DOI:
10.1039/C3AY42038H
(Paper)
Anal. Methods, 2014,
6, 1884-1889
Voltammetric behaviour and quantitative determination of pesticide iminoctadine
Received
15th November 2013
, Accepted 15th January 2014
First published on 16th January 2014
Abstract
Iminoctadine (IOD) was determined in spiked river water samples by square wave voltammetry (SWV) using a cyclic renewable silver amalgam film electrode (Hg(Ag)FE). It was found that the compound can act as an electrocatalyst. In Britton–Robinson buffer at pH 6.5 a signal connected with the catalytic hydrogen evolution reaction was detected at −1.8 V versus Ag/AgCl. Validation of the method was carried out. The LOD and LOQ have been estimated to be 2.6 × 10−9 mol L−1 and 8.5 × 10−9 mol L−1, respectively.
Introduction
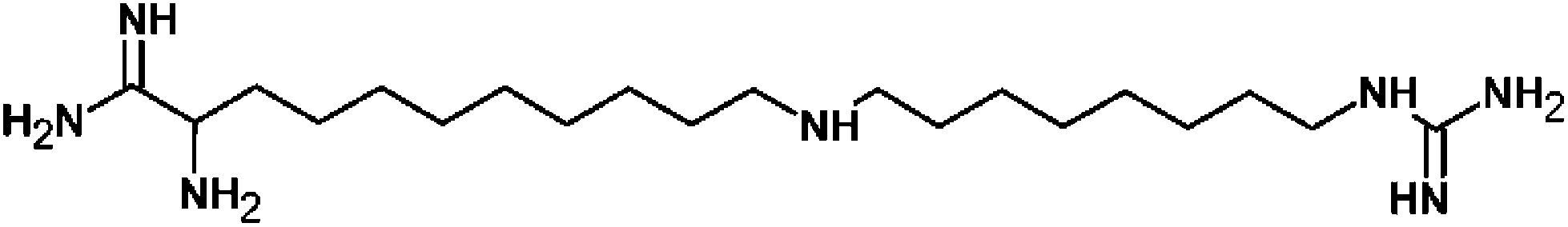
Iminoctadine (1,1′-(iminodioctamethylene)diguanidine) (Fig. 1) is a non-systemic aliphatic nitrogen contact fungicide which impairs functioning membranes in fungi.
 |
| | Fig. 1 Molecular structure of iminoctadine. | |
Iminoctadine (IOD) is widely used on fruits, trees and lawns to control a variety of pathogens including Gloeodes and Alternaria. Frequently it is also sold in a mixture (called guazatine) of products resulting from the amidination of technical iminodi(octamethylene)diamine, containing numerous guanidines and polyamines. Guazatine controls a wide range of seed-borne diseases of cereals, e.g. seedling blight (Fusarium spp.), glume blotch (Septoria), smut (Ustilago), common bunt (Tilletia spp.) and common root rot (Helminthosporium). On citrus fruits, guazatine is used in washing installations to disinfect the process water, as a bulk dip after harvest, and as a spray in the packing line. It controls sour rot (Geotrichum candidum), blue mould (Penicillium italicum) and green mould (Penicillium digitatum).1,2 Iminoctadine contains a guanidine group in its structure so it can act as an electrocatalyst, being protonated and adsorbed at the electrode surface, and then irreversibly reduced, yielding their initial form and hydrogen.3,4 To our knowledge, there is no electrochemical studies based on catalytic properties of IOD. Only a few nonelectrochemical (LC/MS5 and LC/ESI/MS6) methods for quantitative determination of iminoctadine were published to date. Determination at a trace level of guazatine has been performed mainly by gas chromatographic methods (in fruits,7 crops and soil8) and using the LC/ESI/MS method for the quantitative detection of guazatine residues in cereals.9 Moreover, Pang reported that guazatine cannot be determined by GC-MS and LC-MS-MS.10 In the current paper, we present a study of iminoctadine on a Hg(Ag)FE under conditions of square-wave voltammetry (SWV). Square-wave voltammetry11,12 is nowadays one of the most advanced pulse voltammetric techniques for analytical application,13–16 mechanistic17–19 and kinetic studies of electrode processes.20–23 In the last two decades the majority of published voltammetric methods was based on the mercury electrodes. HMDEs are particularly useful due to their high repeatability and sensitivity. Notwithstanding, increasing public awareness and care for the environment caused a decline in the usage of mercury electrodes in analytical practice. Because of these tendencies in this work we propose a cyclic renewable silver amalgam film electrode (Hg(Ag)FE). Construction of an Hg(Ag)FE, its features and the principles of its operation were described in detail formerly.24–26 The use of silver amalgamate enables the electrode to work for a few months in a stable manner.27 This feature of preserving the properties of the mercury electrode with consuming a very small amount of mercury (about 1 μL on 1000 measurement cycles27) is the main advantage of this electrode. As mentioned in our previous papers, Hg(Ag)FEs and other amalgam based electrodes have been widely used in the analysis of metals,27–30 as well as inorganic31,32 and organic compounds.33–35 In the present study, the electrode mechanism is confirmed in the light of recent theories for guanidine compounds. Based on the knowledge of the electrode reaction mechanism, a quantitative method for determination of IOD is proposed by means of square-wave voltammetry.
Experimental
Instrumentation
All voltammetric experiments were performed using μAutolab Type III/GPES (General Purpose Electrochemical System, version 4.9, Eco Chemie, Netherlands) and an M164 electrode stand (Mtm-Anko, Cracow, Poland). Experiments were performed in a three-electrode system consisting of Ag/AgCl (3 mol L−1 KCl) as a reference electrode, Pt wire as a counter electrode, and a silver-based renewable silver amalgam film electrode (Mtm-Anko, Cracow, Poland) as a working electrode. Silver amalgam film was easily made before each experiment. The procedure of refreshing involves two steps: pulling up the silver electrode inside the electrode holder, through the mercury reservoir and then pushing it back outside the electrode holder. Construction of the Hg(Ag)FE gives the possibility of precise regulation of the electrode surface (up to 12 mm2). In these experiments surface area of 12 mm2 was used, due to the fact that the recorded peak current increased linearly with enlargement of the electrode surface area. Measurements of pH were made using a CP-315M pH-meter (Elmetron, Poland) with a combined glass electrode.
Solutions and materials
All chemicals used were of analytical reagent grade. Iminoctadine acetate and other chemicals were purchased from Sigma Aldrich. A fresh stock solution of 1.00 × 10−3 mol L−1 IOD was prepared weekly by dissolving 13.39 mg of the compound in 25 mL of water. The studied supporting electrolytes were 0.2 mol L−1 citrate-phosphate buffers (pH 6.5–8.0), 0.04 mol L−1 Britton–Robinson (BR) buffers (pH 2.0–8.7) and 0.02 mol L−1 phosphate buffers (pH 6.5–8.0). Triply distilled and deionised water was used throughout the experiments. Solutions were purged with pure argon for at least 10 min prior to each voltammetric scan, and argon was passed over the solutions during the measurements. All electrochemical measurements were carried out at the ambient temperature of the laboratory (20–22 °C).
Voltammetric procedure
The general procedure used to obtain voltammograms was as follows: 10 mL of the supporting electrolyte was transferred to the electrochemical cell, deaerated by passing an argon stream for 10 min, and then a voltammogram was registered under the inert atmosphere of the cell. When an initial blank was recorded, the required volumes of the compound were added by means of a micropipette. In the present study, the optimal results for SW experiments were obtained in BR buffer at pH 6.5, an amplitude of 80 mV, a frequency of 16 Hz and a step potential of 7 mV.
Analysis of river water samples
Preparation of spiked river water is as follows: 2.5 nmol (samples 1a and 2a), 5 nmol (samples 1b and 2b) or 10 nmol (samples 1c and 2c) of iminoctadine was placed in a 50 ml flask and filled up to the volume with river water (samples 1a, 1b, and 1c – Bzura, Poland; samples 2a, 2b, and 2c – Labe, Czech Republic). In all experiments, voltammograms were recorded under the same conditions as for pure iminoctadine. The spiked river water was analyzed using the standard addition method and the recoveries obtained after three replicate experiments were calculated.
Results and discussion
Influence of pH and SW parameters
The choice of supporting electrolyte is an important stage in electrochemical studies. The composition of the supporting electrolyte affects the properties of the solution and the solution–electrode interface, which influence the kinetics and thermodynamics of the charge transfer process.36 The effect of various supporting electrolytes such as citrate–phosphate buffer, phosphate buffer and Britton–Robinson buffer solutions on the IOD peak current was investigated using the Hg(Ag)FE electrode (voltammograms not shown). The results showed similar current responses in all the buffer types. However, the best-defined peaks were observed in BR buffer (E [V] = −0.0587pH − 1.27 [V]) with the highest signals at pH 6–7. Similar results were obtained in previous studies for other compounds with a guanidine group.37 Hence, BR buffer pH 6.5 was chosen as the most suitable supporting electrolyte for analytical application in all further voltammetric experiments. The influence of the ionic strength of the supporting electrolyte was also investigated. The best response was obtained when the supporting electrolyte contained 100% of 0.04 mol L−1 BR buffer. As a popular electrochemical method with good discrimination against capacitive current, SWV has been applied to numerous biologically and electrochemically active compounds in the analysis of their trace amounts. The optimization of instrumental SWV parameters, which can influence the current response, is an important stage in the development of an electroanalytical methodology. Hence, the influence of instrumental parameters such as SW frequency (f), the height of SW pulses (amplitude ESW) and the step potential of the staircase waveform (ΔE) were investigated.11 These parameters have an interdependent effect on the peak potential and current, but in this work only the general tendencies were investigated. During adjustment of the mentioned parameters, each parameter was changed while the others were kept constant using a 5 × 10−7 mol L−1 IOD concentration. The studied parameter ranges for the amplitude, step potential and frequency were 5–150 mV, 1–25 mV, 8–1995 Hz, respectively. The optimized values were: ESW = 80 mV, ΔE = 7 mV, f = 16 Hz.
Electrochemical behaviour of iminoctadine
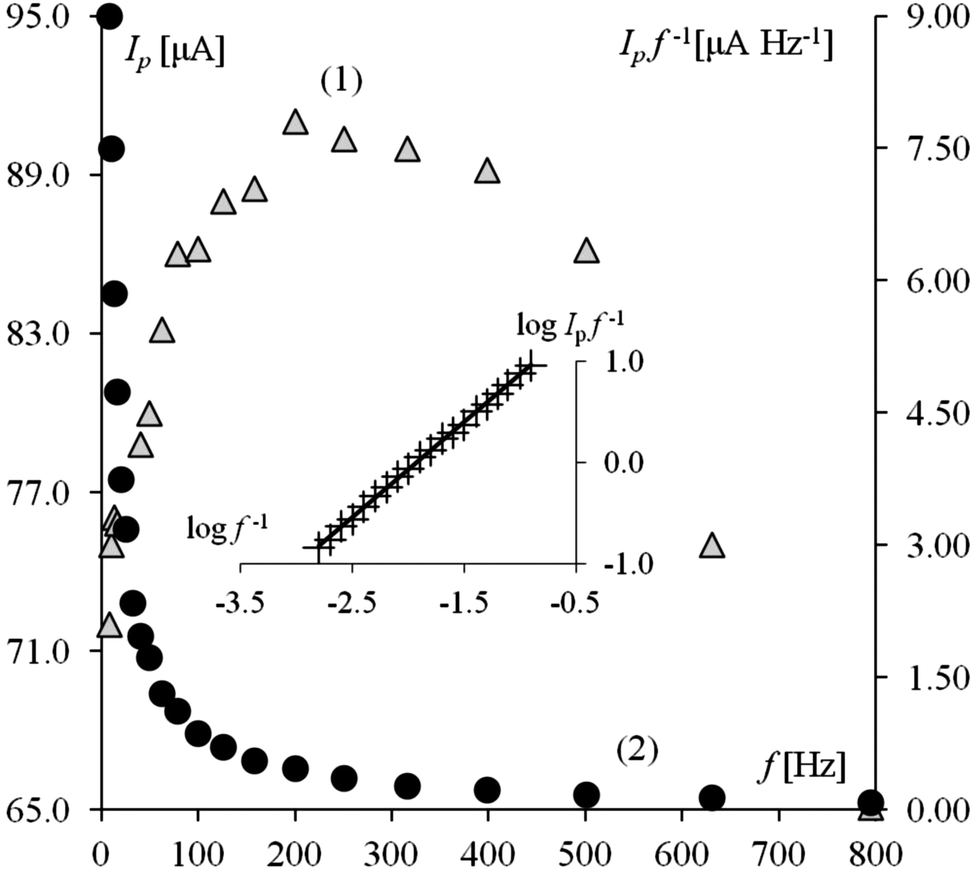
The presence of a guanidine group in the IOD structure suggests a possibility for catalytic hydrogen evolution. The appearance of a peak in the potential range characteristic for this type of mechanism confirms this thesis. Theoretical and experimental studies performed with square wave voltammetry were used to explain the catalytic hydrogen evolution mechanism of the adsorbed catalyst.4 Such a procedure can be used in experiments with working electrodes containing mercury when adsorption takes place and the type of electrode mechanism can be concluded from the influence of frequency on the recorded peak current. The investigation based on the frequency requires the examination of several dependences: Ipf−1 = f(f), Ip = f(f), log(Ip/f) = log(1/f).3,4,38–40 As demonstrated in ref. 3, the overall catalytic effect depends on the κcat = kp/f (dimensionless catalytic parameter), where kp = k'p × CH+ and k'p is the protonation rate constant. Therefore, an uprising frequency causes a decline in the net peak current (NPC). On the other hand by adjusting the time window of the experiment (realized with alteration of frequency) it is possible to influence the height of recorded peak current which is an obvious feature in SWV for all types of electrode mechanisms.12 Then, the overall dependence of the NPC on frequency is a sum of the two contrary actions of frequency, which is demonstrated in Fig. 2 (curve 1). The Ip/f on f dependence shows solely the influence of the catalytic parameter (curve 2 in Fig. 2). Such a course of the curve is typical for all catalytic mechanisms.38,39 By numerical simulations,3 it has been indicated that the most typical feature of the current catalytic mechanism is a linear dependence log(Ip/f) vs. log(κcat), which holds under a large variety of experimental conditions. This type of dependence can be obtained by plotting log(Ip/f) versus log(1/f) (inset in Fig. 2), and its linearity can be regarded as a diagnostic criterion for the catalytic hydrogen evolution reaction in SWV. Therefore, received curves (for the Ip = f(f), Ipf−1 = f(f) and log(Ipf−1) = log(f−1) (Fig. 2) dependences) are consistent with theoretical data3,4 for other guanidine compounds which confirms that IOD plays the role of an electrocatalyst and the electrode mechanism can be featured by two equations. Eqn (1) describes the preceding chemical reaction, in which the adsorbed catalyst undergoes protonation. Eqn (2) refers to the protonated form of the catalyst, which is irreversibly reduced yielding the initial form of the catalyst and atomic hydrogen:| | | IOD(ads) + H(aq)+ ↔ IODH(ads)+ | (1) |
| | | IODH(ads)+ + e → IOD(ads) + H(aq) | (2) |
 |
| | Fig. 2 The influence of the frequency (f) on the net peak current (Ip) (curve 1 left ordinate) and the ratio Ip/f (curve 2, right ordinate) recorded at the Hg(Ag)FE. The inset shows the dependence of log(Ip/f) on the log(1/f). The conditions of the experiments were: BR buffer pH 6.5, c(IOD) = 5 × 10−7 mol L−1, amplitude Esw = 80 mV, step potential ΔE = 7 mV. | |
Analytical application
In order to develop an analytical method for determination of IOD square wave voltammetry at Hg(Ag)FEs was selected as the one which guarantees effective and rapid determination with low background current and low detection limits.41 Quantitative measurements were performed using SWV, BR buffer pH 6.5, and the best conditions for analytical application. The cathodic peak current increased linearly with increasing the concentration of IOD from 1 × 10−8 to 1 × 10−6 mol L−1 (Fig. 3). Calibration curves for the SWV techniques were constructed by plotting the peak current against the IOD concentration.
 |
| | Fig. 3 SW voltammograms of IOD in BR buffer pH 6.5, IOD concentration indicated by each line. The other experimental conditions were: amplitude ESW = 80 mV, step potential ΔE = 7 mV, frequency f = 16 Hz. | |
The characteristics of the calibration plots are provided in Table 1. The limits of quantification (LOQ) and detection (LOD) were calculated from the calibration curves as kSD/b (k = 10 for LOQ, k = 3 for LOD, SD = standard deviation of the intercept, b = slope of the calibration curve).42 The repeatability of the procedure was estimated with 3 measurements at the same IOD concentration. In order to check the correctness of the method (Table 2), the precision and recovery of the method were also calculated for different concentrations in the linear range.
Table 1 Quantitative determination of IOD in BR buffer pH 6.5 by SWV. Basic statistic data of the regression line
| Linear concentration range (mol L−1) |
1.0 × 10−8 – 1.0 × 10−6 |
| Slope of the calibration graph (A L mol−1) |
148.7 |
| SD of the slope |
1.32 |
| Intercept (A) |
4.5 × 10−8 |
| SD of the intercept |
1.27 × 10−7 |
| Correlation coefficient |
0.9996 |
| Number of measurements |
3 |
| LOD (mol L−1) |
2.6 × 10−9 |
| LOQ (mol L−1) |
8.5 × 10−9 |
Table 2 Recovery and precision of the IOD peak currents at various IOD concentrations
| Concentration given [μmol L−1] |
Concentration found [μmol L−1] |
SD [×10−9] |
Precision RSD [×10−2] |
Recoveryb [%] |
|
t(S/n1/2), p = 95%, n = 6.
Recovery = 100% + [(found − added)/added] × 100%.
|
| 0.01000 |
0.00969 ± 0.00038a |
0.34 |
3.49 |
96.9 |
| 0.03000 |
0.02993 ± 0.00039 |
0.35 |
1.15 |
99.8 |
| 0.0500 |
0.0492 ± 0.0015 |
1.30 |
2.64 |
98.4 |
| 0.0700 |
0.0692 ± 0.0011 |
0.95 |
1.38 |
98.9 |
| 0.0900 |
0.0923 ± 0.0029 |
2.54 |
2.75 |
102.6 |
| 0.1000 |
0.0974 ± 0.0029 |
2.55 |
2.61 |
97.4 |
| 0.3000 |
0.3017 ± 0.0133 |
11.78 |
3.91 |
100.6 |
| 0.5000 |
0.4857 ± 0.0054 |
4.79 |
0.99 |
97.1 |
| 0.7000 |
0.7155 ± 0.0078 |
6.86 |
0.96 |
102.2 |
| 0.9000 |
0.8893 ± 0.0067 |
5.96 |
0.67 |
98.8 |
| 1.000 |
1.003 ± 0.014 |
12.85 |
1.28 |
100.3 |
Analysis of spiked river water
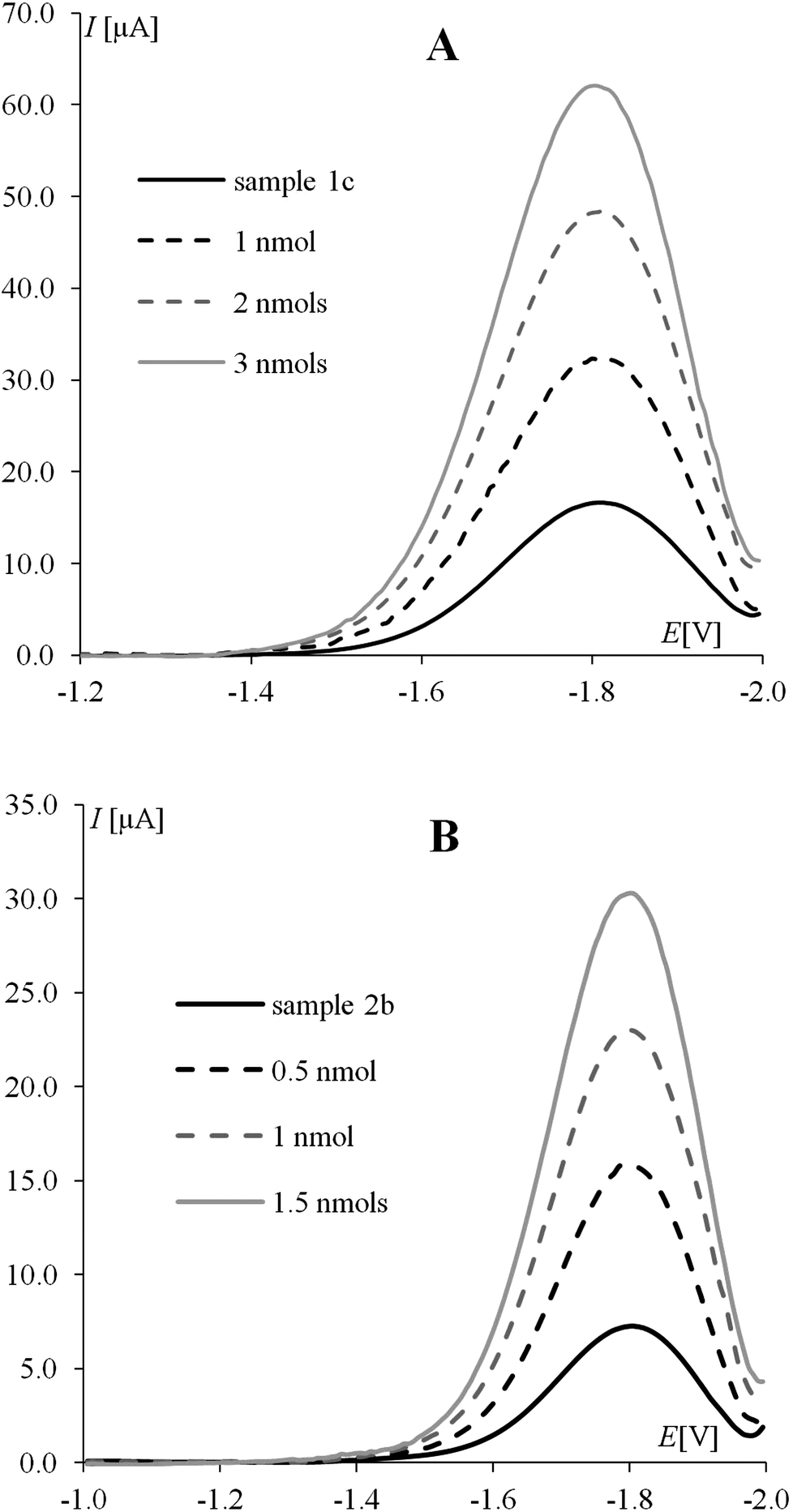
In this work river water samples (samples 1a, 1b, and 1c – Bzura, Poland; samples 2a, 2b, and 2c – Labe, Czech Republic) were selected for application of the proposed procedure. The samples were used without any pre-separation or pre-concentration. Each sample was contaminated by the addition of a specific concentration of the iminoctadine pesticide. An aliquot of the each sample was added into the electrochemical cell and the recovery curves using the optimized parameters were constructed by the standard addition method. Three replicate analyses for each sample were made. To evaluate the recovery percentage, the least squares regression method was used. The obtained SWV responses for samples 1c and 2b are shown in Fig. 4 as examples. As it can be seen the IOD voltammetric peak is free from sample component interference. The data related to the recovery curves in river water samples are shown in Table 3. Values of recovery, calculated for all the samples, pointed that the components of the matrices do not affect the analytical sensitivity. According to that the proposed procedure can be easily and successfully employed in the evaluation of recovery curves.
 |
| | Fig. 4 SW voltammograms of IOD determination in spiked river water (A – sample 1c, B – sample 2b) using the standard addition method; additions indicated by each line. Experimental conditions are the same as in Fig. 3. | |
Table 3 Results of the IOD determination in river water samples by SWV, n = 3
| Sample |
Added [μmol L−1] |
Found [μmol L−1] |
SD [×10−9] |
Precision RSD [×10−2] |
Recoveryb [%] |
|
t(S/n1/2), p = 95%, n = 6.
Recovery = 100% + [(found − added)/added] × 100%.
|
| 1a |
0.0500 |
0.0486 ± 0.0021a |
1.88 |
2.59 |
97.2 |
| 1b |
0.1000 |
0.0986 ± 0.0018 |
1.56 |
1.58 |
98.6 |
| 1c |
0.2000 |
0.1950 ± 0.0148 |
13.1 |
6.72 |
97.5 |
| 2a |
0.0500 |
0.0495 ± 0.0025 |
2.49 |
4.54 |
99.0 |
| 2b |
0.1000 |
0.0995 ± 0.0043 |
3.80 |
3.82 |
99.5 |
| 2c |
0.2000 |
0.2040 ± 0.0078 |
6.88 |
3.36 |
102.1 |
Interferences
The selectivity of the proposed method was evaluated by the addition of possible interferents – heavy metals (lead, zinc, cadmium, and copper) and other pesticides (metham, clothianidin, nitrothal, acibenzolar-S-methyl, and aclonifen) to a 5 × 10−7 mol L−1 iminoctadine solution at the concentration ratios 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, 1:10, 1:20 (Fig. 5). The responses were compared with that obtained using the iminoctadine standard solution. Only the presence of cadmium above the concentration 1 × 10−6 mol L−1 caused fourfold decrease of the iminoctadine signal. Rest of the studied substances do not interfere (signal change < 5%) in the determination of iminoctadine at the used working conditions.
:1, 1:2, 1:10, 1:20 (Fig. 5). The responses were compared with that obtained using the iminoctadine standard solution. Only the presence of cadmium above the concentration 1 × 10−6 mol L−1 caused fourfold decrease of the iminoctadine signal. Rest of the studied substances do not interfere (signal change < 5%) in the determination of iminoctadine at the used working conditions.
 |
| | Fig. 5 SW voltammograms of IOD determination in the presence of chosen interferents: A – cadmium, B – copper, and C – nitrothal; sample – 5 × 10−7 mol L−1 IOD; interferent concentrations: a – 5 × 10−7 mol L−1, b – 1 × 10−6 mol L−1, c – 5 × 10−6 mol L−1, and d – 1 × 10−5 mol L−1. Experimental conditions are the same as in Fig. 3. | |
Conclusions
The electrochemical behaviour of iminoctadine at the Hg(Ag)FE was established and studied for the first time. The electrode mechanism based on frequency investigation was analyzed under conditions of square wave voltammetry. It was established that the voltammetric response in the presence of IOD is a result of catalytic hydrogen evolution. This behaviour provides a useful tool for the detection and quantification of the compound in samples at low concentration levels. The proposed method is simple, sensitive and could be used for routine control of laboratory samples containing IOD. Moreover, the main advantages of the proposed method (LOD = 2.6 × 10−9 mol L−1, LOQ = 8.5 × 10−9 mol L−1) are more pronounced in comparison with the procedures developed previously. The developed procedures based on SW techniques are cheaper, more environmentally friendly and have lower detection and quantification limits (LOD = 9.4 × 10−9 mol L−1, LOQ = 2.8 × 10−8 mol L−1 for ref. 6).
Acknowledgements
Financial support with the Grant for Young Researchers from the University of Lodz agreement no. 545/726 is gratefully acknowledged.
Notes and references
- Pesticide residues in food – 1997, Report (FAO Plant Production and Protection Paper – 145), http://www.fao.org/docrep/w8141e/w8141e0v.htm.
- European Food Safety Authority, Conclusion on the peer review of the pesticide risk assessment of the active substance guazatine, EFSA J., 2010, 8, 1708, DOI:10.2903/j.efsa.2010.1708 , http://www.efsa.europa.eu.
- S. Skrzypek, W. Ciesielski, A. Sokołowski, S. Yilmaz and D. Kazmierczak, Talanta, 2005, 66, 1146 CrossRef CAS PubMed.
- S. Skrzypek, Electroanalysis, 2010, 22, 2339 CrossRef CAS.
- H. Kobayashi, Bunseki Kagaku, 2009, 58, 985 CrossRef CAS.
- T. Kawamoto, M. Yano and N. Makihata, Anal. Sci., 2006, 22, 489 CrossRef CAS.
- A. Neicheva, D. Karageorgiev and T. Konstantinova, Sci. Total Environ., 1992, 123–124, 29 CrossRef CAS.
- V. P. Lynch, Environ. Qual. Saf., Suppl., 1975, 3, 124 CAS.
- E. Dreassi, A. T. Zizzari, A. Zanfini, G. Corbini and M. Botta, J. Agric. Food Chem., 2007, 55, 6850 CrossRef CAS PubMed.
- G.-F. Pang, Y.-Z. Cao, J.-J. Zhang, C.-L. Fan, Y.-M. Liu, X.-M. Li, G.-Q. Jia, Z.-Y. Li, Y.-Q. Shi, Y.-P. Wu and T.-T. Guo, J. Chromatogr., A, 2006, 1125, 1 CrossRef CAS PubMed.
-
V. Mirceski, S. Komorsky-Lovric and M. Lovric, in Square-wave voltammetry: theory and applications, ed. F. Scholz, Springer, Heidelberg, 2007 Search PubMed.
-
M. Lovric, Square-wave voltammetry, in Electroanalytical methods: guide to experiments and applications, ed. F. Scholz, Springer-Verlag, Berlin, 2002 Search PubMed.
- F. G. Banica, D. Guziejewski, S. Skrzypek, W. Ciesielski and D. Kaźmierczak, Electroanalysis, 2009, 21, 1711 CrossRef CAS.
- S. Komorsky-Lovric and I. Novak, J. Food Sci., 2011, 76, C916 CrossRef CAS PubMed.
- M. Trindade and M. Zanoni, Electroanalysis, 2007, 19, 1901 CrossRef CAS.
- M. A. El Mhammedi, M. Bakasse, R. Bachirat and A. Chtaini, Food Chem., 2008, 110, 1001 CrossRef CAS PubMed.
- V. Mirceski, J. Electroanal. Chem., 2001, 508, 138 CrossRef CAS.
- V. Mirceski and M. Lovric, J. Electroanal. Chem., 2004, 565, 191 CrossRef CAS PubMed.
- V. Mirceski, D. Guziejewski and W. Ciesielski, Electroanalysis, 2011, 23, 1365 CrossRef CAS.
- A. Nosal-Wiercińska, Cent. Eur. J. Chem., 2012, 10, 1290 CrossRef PubMed.
- A. Nosal-Wiercińska, Electrochim. Acta, 2010, 55, 5917 CrossRef PubMed.
- V. Mirceski, D. Guziejewski and K. Lisichkov, Electrochim. Acta, 2013, 114, 667 CrossRef CAS PubMed.
- V. Mirceski, E. Laborda, D. Guziejewski and R. G. Compton, Anal. Chem., 2013, 11, 5586 CrossRef PubMed.
-
B. Baś, Polish Pat. P-319984, AGH University of Science and Technology, 1997.
- B. Baś and Z. Kowalski, Electroanalysis, 2002, 14, 1067 CrossRef.
- S. Smarzewska, S. Skrzypek and W. Ciesielski, Electroanalysis, 2012, 24, 1591 CrossRef CAS.
- A. Bobrowski, M. Gawlicki, P. Kapturski, V. Mirceski, F. Spasovski and J. Zarębski, Electroanalysis, 2009, 21, 36 CrossRef CAS.
- R. Piech, B. Baś and W. W. Kubiak, Talanta, 2008, 76, 295 CrossRef CAS PubMed.
- O. Mikkelsen, K. H. Schroeder and T. A. Aarhaug, Collect. Czech. Chem. Commun., 2001, 465 CrossRef CAS.
- O. Mikkelsen and K. H. Schroeder, Anal. Lett., 2000, 33, 3253 CrossRef CAS.
- B. Yosypchuk and L. Novotny, Electroanalysis, 2002, 14, 1733 CrossRef CAS.
- B. Yosypchuk and L. Novotny, Crit. Rev. Anal. Chem., 2002, 32, 141 CrossRef CAS.
- S. Smarzewska, S. Skrzypek and W. Ciesielski, Electroanalysis, 2012, 24, 1966 CrossRef CAS.
- R. Selesovska-Fadrna, M. Fojta, T. Navratil and J. Chylkova, Anal. Chim. Acta, 2007, 582, 344 CrossRef CAS PubMed.
- R. Selesovska-Fadrna, T. Navratil and M. Vlcek, Chem. Anal., 2007, 52, 911 CAS.
-
P. T. Kissinger and W. R. Heinemann, Laboratory Techniques in Electroanalytical Chemistry, Taylor & Francis, New York, 2nd edn, 1996 Search PubMed.
- S. Skrzypek, S. Smarzewska and W. Ciesielski, Electroanalysis, 2012, 24, 1153 CrossRef CAS.
- V. Mirceski and R. Gulaboski, J. Solid State Electrochem., 2003, 7, 157 CAS.
- V. Mirceski and R. Gulaboski, Electroanalysis, 2001, 13, 1326 CrossRef CAS.
- S. Skrzypek, V. Mirceski, S. Smarzewska, D. Guziejewski and W. Ciesielski, Collect. Czech. Chem. Commun., 2011, 76, 1699 CrossRef CAS.
-
J. Wang, Electroanalytical Techniques in Clinical Chemistry and Laboratory Medicine, VCH, New York, 1988 Search PubMed.
- L. B. O. dos Santos, G. Abate and J. C. ;Masini, Talanta, 2004, 62, 667 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence