PCR – the polymerase chain reaction
Analytical Methods Committee, AMCTB No 59
First published on 10th December 2013
Abstract
DNA-based procedures are becoming increasingly common within the analytical laboratory where the polymerase chain reaction (PCR) has become an indispensable technique. Developed in 1985 by Kary B. Mullis, PCR revolutionized the way that deoxyribonucleic acid (DNA) could be copied. Mullis's invention allowed researchers to make millions of copies of a selected DNA region within hours. Today, PCR can take minutes. PCR has widespread analytical applications in the food, environmental, medical and forensic fields. This Technical Brief covers the basics of PCR and some of its variations.
The basic components of PCR include a thermocycler, template DNA, primers, reaction buffer, free nucleotides, polymerase, salt and water (free from nucleases and contaminating DNA). Nearly any DNA region can act as a template for PCR amplification if the base sequences either side of the target region (i.e., the particular DNA sequence sought) are known. AMC Technical Briefs No 35 describes the general structure of DNA and some essential terminology. Within a typical PCR analysis the target region can be copied to almost any extent by repeating a simple three step process (amplification cycle): denaturation, annealing and elongation (Fig. 1). In theory PCR appears simple but, with numerous variables to consider, PCR can be difficult to optimise.

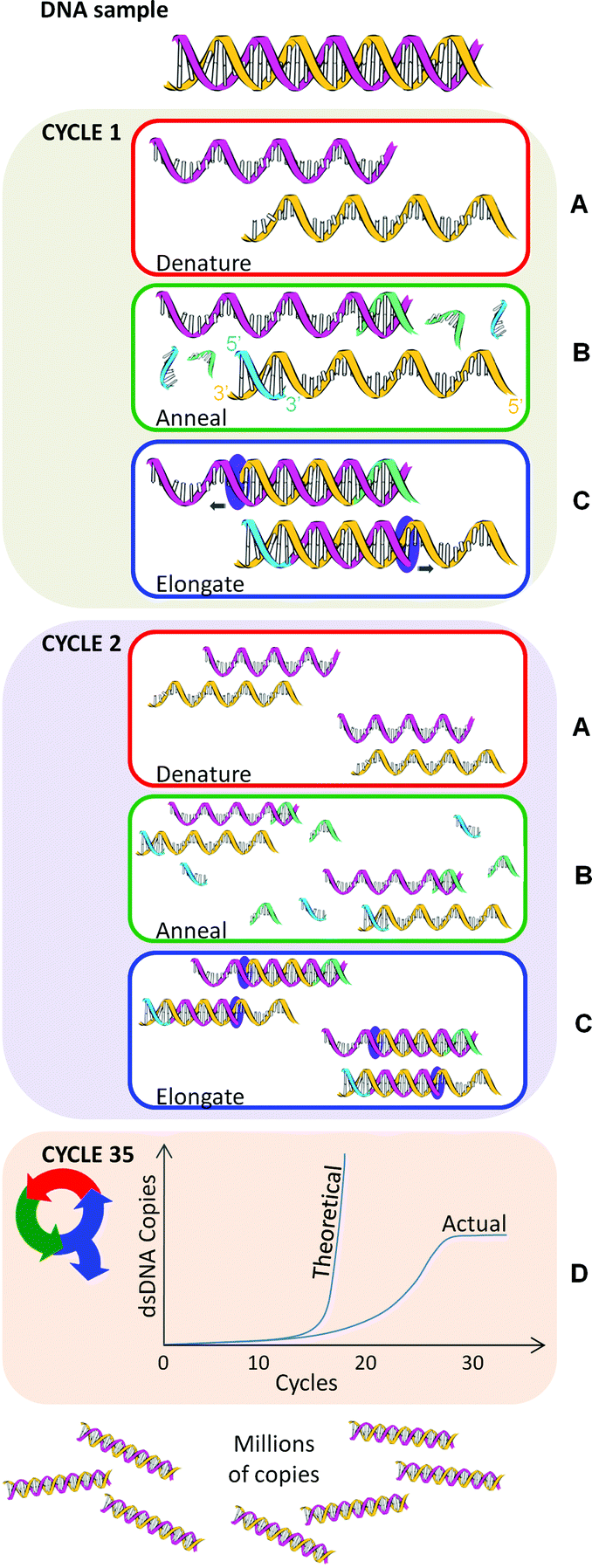 | ||
| Fig. 1 An example PCR repeated for 35 cycles. Yellow and pink helices are template nucleic acid. Green and blue strands are primers. | ||
Step A – denaturation
Here the double strands of the DNA helix (dsDNA) are separated by ‘melting’, a process analogous to pulling apart the opposing teeth of a zipper (Fig. 1A). The denaturation temperature (i.e., 90–94 °C) breaks the hydrogen bonds between the base pairs adenine (A) and thymine (T) and between cytosine (C) and guanine (G). The time and temperature required to denature dsDNA is contingent on the DNA sequence. DNA with a large number of GC base pairs requires a higher melting temperature than that with a high AT content. A higher temperature is required because GC base pairs have three hydrogen bonds whereas AT pairs have two. However, denaturation does not break the bonds between the deoxyribose sugars and the nucleoside bases, or between the sugars and phosphate entities. Thus released, the single-stranded DNA molecules (ssDNA), analogous to zipper halves, are left free to act as templates for the amplification cycle.Step B – annealing
Following denaturation, the temperature is reduced to the range 55–70 °C to allow PCR primers to bind (‘anneal’) to the ssDNA (Fig. 1B). Primers are short single strands of nucleic acid. Two sets of primers are designed to bind to complementary segments on either side of the target DNA, with their 3′ ends facing inwards. These primers serve as starting points for DNA copying to occur.Primer design is crucial to the success of the PCR technique. The PCR may fail if the sets of primers are poorly designed, resulting in no production of copies or the occurrence of mismatches. Some key design considerations here are primer length, melting temperature, GC content, DNA target length, and the formation of secondary structures.
Primer length
Typically, the optimal PCR primer length is 18–22 base pairs. Sequences greater than 17 base pairs essentially eliminate primer mismatching. Primer length influences the rate of primer hybridisation (i.e., binding) to the template DNA. If the primer length is too long the efficiency (the accuracy of reproduction) of the PCR may be reduced. Consequently, primers longer than 30 base pairs are rarely used.Melting temperature
Primer melting temperature (Tm) is the temperature where half of the DNA is in double strands and the remainder in single strands. A temperature 1–2 °C lower than the Tm usually allows the primers to hybridise to the template with minimal opportunity for mismatches to occur. Primer melting temperature is estimated using: Tm = 4[G + C] + 2[A + T] °C, where G, C, A and T are the number of nucleotides in the primer sequence. The two primer sets are designed with similar Tm so that both primers can bond with equal ease.DNA target length
The DNA target length depends on what the copied DNA fragment (otherwise known as the ‘amplicon’ or ‘PCR product’) will be used for. Obtaining large amplicons may be difficult because of poor amplification efficiency. In such cases, the PCR amplification cycle may need to be redesigned.GC content
GC content should be 40–60% to allow stable annealing of the primers to the ssDNA templates because GC bonds more strongly than AT. For similar reasons, a stretch of GC-rich sequences (a ‘GC clamp’) at the 3′ end of primers assists in the reduction of false binding. However, a balance still needs to be struck between the GC and AT content in the clamp.Formation of secondary structures
It is important to reduce the chances of the formation of secondary structures, such as ‘hairpins’ (where the primer folds on itself), ‘self-dimers’ (where the primer binds with an identical primer) or ‘cross dimers’ (where one primer binds to the other). It's also vital to design primers that do not bind to areas of ssDNA template that form secondary structures with itself. These secondary structures may prevent primers from binding to the ssDNA template.The above mentioned are not the only considerations to take into account in PCR design. It is crucial that the primers do not possess multiple sequences or cross-homology with the target. The template itself may present a challenge if it has a high GC content, or contains repetitive sequences or secondary structures, and some of the reagents used in DNA isolation may inhibit or reduce the efficiency the PCR. The pH and salt concentration may need adjusting. Getting the conditions just right is often easy but sometimes very difficult.
Step C – elongation
Once primers have annealed to the template, the temperature is increased to ∼70–75 °C. A DNA-polymerase begins the synthesis of new DNA strands from the annealed primers (Fig. 1C). Nucleotides (deoxyribonucleotide triphosphates, dNTPs) are individually added to the growing DNA strand in the 5′→3′ direction. The standard dNTPS are:• 2′-deoxyadenosine-5′-triphosphate (dATP),
• 2′-deoxycytidine 5′-triphosphate (dCTP),
• 2′-deoxyguanosine-5′-triphosphate (dGTP), and
• 2′-deoxythymidine-5′-triphosphate (dTTP).
For several reasons, however, other types of dNTPs can be used including:
• 2′-deoxyuridine-5′-triphosphate (dUTP) and
• 2′-deoxyinosine-5′-triphosphate (dITP).
For example, dUTP can be used instead of dTTP to prevent carryover contamination by amplicons from previous PCR amplifications. dITP is used in place of dGTP to improve sequencing as dGTP tends to form undesirable secondary structures that bring sequencing to a halt.
Repetition of PCR amplification cycle
The PCR amplification cycle (denaturation, annealing and elongation) is repeated in a thermocycler, usually 25–35 times for a satisfactory amount of DNA for subsequent analysis (Fig. 1D). In theory, PCR amplifies exponentially: starting from a single DNA molecule, after one cycle there will be 2 copies, after two cycles, there will be 4 copies, three cycles will result in 8 copies and so on. However, in reality an exact exponential doubling of the DNA target template doesn't immediately occur unless the primers are specifically designed to target the end of each template strand. In other words, in the first two cycles of PCR, the DNA strands are of a variable length and it's not until cycle 3 before uniform length dsPCR product is produced (i.e. 2 out of 8 total pieces of dsDNA starting from one copy).As more and more dsPCR products are amplified, they become the dominant template in the PCR. Ultimately the presence of variable length DNA is negligible. After thirty-five rounds of thermal cycling there should theoretically be approximately 3.44 × 1010 copies of that single dsDNA target. In practice, however, the polymerase, dNTPs, and primers become limiting factors that cause the reaction to plateau (Fig. 1D). The PCR plateaus even if an excess of primers and dNTPs are added, for a number of reasons including the inhibitory effect of amplicon accumulation. A final ∼72 °C incubation ends the PCR.
Polymerase
The original Mullis PCR technique was a long and costly process. After each amplification cycle, the original DNA polymerase (isolated from Escherichia coli) was irreversibly inactivated by the denaturation temperature, so the polymerase had to be refreshed at the start of each cycle. However, in 1988 Saki et al. greatly improved the PCR process by using a thermostable DNA polymerase (Taq), which was isolated from Thermus aquaticus bacteria that thrive in hot springs. Taq eliminated the need to refresh the polymerase after each amplification cycle. In addition, Taq copied at a higher accuracy than the original Mullis invention. There are now available many alternatives to Taq polymerase.PCR variations
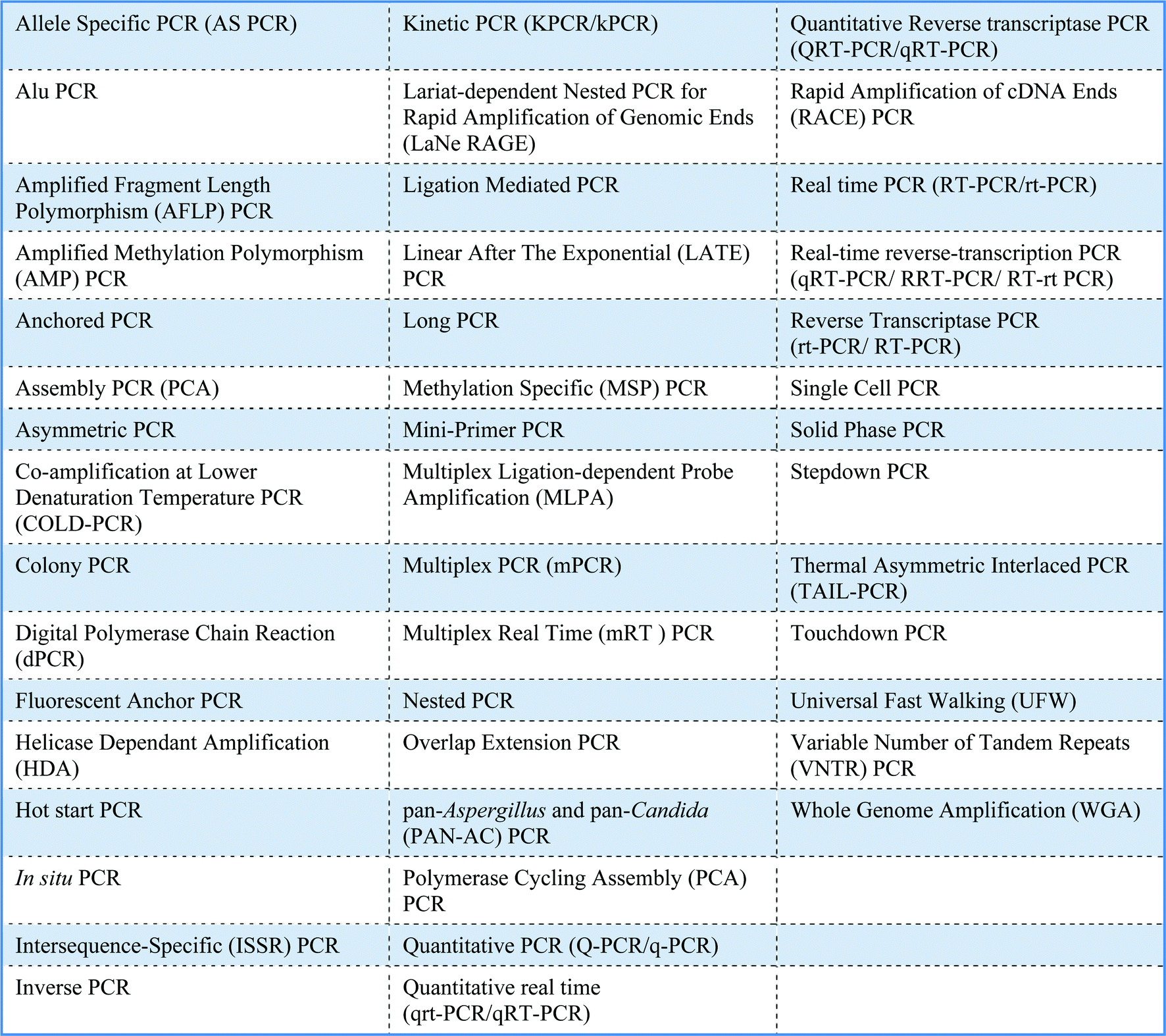
There are many different applications for PCR, which are in part reflected by the large number of variations of PCR (Table 1). No single PCR protocol fits every application. The number of acronyms for PCR variations is also huge and causes much confusion. For example, the acronyms for real-time PCR, reverse transcriptase PCR and real-time reverse transcriptase PCR are all similar and can be perplexing! Moreover, real-time PCR has a variety of alternative names including kinetic PCR (original name), quantitative PCR and quantitative real-time PCR. A standardised nomenclature system has yet to be agreed.
|
Gimara H. Duncan and Andrew P. Damant [Food Standards Agency]
This Technical Brief was prepared for the Analytical Methods Committee and approved on 14/11/13.

Further reading
- DNA – an analytical chemist's view, AMC Technical Briefs, No 35 Search PubMed.
- Specific Synthesis of DNA in vitro via a Polymerase-Catalyzed Chain Reaction, Methods Enzymol., 1987, 155, 335–350. (The original paper describing the invention of PCR published in Methods of Enzymology. The invention PCR afforded its creator, K. Mullis, the Nobel Prize in 1993) Search PubMed.
- Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase, Science, 1988, 239, 487–491 Search PubMed. The seminal paper to describe PCR using Thermus aquaticus polymerase.
-
T. Nolan, J. Huggett and E. Sanchez, Good Practice Guide for the Application of Quantitative PCR (qPCR), 2013 Search PubMed
, National Measurement System Chemical and Biological Metrology Website, http://www.nmschembio.org.uk/PublicationArticle.aspx?m=115%26amid=7994.
| This journal is © The Royal Society of Chemistry 2014 |