
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetallophore mapping in complex matrices by metal isotope coded profiling of organic ligands†
Michael
Deicke
a,
Jan Frieder
Mohr
a,
Jean-Philippe
Bellenger
b and
Thomas
Wichard
*a
aFriedrich Schiller University Jena, Institute for Inorganic and Analytical Chemistry, Lessingstr. 8, 07743 Jena, Germany. E-mail: Thomas.Wichard@uni-jena.de; Fax: +49 3641 948172; Tel: +49 3641 948184
bDépartement de Chimie, Université de Sherbrooke, Sherbrooke, QC J1K2R1, Canada
First published on 26th September 2014
Abstract
Metal isotope coded profiling (MICP) introduces a universal discovery platform for metal chelating natural products that act as metallophores, ion buffers or sequestering agents. The detection of cation and oxoanion complexing ligands is facilitated by the identification of unique isotopic signatures created by the application of isotopically pure metals.
Metal chelators are essential for fundamentally important chemical mediated processes in plants, microorganisms and animals. In particular, about 500 siderophores (i.e. iron carriers) were discovered during the last decades demonstrating their unique importance in bacterial iron acquisition and metal cycling.1–3 Apart from iron, microorganisms need to acquire further trace metals for the specific needs of e.g. enzymatic cofactors. Owing to the potential multiple functions of these chelators the more general term “metallophores” for ligands complexing any metal ion e.g. Fe, Cu, Zn, Mo or V was proposed.4 Microorganisms release these metallophores (e.g., 1 and 2 in Fig. 1 and 3–9 in Fig. S1†) especially under trace metal deficient conditions or for detoxification at adverse concentrations of metal ions in their vicinity.4,5
 | ||
| Fig. 1 Catecholate metallophores protochelin (1) and anachelin (2) produced by Azotobacter vinelandii and by Anabaena cylindrica. | ||
Several identification techniques were developed to determine potential metallophores including fast unspecific colorimetric surveys based on reactions with e.g. chrome azurol S (CAS),6 state-of-the-art high resolution mass spectrometry (HRMS)7,8 and labelling with radioisotopes. In this study, we propose a novel concept for the mass spectrometric determination of (unknown) metal binding compounds independent from their structure, from the type of employed mass spectrometer, and from the producing organisms. The proposed approach, metal isotope coded profiling (MICP), utilizes stable pairs of metal isotopes creating unique isotopic signatures that can be read out using automated evaluation routines. Two different strategies were followed to achieve metallophore mapping in complex biological matrices, and these are exemplified with the isotope pairs 54Fe/58Fe and 95Mo/98Mo:
-Target analysis where pairs of isotopes 54Fe/58Fe or 95Mo/98Mo are administered directly to e.g. growth media or culture extracts for labelling metal complexing ligands. Metallophores are directly identified by their unique isotopic pattern (Fig. 2A and S2A and C†).
 | ||
| Fig. 2 Analytical process of the metal isotope coded profiling (MICP) of organic ligands for metallophore mapping. Growth media are spiked with isotopically pure Mo and Fe and subsequently analyzed by (A) target analysis or (B) multivariate comparative analysis. | ||
-Multivariate comparative analysis where growth media are divided into two equal fractions and spiked (i) with 95Mo/54Fe and (ii) with 98Mo/58Fe. Here, metal complexing ligands of the isotopically pure metal define the differences of the respective two groups and can be subsequently identified by chemometric discriminant analyses (Fig. 2B and S2B and D†).
In both cases, complexes with unique isotopic patterns are generated corresponding to the adjusted isotopic ratio in the matrix. The target analysis is constrained to these specific isotopic signatures characterized by defined mass difference and intensity. These unique signatures provide then a highly sensitive and distinctive tool for mapping chromatographically unknown chelators (see the ESI for details†).
In aquatic and soil environments, metal speciation and metal bioavailability are dependent on a variety of factors such as organic matter content, mineralogical composition, and pH value. Thus, the coordination chemistry and binding constants cannot always be predicted. To minimize de- or trans-complexation during the reverse-phase chromatographic process, ultrahigh performance liquid chromatography (UHPLC) was conducted under conditions, which mimicked the physiological pH of the environmental habitat of the studied organisms. Therefore, naturally abundant metal complexes are supposed to endure the chromatographic process.9 To establish the methodology, we selected the nitrogen fixing soil bacterium Azotobacter vinelandii, which became a model organism for investigations in metal acquisition of molybdenum and iron.4 Here, both Mo and Fe are recruited by the same catecholate metallophore 1 (Fig. 1). Mo and Fe are cofactors for the nitrogenase, which reduces atmospheric nitrogen into bioavailable ammonium.10 They have to be acquired by the bacterium in order to sustain nitrogen fixation.11 Hereby, all Fe- and Mo-sources were replaced with the isotopes 54Fe–EDTA/58Fe–EDTA and 95MoO42−/98MoO42− adjusted exactly to the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of each isotope pair (pre-labelling, Fig. 2). Upon harvesting and filtration, the potential metallophores can be directly determined by UHPLC-ESI-ToF-MS analyses (Fig. 2). Optionally, solid phase extracts can be spiked by the isotopically pure metals upon adjusting the physiological pH (post-labelling, Fig. S2†). We have applied the built-in algorithm of e.g. the data analysis software MarkerLynxV4.1 (Waters, UK) that collects automatically mass retention time pairs, if the given mass difference of Δ4 for Fe (54Fe/58Fe) and Δ3 for Mo (95Mo/98Mo) is detected with the adjusted ratio of 1:1 (Table S1†). Indeed, the extracted chromatogram demonstrates that isotope labelling is a valuable tool for reliable determination of metallophores such as Fe–protochelin [FeIII(protochelin) + 2H]− and Mo–protochelin [MoO2(H2–protochelin) + H]− in bacterial cultures (Fig. 3). Besides the key settings Δ4 (Fe) and Δ3 (Mo) for mass differences, the parameters used by the algorithm proved as robust throughout all measurements. Depending on the culture conditions and growth phase, Fe–azotochelin and Mo–azotochelin could be detected along with several non-identified putative derivatives of 1 and azotochelin (3) in the late exponential phase (Fig. S3†).9 The fact that besides Fe- also Mo-signatures are picked up indicated that even relatively weak cis-dioxido-MoVI complexes with 2,3-dihydroxy-benzamide-containing ligands (logK = 7.6)12 can be analyzed by this approach.
:1 of each isotope pair (pre-labelling, Fig. 2). Upon harvesting and filtration, the potential metallophores can be directly determined by UHPLC-ESI-ToF-MS analyses (Fig. 2). Optionally, solid phase extracts can be spiked by the isotopically pure metals upon adjusting the physiological pH (post-labelling, Fig. S2†). We have applied the built-in algorithm of e.g. the data analysis software MarkerLynxV4.1 (Waters, UK) that collects automatically mass retention time pairs, if the given mass difference of Δ4 for Fe (54Fe/58Fe) and Δ3 for Mo (95Mo/98Mo) is detected with the adjusted ratio of 1:1 (Table S1†). Indeed, the extracted chromatogram demonstrates that isotope labelling is a valuable tool for reliable determination of metallophores such as Fe–protochelin [FeIII(protochelin) + 2H]− and Mo–protochelin [MoO2(H2–protochelin) + H]− in bacterial cultures (Fig. 3). Besides the key settings Δ4 (Fe) and Δ3 (Mo) for mass differences, the parameters used by the algorithm proved as robust throughout all measurements. Depending on the culture conditions and growth phase, Fe–azotochelin and Mo–azotochelin could be detected along with several non-identified putative derivatives of 1 and azotochelin (3) in the late exponential phase (Fig. S3†).9 The fact that besides Fe- also Mo-signatures are picked up indicated that even relatively weak cis-dioxido-MoVI complexes with 2,3-dihydroxy-benzamide-containing ligands (logK = 7.6)12 can be analyzed by this approach.
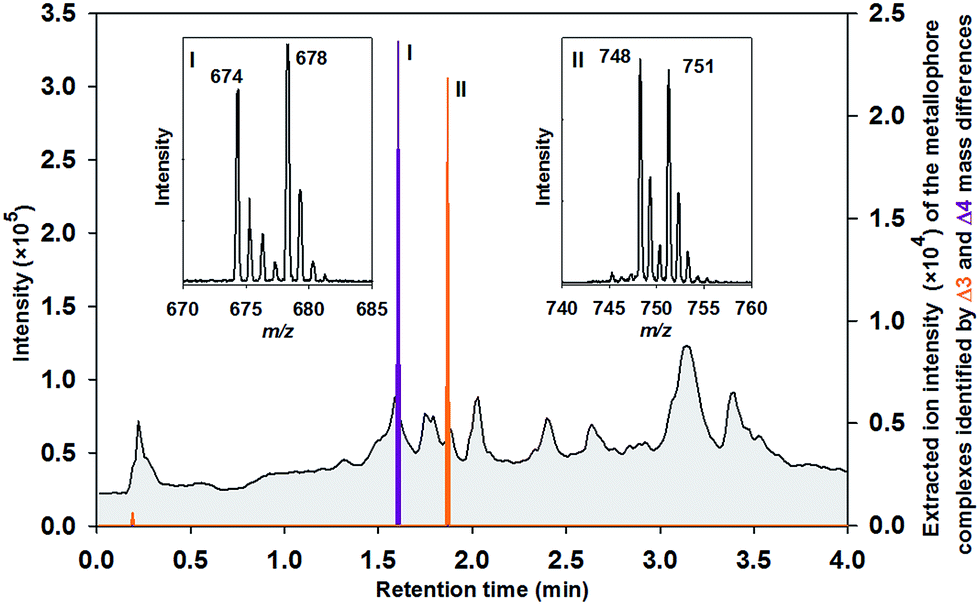 | ||
| Fig. 3 Total ion current chromatogram of a medium collected at an early exponentially growth stage of an A. vinelandii culture which was spiked with 54Fe/58Fe and 95Mo/98Mo. The extracted chromatograms indicate metallophore complexes based on the simultaneous identification of Δ4 (siderophores: purple) and Δ3 (molybdophores: orange) mass differences in the isotopic signature with an intensity ratio of 1:1. Inserts present the corresponding negative ion mass spectra of (I) 54Fe/58Fe–protochelin and (II) 95Mo/98Mo–protochelin. | ||
For validation of the experimental routine, MICP was also applied to cultures of the nitrogen fixing cyanobacterium Anabaena cylindrica13,14 (CCAP 1403/2A) producing the catecholate siderophore anachelin (2) as well as to organisms releasing other classes of metallophores (Fig. S1†). In A. cylindrica, it was not only feasible to identify the iron complex of 2, but also the hitherto unidentified Mo–anachelin complex that suggests a dual function of anachelin in recruitment of both metals (Fig. S4†). Besides catecholate metallophores, the Fe and Mo complexes of azotobactin (5) as well as Fe–pyoverdine (6) could be detected in A. vinelandii (F196) and Pseudomonas fluorescens, respectively (Fig. S5†). From the class of hydroxamate metallophores, ferricrocin (4) was exemplarily determined in the fungi15Cladosporium cladosporioides (Fig. S6†).
In an alternative approach, MICP can also be used with an unbiased multivariate comparative analysis. Here, growth media or already prepared extracts were divided into two fractions and spiked with (i) 54Fe/95Mo and (ii) with 58Fe/98Mo, respectively (Fig. S2B and D†). An automated peak extraction routine was used for data mining that delivers mass retention time pairs for every compound (Table S2†). The two groups defined by the applied isotopes were evaluated using canonical analysis of principal coordinates (CAP).16 The output of this discriminant analysis revealed highly significantly separated groups due to the identified organic ligands which were complexed with 95Mo or 54Fe (group 1) and with 98Mo or 58Fe (group 2). Vectors can be constructed in the respective direction of these groups and thus be traced back to the corresponding biomarkers (= candidates for metallophores). The score plots of the analyses illustrate clearly the two separated groups based on mass retention time pairs with high Pearson's correlation coefficient (P > 0.98) in samples harvested from A. vinelandii and other cultures (Fig. 4 and S7†). These pairs characterize the separation of the two groups most significant in A. vinelandii. Moreover, the approach can also be facilitated for the identification of doubly charged complexes of the same ligands: indeed, Fe–protochelin as well as Mo–protochelin forms doubly charged complexes with m/z 338.5 [FeIII(protochelin) + H]2− and m/z 375.0 [MoO2(H2–protochelin)]2− indicated by the corresponding masses at the same retention time (Fig. 4).9 The delta values Δ2 (doubly charged) and Δ4 (singly charged) correspond to Fe–protochelin. In analogy, mass differences of Δ1.5 and Δ3 are the doubly and singly charged Mo-complex of protochelin (Fig. 4). The mass retention time pairs without a specific delta value, although indicating a high Pearson’s correlation coefficient, cannot be assigned to metabolites chelating Fe or Mo, because the respective corresponding mass retention time pair was missing in the other group (complete data set can be found in Fig. S7A–D†).
 | ||
| Fig. 4 Score plot of the discriminant analysis illustrates the separated groups based on the organic ligands bound to either (54Fe/95Mo) or (58Fe/98Mo). Table lists the best identified candidates for metallophores. | ||
The limit of detection (LOD) of MICP was exemplarily determined for the iron and molybdenum complexes of the catecholate metallophores protochelin and azotochelin based on signal to noise measurements:9 the LODs were 9 × 10−8 mol L−1 for Mo–protochelin, 2 × 10−7 mol L−1 for Fe–protochelin, 3 × 10−7 for Mo–azotochelin, and 5 × 10−7 mol L−1 for Fe–azotochelin.
Metallophore mapping was applied to the understudied Anabaena variabilis (ATCC 29413) (Fig. 5). A set of potential novel metallophores could be detected for iron complexation identified by the Δ4 mass differences of the corresponding mass spectra, which show clearly the adjusted intensities of 54Fe and 58Fe (ratio 1:1) (Fig. 5). It highlights the strength of MICP and its targeted analysis as a valuable tool identifying potential metal chelators in complex matrices. The same mass retention pairs were found by the approach using chemometric data analyses (Fig. S7B†). Masses of the six potential 54Fe-labeled metallophores were as follows: 472 m/z (I), 656 m/z (II), 584 m/z (III), 612 m/z (IV), 654 m/z (V) and 640 m/z (VI) (ESI-MS measurement in positive-ion mode). Besides the iron complex of schizokinen (7) (472/476 m/z), which is produced by various strains of A. variabilis,17 HR-MS analyses (Tables S3 and 4†) revealed peak matching with the siderophore synechobactin B and A (8 and 9).18 Identity was further proven by co-injections with extracts from the reference strain Synechococcus sp. (PCC 7002) producing 8 and 9 as well as by MS2 experiments of the molecular ions (Fig. S8†). Mo-complexing ligands were not found either in positive or in negative-ion mode (Fig. 5, orange line).
 | ||
| Fig. 5 Mapping of iron binding metallophores in A. variabilis cultures. The extracted peaks I–VI reveal masses with an exact mass difference of Δ4 indicating the binding of iron (purple line). Positive iron mass spectra exhibit hence the unique introduced isotopic pattern upon complexation of the isotopes 54Fe and 58Fe. Peaks I, III and IV correspond to the iron complexes of 7, 8 and 9. Three unknown siderophores (II, V, VI) were determined. No molybdophore was detected (orange line). | ||
As in biological systems metal ions might be complexed to any organic ligands e.g., to polyphenols,19,20 we also tested the MICP approach with a tannic acid enriched matrix (Fig. 6). Polyphenolic compounds provide numbers of binding sides for cations and oxoanions which hence affect the bioavailability of those trace elements. For instance, binding of Mo to leaf organic matter including plant polyphenols reduces Mo leaching rates from the soil, keeping Mo in the soil environment. Such undefined organic matter works as a metal ion buffer where bacteria can then acquire these metal ions via metallophores.19 The typical pattern of a RP-HPLC separated tannic acid sample with overlapping peaks is shown in Fig. 6. Indeed, several Mo–tannin complexes [MoO2(tannin) + H]− were identified by the Δ3 mass differences between tR = 1.0–2.6 min. Electrospray ionization of Mo–tannic acid revealed strong fragmentation of the complexes, but each fragment was singly charged as the Δ3 mass difference indicated. Similar results were obtained upon 54Fe/58Fe labelling (Fig. S9†).
 | ||
| Fig. 6 Mapping of Mo-binding ability in an aquatic matrix enriched with tannic acids (A). Negative ion mass spectra of the artificial (B) and the natural Mo-isotopic pattern (C) of a selected group of tannin complexes are shown for comparison. | ||
Conclusions
In summary, a novel unbiased UHPLC-MS approach has been employed to identify metal-isotope labelled metallophores and ligands in complex biological matrices or various extracts that allow complexation of metal-cations or oxoanions. Extracts of bacterial and fungal cultures were spiked with the isotope pairs of 54Fe/58Fe and 95Mo/98Mo. To avoid contamination issues regarding the naturally abundant isotope 56Fe, we recommend utilizing the pairs of 54Fe/58Fe rather than 54Fe/56Fe. Mass spectra can be easily scanned for mass differences of Δ4 (siderophores) and of Δ3 (molybdophores) by implemented search algorithms in specialized software or by general available chemometric data analysis software programs with high sensitivity and robustness across a wide range of tested metallophores.Certainly, various other pairs of stable metal isotopes including 63Cu/65Cu, 64Zn/66Zn or 110Cd/112Cd can also be applied. Both introduced methods provide equally an exploratory approach using an automated mining of complex data sets to selectively identify metallophore complexes. The targeted analysis is a very valuable approach for convenient, selective and fast screening. In particular, when e.g. delta values cannot be pre-assigned, we recommend the universal multivariate comparative analysis for identification of chelators.
Acknowledgements
We gratefully thank the “Hans-Böckler-Stiftung” (M.D.), the “Fonds der Chemischen Industrie” (T.W.), the “Deutscher Akademischer Austauschdienst” (T.W., M.D.) and the Canadian Research Chair in Terrestrial Biogeochemistry (J.B.) for funding.Notes and references
- R. C. Hider and X. L. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- H. Boukhalfa and A. L. Crumbliss, BioMetals, 2002, 15, 325–339 CrossRef CAS.
- G. Winkelmann and H. Drechsel, in Biotechnology Set, Wiley-VCH Verlag GmbH, 2008, pp. 199–246 Search PubMed.
- A. M. L. Kraepiel, J. P. Bellenger, T. Wichard and F. M. M. Morel, BioMetals, 2009, 22, 573–581 CrossRef CAS PubMed.
- A. K. Duhme, Eur. J. Inorg. Chem., 2009, 9, 3689–3701 CrossRef.
- B. Schwyn and J. B. Neilands, Anal. Biochem., 1987, 160, 47–56 CrossRef CAS.
- S. M. Lehner, L. Atanasova, N. K. N. Neumann, R. Krska, M. Lemmens, I. S. Druzhinina and R. Schuhmacher, Appl. Environ. Microbiol., 2013, 79, 18–31 CrossRef CAS PubMed.
- I. Velasquez, B. L. Nunn, E. Ibisanmi, D. R. Goodlett, K. A. Hunter and S. G. Sander, Mar. Chem., 2011, 126, 97–107 CrossRef CAS PubMed.
- M. Deicke, J. P. Bellenger and T. Wichard, J. Chromatogr. A, 2013, 1298, 50–60 CrossRef CAS PubMed.
- J. P. Bellenger, T. Wichard, A. B. Kustka and A. M. L. Kraepiel, Nat. Geosci., 2008, 1, 243–246 CrossRef CAS.
- E. I. Stiefel, ACS Symp. Ser., 1993, 535, 1–19 CrossRef CAS PubMed.
- J. P. Bellenger, F. Arnaud-Neu, Z. Asfari, S. C. B. Myneni, E. I. Stiefel and A. M. L. Kraepiel, J. Biol. Inorg. Chem., 2007, 12, 367–376 CrossRef CAS PubMed.
- H. Beiderbeck, K. Taraz, H. Budzikiewicz and A. E. Walsby, Z. Naturforsch., C: Biosci., 2000, 55, 681–687 CAS.
- K. Gademann and C. Portmann, Curr. Org. Chem., 2008, 12, 326–341 CrossRef CAS.
- N. Pourhassan, R. Gagnon, T. Wichard and J.-P. Bellenger, Nat. Prod. Commun., 2014, 9, 539–540 CAS.
- M. Rempt and G. Pohnert, Angew. Chem., Int. Ed. Engl., 2010, 49, 4755–4758 CrossRef CAS PubMed.
- F. B. Simpson and J. B. Neilands, J. Phycol., 1976, 12, 44–48 Search PubMed.
- Y. Ito and A. Butler, Limnol. Oceanogr., 2005, 50, 1918–1923 CrossRef CAS.
- T. Wichard, B. Mishra, S. C. B. Myneni, J. P. Bellenger and A. M. L. Kraepiel, Nat. Geosci., 2009, 2, 625–629 CrossRef CAS.
- S. Hattenschwiler and P. M. Vitousek, Trends Ecol. Evol., 2000, 15, 238–243 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) is available: See DOI: 10.1039/c4an01461h |
| This journal is © The Royal Society of Chemistry 2014 |