first voltammetric method that has been elaborated upon for the determination of the selected iodinated X-ray contrast agents in standard solutions and in artificial urine samples. No voltammetric methods for the determination of iopromide (IOP), iodixanol (ION) and amidotrizoic acid (DTZA) have previously been described in the literature. This method may be considered as a suitable alternative to the existing chromatographic methods. Preparation of the sample was easy and the method was neither time consuming nor expensive.
Although the disadvantages of voltammetric techniques are the relatively small selectivity and the fact that the analyzed compounds must be electroactive, there are significant advantages for the proposed method. They include the easy sample preparation, and the relatively simple and inexpensive measuring apparatus compared with chromatographic techniques. Moreover, the analysis time is very short (two to three minutes) compared with the chromatographic analysis (usually a few minutes).
References
- M. V. Rocco, V. M. Buckalew, L. C. Moore and Z. K. Shihabi, Am. J. Kidney Dis., 1996, 28, 173 CAS.
- V. Meucci, A. Gasperini, G. Soldani, G. Guidi and M. Giorgi, J. Chromatogr. Sci., 2004, 42, 107 CAS.
- P. B. Jacobsen, L. Blindheim and T. Scotland, Acta Radiol., Suppl., 1995, 399, 61 CAS.
- E. Krutzén, S. E. Back, I. Nilsson-Ehle and P. Nilsson-Ehle, J. Lab. Clin. Med., 1984, 104, 955 Search PubMed.
- M. A. Jenkins, C. Houlihan, S. Ratnaike, G. Jerums and J. Des Parkin, Ann. Clin. Biochem., 2000, 37, 529 CAS.
- Z. K. Shihabi and M. S. Constantinescu, Clin. Chem., 1992, 38, 2117 CAS.
- T. Grönberg, S. Sjöberg, T. Almén and K. Goleman, Invest. Radiol., 1983, 18, 445 Search PubMed.
- S. E. Back, E. Krutzén and P. Nilsson-Ehle, J. Pharm. Sci., 1988, 77, 765 CAS.
- P. Olofsson, E. Krutzén and P. Nilsson-Ehle, Eur. J. Obstet. Gynecol. Reprod. Biol., 1996, 64, 63 CAS.
- A. Gleadhill and A. R. Michell, Res. Vet. Sci., 1996, 60, 117 CAS.
- D. Farthing, D. A. Sica, I. Fakhry, T. Larus, S. Ghosh, C. Farthing, M. Vranian and T. Gehr, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 826, 267 CAS.
- M. Pöytäkangas, E. Saario-Paunio, T. Putkonen, I. Saastamoinen, R. Frias, T. Spillmann and H. Saloniemi, Chromatographia, 2010, 71, 211 Search PubMed.
- M. Miura, N. Yamagishi, K. Sasaki, D. Kim and B. Devkota, Res. Vet. Sci., 2012, 93, 378 CAS.
- C. M. Spencer and K. L. Goa, Drugs, 1996, 52, 899 CAS.
- G. Deray, J. Invasive Cardiol., 2006, 18, 84 Search PubMed.
- H. Vik-Mo, R. Danielsen, K. Skinningsrud, T. Haider and A. Bjørkhaug, Eur. Radiol., 1997, 7, 156 Search PubMed.
- P. B. Jacobsen, Am. J. Neuroradiol., 1992, 13, 1521 CAS.
- H. Nomura, E. Teshima and H. Hakusui, J. Chromatogr., 1991, 572, 333 CAS.
- S. D. Chitnis and F. Akhlaghi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 869, 133 CAS.
- P. B. Jacobsen, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 749, 135 CAS.
- M. C. Denis, K. Venne, D. Lesiège, M. Francoeur, S. Groleau, M. Guay, J. Cusson and A. Furtos, J. Chromatogr. A, 2008, 1189, 410 CrossRef CAS PubMed.
- S. E. Bäck, P. Masson and P. Nilsson-Ehle, Scand. J. Clin. Lab. Invest., 1988, 48, 825 CrossRef.
- B. O. Axelsson, M. Jörnten-Karlsson, P. Michelsen and F. Abou-Shakra, Rapid Commun. Mass Spectrom., 2001, 15, 375 CrossRef CAS PubMed.
- J. L. Encina, L. Marti-Bonmati, C. L. Ronchera-Oms and V. Rodiguez, Eur. Radiol., 1997, 7, 115 CrossRef.
- P. Lanning and O. Brekke, Eur. Radiol., 1997, 7, 120 CrossRef.
- W. Seitz, W. H. Weber, J. Q. Jiang, B. J. Lloyd, M. Maier, D. Maier and W. Schultz, Chemosphere, 2006, 64, 1318 CrossRef CAS PubMed.
- A. Putschew, S. Schittko and M. Jekel, J. Chromatogr. A, 2001, 930, 127 CrossRef CAS.
- F. Busetti, K. L. Linge, J. W. Blythe and A. Heitz, J. Chromatogr. A, 2008, 1213, 200 CrossRef CAS PubMed.
- M. Kostopoulou and A. Nikolaou, Trends Anal. Chem., 2008, 27, 1023 CrossRef CAS PubMed.
- F. Sacher, F. T. Lange, H. J. Brauch and I. Blankenhorn, J. Chromatogr. A, 2001, 938, 199 CrossRef CAS.
- L. J. Fono and D. L. Sedlak, Water Res., 2007, 41, 1580 CrossRef CAS PubMed.
- S. A. Farag and C. E. Wells, Mikrochim. Acta, 1997, 126, 141 CrossRef CAS.
- R. S. Soman, H. Zahir and F. Akhlaghi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 816, 339 CrossRef CAS PubMed.
- S. De Baere, P. Smets, N. Finch, R. Heiene, P. De Backer, S. Daminet and S. Croubels, J. Pharm. Biomed. Anal., 2012, 61, 50 CrossRef CAS PubMed.
- V. Laroute, H. P. Lefebvre, G. Costes and P. L. Toutain, J. Pharmacol. Toxicol. Methods, 1999, 41, 17 CrossRef CAS.
- T. A. Annesley and L. T. Clayton, Clin. Chem., 2009, 55, 1196 CAS.
- J. Edelson, G. Palace and G. Park, J. Chromatogr., 1983, 274, 428 CrossRef CAS.
- S. Klenner, C. Bergmann, K. Strube, W. Ternes and T. Spillmann, Chromatographia, 2007, 65, 733 CAS.
- T. Agasøster, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 716, 293 CrossRef.
- E. Cavalier, E. Rozet, N. Dubois, C. Charlier, P. Hubert, J. P. Chapelle, J. M. Krzesinski and P. Delanaye, Clin. Chim. Acta, 2008, 396, 80 CrossRef CAS PubMed.
- P. Konieczka and J. Namieśnik, J. Chromatogr. A, 2010, 1217, 882 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |