 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation and purification of ent-pimara-8(14),15-diene from engineered Aspergillus nidulans by accelerated solvent extraction combined with HPLC†
K.
Bromann
*a,
K.
Viljanen
a,
V. M.
Moreira
b,
J.
Yli-Kauhaluoma
b,
L.
Ruohonen
a and
T.
Nakari-Setälä
a
aVTT Technical Research Centre of Finland, FI-02044 VTT, Tietotie 2, Finland. E-mail: kirsi.bromann@vtt.fi; Fax: +358 20 722 7071; Tel: +358 40 067 5071
bDivision of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Helsinki, FI-00014 University of Helsinki, Viikinkaari 5E, Finland
First published on 11th December 2013
Abstract
We recently engineered Aspergillus nidulans to produce a pimarane-type diterpene, ent-pimara-8(14),15-diene. Here, we describe methods for its isolation and purification from the engineered A. nidulans production strain and extend these findings with structural confirmation for the diterpene from this fungal source. The extraction protocol was optimized using accelerated solvent extraction (ASE) with three varying parameters: solvent composition, pressure, and temperature. This ASE method using 100% ethyl acetate at 90 °C with 12.07 MPa was more efficient and faster compared to ultrasonic-assisted extraction. Using both analytical and preparative C18 columns at isocratic elution of acetonitrile developed an HPLC separation; and, the diterpene was well separated within 28 min and 35 min, respectively. Also, TLC of the fungal culture extracts was performed with visualization using rhodamine. The reproduction and efficiency of the purification methods were evaluated by GC-MS; and, the structure of the compound was verified by NMR. The method enabled the isolation of this very lipophilic diterpene in pure form, allowing its testing in a number of bioactivity assays. Notably, we demonstrate for the first time the antioxidant activity for ent-pimara-8(14),15-diene using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay.
Introduction
Aspergillus nidulans is a filamentous fungus widely used as a model organism for studies of cell biology and gene regulation. It is a close relative to other Aspergillus species with industrial and medical significance – e.g., A. niger, A. oryzae, A. flavus, and A. fumigatus. The genus Aspergillus produces a variety of natural products, and offers a model system to study secondary metabolism in eukaryotes.1 Secondary metabolites are microbial organic compounds that are not crucial for the survival of the organism but many times exhibit interesting or useful bioactivities. These are also referred to as natural products. Major groups of secondary metabolites include polyketides, ribosomal and non-ribosomal peptides, and terpenoids. In fungi, the biosynthetic genes of secondary metabolism are organized into clusters.2 The clustering of the genes along with the sequenced genomes of many Aspergillus species has accelerated the discovery of novel natural products.3 Of the numerous secondary metabolites produced by Aspergilli, the most notorious ones include mycotoxins, such as A. flavus aflatoxin4,5 and A. carbonarius ochratoxin.6,7 On the other hand, many of the Aspergillus natural products also exhibit beneficial properties and include the antibiotic penicillin8 and the cholesterol reducing agent lovastatin.9 Although the functional role of secondary metabolites in the producing organism is often unclear, it has been speculated that secondary metabolites function as chemical signals in communication and defense to enhance the survival of the organism within its ecological environment.10Terpenoids are particularly interesting secondary metabolites because of their many pharmaceutical and toxic bioactivities.11–13 Specifically, oxygenated derivatives of pimaradiene and kaurene have vasorelaxant, anti-inflammatory, anti-microbial and trypanocidal properties.14–19 Biosynthesis of fungal natural products is regulated by environmental as well as genetic factors.8 Recently, we reported that overexpression of a Zn(II)2Cys6-type transcription factor in A. nidulans, called pimaradiene biosynthetic cluster regulator (pbcR), led to production of a diterpene hydrocarbon, ent-pimara-8(14),15-diene.20ent-Pimara-8(14),15-diene has previously been identified as a product of the rice (Oryza sativa subspecies japonica) pimaradiene synthase, OsKSL5j.21 Many of the diterpene hydrocarbons in rice, for example, ent-sandaracopimaradiene, ent-cassa-12,15-diene, 9βH-pimara-7,15-diene, and stemar-13-ene groups function as pathway intermediates for diterpene phytoalexins, which have been suggested to act as antibiotics.22 Also, ent-kaur-16-ene is a precursor of a diterpene phytohormone gibberellic acid that regulates many aspects of development in higher plants. However, the biological activity for ent-pimara-8(14),15-diene in rice is still unknown, since OsKSL5 is not involved in the biosynthesis of diterpenoid phytoalexins or gibberellins.23
In addition to higher plants, diterpene hydrocarbons have also been found in fungi.20,24–26 For example, sandaracopimaradiene, isopimara-8,15-diene and pimara-8(14),15-diene have previously been identified among other terpene hydrocarbons in crude extracts of the fungus Phomopsis amygdali.25 Also, many pimarane-type diterpene hydrocarbons are found in the fungus Phoma betae, where 8β-pimara-9(11),15-diene has been proposed to be an intermediate in a biosynthetic pathway of aphidicolin, a specific inhibitor of DNA polymerase α.26 The identification of different diterpene hydrocarbons is many times based on the analysis of terpene synthase products of in vitro enzyme assays;27–29 and, the few reports on the identification of fungal diterpenes do not report bioactivity assay data for these compounds.
Several techniques are used for isolation of oxygenated terpenoids from crude plant and fungal extracts, such as silica gel chromatography, reverse phase chromatography, vacuum liquid chromatography, flash chromatography, preparative thin-layer chromatography, capillary electrophoresis, and recrystallization with methanol.11,30–32 These methods are most suitable for terpenoids with functional groups, which add hydrophilic properties to the compound and provide UV-absorbent groups. ent-Pimara-8(14),15-diene is a very lipophilic compound, and it does not have any functional groups or bonds that have UV absorbance. The compound is not detectable directly by UV fluorescence. In this report we describe the methods for isolation and purification of ent-pimara-8(14),15-diene from A. nidulans by using ASE combined with reverse phase HPLC. Compared to ultrasonic extraction, ASE was more efficient and was further optimized for A. nidulans mycelia. GC-MS was used to analyze the efficiency of the extraction methods and the HPLC separation. We also describe a TLC separation and rhodamine staining detection method, as well as structural confirmation for A. nidulans ent-pimara-8(14),15-diene by NMR.
The purification methods enabled us to study the activity of pure ent-pimara-8(14),15-diene in a number of bioactivity assays. In DPPH radical scavenging assays, purified ent-pimara-8(14),15-diene had higher activity than the known tetraterpene antioxidant, beta-carotene, demonstrating for the first time the antioxidant activity of ent-pimara-8(14),15-diene. With our production strain and purification method larger scale production of ent-pimara-8(14),15-diene is possible.
Experimental
Aspergillus nidulans strains and culture media
A. nidulans FGSC A4 (Glasgow wild type, veA+)33 was obtained from the Fungal Genetics Stock Center (Kansas City, Missouri, USA) and used as a wild type control strain. ent-Pimara-8(14),15-diene producing strain, oe:PbcR, has been described earlier.20 Briefly, the genomic sequence of the Zn(II)2Cys6-type transcription factor PbcR (AN1599.4, GenBank:BN001307) was cloned into a fungal expression vector pKB1, where the expression of pbcR is under the A. nidulans gpdA promoter. 20 mg of linearized expression plasmid DNA was transformed into A. nidulans FGSC A4 by protoplasting, and the positive colonies were selected on minimal medium agar plates with 200 mg mL−1 of glufosinate ammonium. Strains were grown in liquid YES-media (2% yeast extract, 4% sucrose) supplemented with 3% gelatin at 30 °C with 250 rpm.Extraction
Extraction conditions of the sonication method have been described earlier.20 Filtered and freeze-dried A. nidulans mycelia were homogenized with a Retsch MM301 Ball Mill at 29 Hz for 30–60 s. The homogenized powder was mixed with diatomaceous earth in ASE sample cells (Dionex) and extracted with ASE 200 (Dionex) using hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (Hex:EtOAc) (1:1), EtOAc, or acetonitrile (ACN). Extraction conditions were as follows: pressure, 12.07 MPa or 15.51 MPa; temperature, 70 °C, 80 °C, or 90 °C; preheat time, 1 min; heat time, 5 min; static time, 10 min; flush volume, 30%; and purge time, 90 seconds. When assessing the efficiency of the sonication and ASE methods, one batch of mycelia was extracted three times with EtOAc.
:ethyl acetate (Hex:EtOAc) (1:1), EtOAc, or acetonitrile (ACN). Extraction conditions were as follows: pressure, 12.07 MPa or 15.51 MPa; temperature, 70 °C, 80 °C, or 90 °C; preheat time, 1 min; heat time, 5 min; static time, 10 min; flush volume, 30%; and purge time, 90 seconds. When assessing the efficiency of the sonication and ASE methods, one batch of mycelia was extracted three times with EtOAc.
Thin layer chromatography
An aliquot (200 μL) of the Hex:EtOAc (1:1) crude extract of FGSC A4 (wild type) and oe:PbcR (pimaradiene producing strain) was applied to silica plates (Silica Gel 60, Pre-coated TLC Plates, Merck, Germany) and purified by thin-layer chromatography. The separation was done with EtOAc:heptane:acetic acid (9:90:1), and the different compound groups were visualized under UV light after spraying the silica plates with 0.01% rhodamine 6B GO (Merck, Germany). The areas with different compounds were marked with a pencil, and silica was scraped and extracted with Hex:EtOAc (1:1). The purity of obtained fractions was analyzed by GC-MS.
Gas chromatography-mass spectrometry
GC-MS analysis has been previously described by Bromann et al. (2012).20 The separations were performed using an Agilent 6890 Series GC combined with a 5973 Network MS detector (Agilent, USA) and a Combipal injector (Varian Inc., USA). Samples were dissolved in EtOAc, injected in split mode (10:1) and separated on a 25 m × 0.2 mm × 0.33 μm HP-1 capillary column (Agilent, USA) with a helium flow rate of 1.3 mL min−1. The ion source temperature was 230 °C, the interface was 280 °C, and the oven temperature was equilibrated at 100 °C for 30 seconds and then increased to 320 °C at a rate of 10 °C min−1 and held at that temperature for 25 minutes. The MS detector was operated in EI mode at 70 eV with a mass range of 40–550. Compounds were identified with the Palisade Complete 600K Mass spectral library (Palisade Mass Spectrometry, USA).
High-performance liquid chromatography
A. nidulans oe:PbcR cell extracts were evaporated to dryness and resuspended in HPLC grade ACN. Analytical HPLC was performed using an XTerra MS C18 column (250 × 4.6 mm; 3.5 μm; Waters) with a Waters 616 pump with a Waters 600 S system controller equipped with a Waters in-line degasser AF, a Waters 717plus autosampler, a Waters 2996 photodiode array detector (200–600 nm), and Waters Empower Pro chromatography software for data processing (Milford, MA). Samples were eluted with a flow rate of 0.5 mL min−1. The injected volume of the crude extract per run was 150 μL and the total analysis time was set to 60 min. Preparative scale purification was done using a Waters XTerra Prep MS C18 column (300 × 10 mm; 10 μm; Waters) with a 2 mL sample loop and a 2.5 mL syringe (Waters). The injected volume was 2 mL of the crude extract and the flow rate was set to 2.5 mL min−1. The total analysis time was set to 60 min. The elution of the different fractions was monitored at 225 nm, and the collected fractions were evaporated to dryness under N2 flow at 30 °C. Fractions were dissolved in a small volume of EtOAc and the purity was analyzed by GC-MS.DPPH radical scavenging assay
DPPH radical scavenging assay was performed according to Kähkönen et al.34 with slight modifications. Methanolic 0.1 mM DPPH radical solution was mixed with ent-pimara-8(14),15-diene or beta-carotene solution. The concentrations of pure compounds were either 1 mM, 2.5 mM or 5 mM. The absorption at 517 nm was monitored up to 120 min using a UV-1800 UV-Vis spectrophotometer (Shimadzu). The results were calculated as the percentage of radicals scavenged at four different time-points: 20, 60, 90, and 120 min.IR, NMR, HRMS experimental
The IR spectrum was obtained using a Vertex 70 (Bruker Optics Inc., MA, USA) FTIR instrument. The FTIR measurements were made directly in solids with a horizontal Attenuated Total Reflectance (ATR) accessory (MIRacle, Pike Technology, Inc, WI, USA). The transmittance spectrum was recorded at a 4 cm−1 resolution between 4000 and 600 cm−1 using the OPUS 5.5 software (Bruker Optics Inc., MA, USA). ESI-MS was performed by direct injection using a Synapt G2 HDMS (Waters, MA, USA) instrument. NMR spectra were obtained using a Varian Mercury Plus 300 spectrometer, in CDCl3 with tetramethylsilane (TMS) as the internal standard. The chemical shifts were reported in parts per million (ppm) and on the δ scale from TMS as an internal standard. The coupling constants J are quoted in hertz (Hz).Results and discussion
Previously, we reported the identification of the transcription factor, PbcR, in A. nidulans. In the strain overexpressing pbcR (oe:PbcR), transcription of seven genes within a cryptic secondary metabolite gene cluster is activated; and, this results in the production of a pimarane-type diterpene that we identified as ent-pimara-8(14),15-diene.20Comparison of extraction methods
In the current study, oe:PbcR mycelia were collected at the exponential growth phase and then freeze-dried and homogenized. To compare the previously reported sonication extraction with ASE, we performed three serial extractions of the same batch of mycelia using two methods. In GC-MS analysis of three serial sonication extractions of manually ground mycelia with Hex:EtOAc (1:1), 48% of ent-pimara-8(14),15-diene was recovered in the first extraction, 19% in the second extraction, and 33% in the third extraction (Fig. 1A). In contrast, ASE efficiently recovered 100% of ent-pimara-8(14),15-diene using one extraction (Fig. 1B). Thus, automated homogenization of freeze-dried mycelia and subsequent ASE proved to be superior for the recovery of ent-pimara-8(14),15-diene. An additional benefit in using ASE is that weighing of the freeze-dried sample also makes it possible to quantitate the extraction yield and method efficiency.
 | ||
| Fig. 1 Comparison of relative yields of ent-pimara-8(14),15-diene from two different extraction techniques. The sonication extraction (A) recovers only 48.5% of ent-pimara-8(14),15-diene in the first extraction, whereas ASE (B) efficiently recovers 100% of the ent-pimara-8(14),15-diene from A. nidulans mycelia in the first extraction. | ||
The ASE method was optimized for the powdered sample by using different solvent compositions, temperatures, and pressures. The recovery of ent-pimara-8(14),15-diene was monitored by analyzing GC-MS peak areas. We tested three different solvents: Hex:EtOAc (1:1), 100% EtOAc, and ACN. The highest relative recovery was marked as 100% extraction efficiency and other peak areas were calculated as a percentage of this value. The recovery of ent-pimara-8(14),15-diene from the fungal mycelia was highest by using 100% EtOAc (Fig. 2A). Hex:EtOAc was nearly as efficient (92%) in extracting ent-pimara-8(14),15-diene as EtOAc, whereas ACN could extract only 52% of the ent-pimara-8(14),15-diene compared to EtOAc (Fig. 2A).
 | ||
| Fig. 2 Influence of different solvents (A) and temperatures (B) on recovery of ent-pimara-8(14),15-diene from oe:PbcR mycelia. The error bars represent the standard error of the mean (SEM, n = 3). | ||
The extraction method was further optimized with EtOAc by testing at two different pressures: 12.07 MPa and 15.51 MPa. Recovery of ent-pimara-8(14),15-diene was efficient at 12.07 MPa, and increasing pressure slightly lowered the yield (data not shown), suggesting that pressure did not exert a great influence on extraction efficiency. Extraction with EtOAc at three different temperatures (70 °C, 80 °C, and 90 °C), with 12.07 MPa pressure, demonstrated that the highest recovery was achieved at 90 °C (Fig. 2B). In a previous study, Kawamura et al.35 reported that increasing temperature resulted in higher recovery of paclitaxel from bark, although pressure was less important for recovery. In their study, the temperature range used for paclitaxel was 100–150 °C, so it is possible that we could have achieved higher recovery of ent-pimara-8(14),15-diene using temperatures over 100 °C. However, to prevent the possible degradation of the compound, we did not test the extraction at temperatures above 90 °C. Taken together, our data suggest optimal extraction conditions for ent-pimara-8(14),15-diene from mycelia of oe:PbcR.
Purification of ent-pimara-8(14),15-diene with TLC and HPLC
In order to isolate ent-pimara-8(14),15-diene from crude extracts of oe:PbcR, a TLC method was developed. Mycelia from wild-type A. nidulans (FGSC A4) and oe:PbcR were extracted, and the extracts were separated on silica plates. ent-Pimara-8(14),15-diene was visualized by rhodamine staining of the developed silica plates (Fig. 3A), since the compound was not detectable directly by UV fluorescence. ent-Pimara-8(14),15-diene was separated by using EtOAc:heptane:acetic acid (9:90:1). Using this eluent composition, the compound moved along the solvent front (Fig. 3A, lane 1). ent-Pimara-8(14),15-diene was then extracted from the silica, and the purity of the compound was analyzed by GC-MS (Fig. 3B and C). Many fatty acids and other unknown interfering compounds that were present in the crude extract (Fig. 3B) were not detected in the TLC purified fractions (Fig. 3C). However, the TLC-purified compound still contained some non-polar alkanes that interfered with identification by NMR analysis (Fig. 3C).
 | ||
| Fig. 3 (A) TLC of crude Hex:EtOAc extracts of A. nidulans FGSC A4 (wild-type) and oe:PbcR. Separation was done using EtOAc:heptane:acetic acid (9:90:1), and the developed silica plate was visualized with rhodamine staining. Compounds separated with TLC in oe:PbcR were (1) ent-pimara-8(14),15-diene fraction, (2) organic acids, fatty acids, (3) organic acids, sugars, (4) aromatic carboxylic acids, fatty acids and alkanes, (5) aromatic carboxylic acids and fatty acids. (B) GC-MS analysis of the oe:PbcR crude extract and (C) TLC purified fraction showed that some higher alkanes were still present in the purified fraction. Fatty acids and major impurities can be separated from the crude extracts of oe:PbcR with TLC. | ||
To further purify ent-pimara-8(14),15-diene, we developed an analytical scale HPLC method. In HPLC, both the interactions between the purified compound and the stationary phase as well as the solvent play a role in the separation process. Specifically, ent-pimara-8(14),15-diene was purified from crude extracts of oe:PbcR by reversed phase HPLC. The purification method was tested by using three isocratic eluent compositions of 85%, 90%, and 100% ACN; and efficient separation was achieved with 100% ACN in one hour. Low wavelengths (190 nm, 210 nm, 225 nm, and 250 nm) were used to monitor fractionation, because this diterpene hydrocarbon does not have any functional groups or bonds that have UV absorbance; and, the peak corresponding to ent-pimara-8(14),15-diene was not detectable at wavelengths 250 nm and greater. To identify the compound peak, eluent fractions were collected in six 10 minute intervals. ent-Pimara-8(14),15-diene was detected in the third interval (20–30 minutes), and the individual peaks were then collected and analyzed by GC-MS. The UV absorption maximum for the peak corresponding to ent-pimara-8(14),15-diene was 222.1 nm (Fig. 4A), and monitoring of the following purifications was done at 225 nm.
 | ||
| Fig. 4 (A) UV absorption maximum for ent-pimara-8(14),15-diene. (B) Chromatograms for analytical (upper panel) and preparative (lower panel) HPLC purification for ent-pimara-8(14),15-diene. The peak corresponding to ent-pimara-8(14),15-diene eluted at 25 to 28 minutes in analytical scale HPLC and at 30 to 35 minutes in the preparative scale HPLC. Shoulder seen at the preparative HPLC peak was not collected. (C) Gas chromatograms for the crude extract of A. nidulans oe:PbcR (upper panel) and the HPLC purified ent-pimara-8(14),15-diene fraction (lower panel). The yield of HPLC purified ent-pimara-8(14),15-diene from the crude extracts was over 90% (lower panel, small figure, error bar represents the SEM, n = 9). | ||
In the analytical scale HPLC, ent-pimara-8(14),15-diene was eluted at 25 to 28 minutes after the sample injection (Fig. 4B, upper panel). Preparative scale HPLC was performed in order to accommodate larger injection volumes. The chromatograms of preparative scale and analytical scale HPLC were similar (Fig. 4B), and the total analysis time was kept at 60 minutes. In the preparative scale HPLC, the ent-pimara-8(14),15-diene peak was detected at 30 to 35 minutes after injection (Fig. 4B, lower panel). The shoulder seen at the preparative scale HPLC peak was not collected. Isolated ent-pimara-8(14),15-diene fractions were combined, evaporated to dryness, and dissolved in EtOAc prior to GC-MS analysis. Comparison of the GC-MS peak areas for the crude extract and for the HPLC purified ent-pimara-8(14),15-diene (Fig. 4C) indicated that there were no impurities in the collected fraction; and, the recovery of the diterpene was consistently over 90% (Fig. 4C, small panel). After multiple rounds of preparative scale HPLC, 25 mg of pure compound was collected.
Bioactivity testing of ent-pimara-8(14),15-diene
With its isolation and purification, we could now test ent-pimara-8(14),15-diene in a number of bioactivity assays. In Aspergilli, the production of secondary metabolites has been linked to oxidative stress.36,37 For example, lovastatin biosynthesis in A. terreus, and aflatoxin biosyntheses in A. parasiticus and A. flavus respond to accumulation of reactive oxygen species (ROS) in these fungi.38,39 Because oxidative stress has also been shown to activate secondary metabolite biosynthesis in A. nidulans, we tested the anti-oxidant activity of pure ent-pimara-8(14),15-diene hydrocarbon in a DPPH radical scavenging assay with three different concentrations and four time-points. We compared its activity to a known tetraterpene antioxidant, beta-carotene. Pure ent-pimara-8(14),15-diene had significantly higher DPPH radical scavenging activity than beta-carotene in all test conditions (Fig. 5). | ||
| Fig. 5 DPPH radical scavenging activities of ent-pimara-8(14),15-diene (dark gray) and beta-carotene (light gray). The concentrations of the pure compounds were 1 mM, 2.5 mM and 5 mM in the DPPH assay with time points 20, 60, 90 and 120 minutes. ent-Pimara-8(14),15-diene had higher antioxidant activity than beta-carotene. The error bars represent the SEM (n = 3). | ||
Although the function of this compound in A. nidulans is not known, the data suggest that one potential role of ent-pimara-8(14),15-diene may be to function as an antioxidant. The antioxidant activity of ent-pimara-8(14),15-diene could be one way for A. nidulans to protect itself against stressful situations where accumulation of free radicals inside the cells occurs. We also tested purified ent-pimara-8(14),15-diene for antimicrobial activity against Staphylococcus aureus, and for inhibition of axenic amastigotes of Leishmania donovani. However, even relatively high concentrations of the compound (50 μM) were ineffective in both of these tests (data not shown). Additional bioactivity and antioxidant assays will give a more complete picture of the potential bioactivity of the compound, and this will be addressed in future studies.
Structural analysis
To verify the structure of HPLC-purified ent-pimara-8(14),15-diene, infra-red (IR), MS, and 1D and 2D NMR experiments were performed (Table 1). Inspection of the 1H NMR spectrum showed the four methyl group signals as singlets at 0.80, 0.85, 0.88, and 1.04 ppm. The downfield double doublet at 5.78 ppm was assigned to 15-H whereas the double doublets at 4.88 and 4.91 ppm were assigned to 16-Hb and 16-Ha, respectively. Thus, the signal at 4.88 ppm displayed the cis vicinal coupling constant of 10.6 Hz between 16-Hb and 15-H and the geminal coupling constant of 1.5 Hz. The trans vicinal coupling constant for 16-Ha and 15-H was found to be 17.5 Hz.| Position | δ C, type | δ H (J in Hz) |
|---|---|---|
| 4 | 33.2, C | |
| 5 | 54.8, CH | 1.01, m |
| 7α | 36, CH2 | 2.26, m |
| 7β | 2.05, m | |
| 8 | 137.2, C | |
| 9 | 50.6, CH | 1.70, m |
| 10 | 38.2, C | |
| 13 | 37.3, C | |
| 14 | 128.4, CH | 5.21, s |
| 15 | 149.1, CH | 5.78, dd (10.6, 17.5) |
| 16a | 109.8, CH2 | 4.91, dd (1.5, 17.5) |
| 16b | 4.88, dd (1.5, 10.6) | |
| 17 | 25.9, CH3 | 1.04, s |
| 20 | 14.9, CH3 | 0.80, s |
The COSY spectrum displayed correlations between the neighboring 15-H, 16-Ha, 16-Hb proton signals. The assignment of C15 (149.1), C8 (137.2), C14 (128.4), and C16 (109.8) was made by observation of their respective correlations on the HSQC experiment. The signal at 1.70 ppm displayed correlation to the signal at 50.6 ppm, belonging to a CH carbon, and was assigned to 9-H.40 The remaining CH/CH3 signals to assign correlated to the methyl group protons and to 5-H (C5 at 54.8 ppm). Assignment of the 17-methyl group to the signal at 1.04 ppm was based on the HMBC correlations with the signals of C15, C14, and the quaternary carbon C13 at 37 ppm. The remaining methyl group signals all displayed HMBC correlations to C5. The signal at 0.80 ppm was assigned to the 20-methyl protons based on the HMBC correlations to C9 and quaternary C10 (38.2 ppm). The signals at 0.85 and 0.88 ppm, corresponding to the 19 and 18-methyl protons, both displayed HMBC correlations to quaternary C4 (33.2 ppm). The stereochemistry of ent-pimara-8(14),15-diene was verified by the NOESY experiment. Selected NOESY correlations and a 3-D presentation of ent-pimara-8(14),15-diene are depicted in Fig. 6A and B, respectively. The axial position of the 13-methyl group and β-orientation for 5-H and 9-H was confirmed by the observed NOESY cross peaks of 14-H/17-CH3, 14-H/7α-H, and 16a-H/17-CH3, and is consistent with those found in the literature.41 Cross-peaks were also observed for the signals of 9-H and 5-H. The assignment of C17 to the signal at 25.9 ppm is also consistent with the axial position of this methyl group, as opposed to the reported values of about 30 ppm for equatorial orientation.42 These data confirm the structure and stereochemistry of ent-pimara-8(14),15-diene isolated from A. nidulans strain oe:PbcR, and are consistent with our earlier report.20 The structure and the mass spectrum of pure ent-pimara-8(14),15-diene are shown in Fig. 6C. NMR spectra are presented in the ESI (Fig. 1S–6S).†
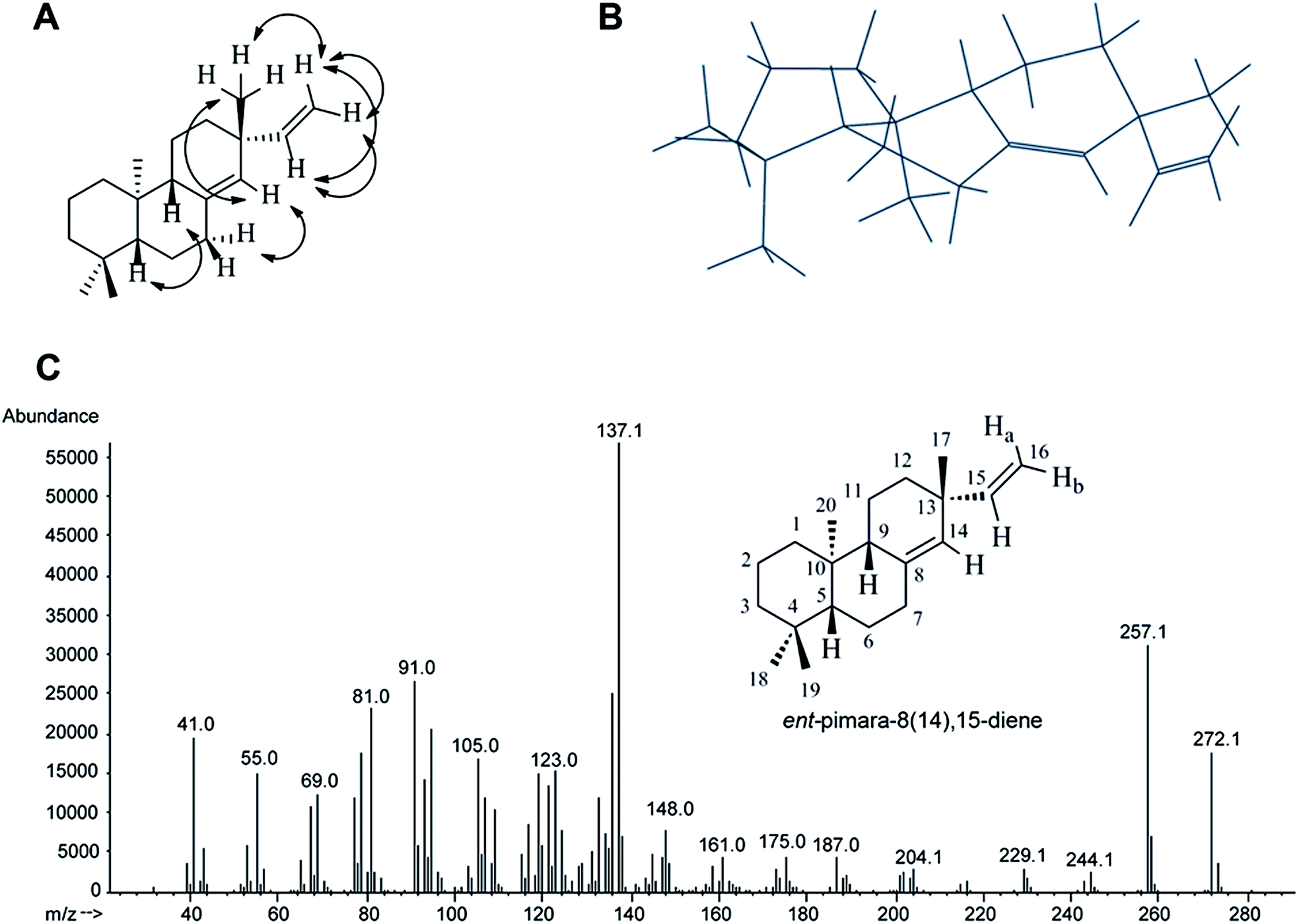 | ||
| Fig. 6 (A) Selected NOESY correlations and (B) a 3D representation of ent-pimara-8(14),15-diene. (C) Structure and the mass spectrum of pure ent-pimara-8(14),15-diene. | ||
Conclusions
We developed isolation and purification methods for a lipophilic tricyclic diterpene, ent-pimara-8(14),15-diene, produced in fungi. The optimized ASE and HPLC methods developed here were very reproducible and could be scaled up to accommodate larger sample volumes. Biotechnological production of this particular diterpene in A. nidulans and the purification methods described here should enable production of large quantities of pure ent-pimara-8(14),15-diene. This is necessary for more systematic bioactivity screening of this compound. Finally, the methods described are relevant for purification of other diterpenes with similar properties.Conflict of interest
The authors declare no conflict of interest.Acknowledgements
We thank Andreas Helfenstein and Päivi Tammela for anti-microbial testing, Charles Jaffe for anti-leishmanial testing, and Paul A. Bromann for his critical reading of the manuscript. This work was supported by Fibic Oy, Future Biorefinery Joint Research 2 program.References
- D. Hoffmeister and N. P. Keller, Nat. Prod. Rep., 2007, 24, 393–416 RSC
.
- M. R. Andersen, J. B. Nielsen, A. Klitgaard, L. M. Petersen, M. Zachariasen, T. J. Hansen, L. H. Blicher, C. H. Gotfredsen, T. O. Larsen, K. F. Nielsen and U. H. Mortensen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E99–E107 CrossRef CAS PubMed
- A. A. Brakhage and V. Schroeckh, Fungal Genet. Biol., 2011, 48, 15–22 CrossRef CAS PubMed
- M. Reverberi, M. Punelli, V. Scala, M. Scarpari, P. Uva, W. I. Mentzen, A. L. Dolezal, C. Woloshuk, F. Pinzari, A. A. Fabbri, C. Fanelli and G. A. Payne, PLoS One, 2013, 8, e68735 CAS
- M. R. Andersen and J. Nielsen, Fungal Genet. Biol., 2009, 46(suppl. 1), S180–S190 CrossRef CAS PubMed
- A. Zinedine, Toxins, 2010, 2, 1121–1133 CrossRef CAS PubMed
- A. Rhouati, C. Yang, A. Hayat and J. L. Marty, Toxins, 2013, 5, 1988–2008 CrossRef PubMed
- A. A. Brakhage, Nat. Rev. Microbiol., 2013, 11, 21–32 CrossRef CAS PubMed
- M. Manzoni and M. Rollini, Appl. Microbiol. Biotechnol., 2002, 58, 555–564 CrossRef CAS PubMed
- E. M. Fox and B. J. Howlett, Curr. Opin. Microbiol., 2008, 11, 481–487 CrossRef CAS PubMed
- T. S. Porto, N. A. Furtado, V. C. Heleno, C. H. Martins, F. B. Da Costa, M. E. Severiano, A. N. Silva, R. C. Veneziani and S. R. Ambrosio, Fitoterapia, 2009, 80, 432–436 CrossRef CAS PubMed
- S. R. Ambrosio, N. S. Arakawa, V. R. Esperandim, S. de Albuquerque and F. B. Da Costa, Phytother. Res., 2008, 22, 1413–1415 CrossRef CAS PubMed
- K. O. Rayanil, S. Limpanawisut and P. Tuntiwachwuttikul, Phytochemistry, 2013, 89, 125–130 CrossRef CAS PubMed
- C. R. Tirapelli, S. R. Ambrosio, F. B. Da Costa and A. M. De Oliveira, Fitoterapia, 2002, 73, 56–62 CrossRef CAS
- C. R. Tirapelli, S. R. Ambrosio, S. T. Coutinho, D. C. de Oliveira, F. B. da Costa and A. M. de Oliveira, J. Pharm. Pharmacol., 2005, 57, 997–1004 CrossRef CAS PubMed
- H. Lim, H. A. Jung, J. S. Choi, Y. S. Kim, S. S. Kang and H. P. Kim, Arch. Pharmacal Res., 2009, 32, 1237–1243 CrossRef CAS PubMed
- F. Ali, P. L. Sangwan, S. Koul, A. Pandey, S. Bani, S. T. Abdullah, P. R. Sharma, S. Kitchlu and I. A. Khan, Eur. J. Clin. Microbiol. Infect. Dis., 2012, 31, 149–159 CrossRef CAS PubMed
- J. Rubio, J. S. Calderon, A. Flores, C. Castroa and C. L. Cespedes, Z. Naturforsch., C: J. Biosci., 2005, 60, 711–716 CAS
- R. Batista, J. L. Humberto, E. Chiari and A. B. de Oliveira, Bioorg. Med. Chem., 2007, 15, 381–391 CrossRef CAS PubMed
- K. Bromann, M. Toivari, K. Viljanen, A. Vuoristo, L. Ruohonen and T. Nakari-Setala, PLoS One, 2012, 7, e35450 CAS
- M. M. Xu, P. R. Wilderman, D. Morrone, J. J. Xu, A. Roy, M. Margis-Pinheiro, N. M. Upadhyaya, R. M. Coates and R. J. Peters, Phytochemistry, 2007, 68, 312–326 CrossRef CAS PubMed
- T. Toyomasu, Biosci., Biotechnol., Biochem., 2008, 72, 1168–1175 CrossRef CAS
- Y. Kanno, K. Otomo, H. Kenmoku, W. Mitsuhashi, H. Yamane, H. Oikawa, H. Toshima, M. Matsuoka, T. Sassa and T. Toyomasu, Biosci., Biotechnol., Biochem., 2006, 70, 1702–1710 CrossRef CAS
- T. Sassa, H. Kenmoku, K. Nakayama and N. Kato, Biosci., Biotechnol., Biochem., 2004, 68, 1608–1610 CrossRef CAS
- H. Kenmoku, M. Tanaka, K. Ogiyama, N. Kato and T. Sassa, Biosci., Biotechnol., Biochem., 2004, 68, 1574–1577 CrossRef CAS
- H. Oikawa, H. Toshima, S. Ohashi, W. A. Konig, H. Kenmoku and T. Sassa, Tetrahedron Lett., 2001, 42, 2329–2332 CrossRef CAS
- K. Hayashi, H. Kawaide, M. Notomi, Y. Sakigi, A. Matsuo and H. Nozaki, FEBS Lett., 2006, 580, 6175–6181 CrossRef CAS PubMed
- C. I. Keeling, L. L. Madilao, P. Zerbe, H. K. Dullat and J. Bohlmann, J. Biol. Chem., 2011, 286, 21145–21153 CrossRef CAS PubMed
- H. Kawaide, R. Imai, T. Sassa and Y. Kamiya, J. Biol. Chem., 1997, 272, 21706–21712 CrossRef CAS PubMed
- M. Isaka, S. Palasarn, W. Prathumpai and P. Laksanacharoen, Chem. Pharm. Bull., 2011, 59, 1157–1159 CrossRef CAS
- H. S. Kang, Y. H. Kim, C. S. Lee, J. J. Lee, I. Choi and K. H. Pyun, Cell. Immunol., 1996, 170, 212–221 CrossRef CAS
- N. T. Phuong, K. A. Lee, S. J. Jeong, C. X. Fu, J. K. Choi, Y. H. Kim and J. S. Kang, J. Pharm. Biomed. Anal., 2006, 40, 56–61 CrossRef CAS PubMed
- G. Pontecorvo, J. A. Roper, L. M. Hemmons, K. D. Macdonald and A. W. Bufton, Adv. Genet., 1953, 5, 141–238 CAS
- M. P. Kahkonen and M. Heinonen, J. Agric. Food Chem., 2003, 51, 628–633 CrossRef PubMed
- F. Kawamura, Y. Kikuchi, T. Ohira and M. Yatagai, J. Nat. Prod., 1999, 62, 244–247 CrossRef CAS PubMed
- S. R. Hong, L. V. Roze and J. E. Linz, Toxins, 2013, 5, 683–702 CrossRef CAS PubMed
- S. R. Hong, L. V. Roze, J. Wee and J. E. Linz, MicrobiologyOpen, 2013, 2, 144–160 CrossRef CAS PubMed
- R. U. Miranda, L. E. Gomez-Quiroz, A. Mejia and J. Barrios-Gonzalez, Fungal Biol., 2013, 117, 85–93 CrossRef CAS PubMed
- L. V. Roze, S. Y. Hong and J. E. Linz, Annu. Rev. Food Sci. Technol., 2013, 4, 293–311 CrossRef CAS PubMed
- L. Han, X. S. Huang, I. Sattler, H. M. Dahse, H. Z. Fu, S. Grabley and W. H. Lin, Pharmazie, 2005, 60, 705–707 CAS
- T. L. Meragelman, G. L. Silva, E. Mongelli and R. R. Gil, Phytochemistry, 2003, 62, 569–572 CrossRef CAS
- A. M. L. Seca, A. M. S. Silva, I. L. Bazzocchi and I. A. Jimenez, Phytochemistry, 2008, 69, 498–505 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra (Fig. 1S–6S). See DOI: 10.1039/c3ay41640b |
| This journal is © The Royal Society of Chemistry 2014 |