
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface-assisted laser desorption/ionization mass spectrometry using ordered silicon nanopillar arrays†
Hashim Z.
Alhmoud‡
a,
Taryn M.
Guinan‡
a,
Roey
Elnathan
a,
Hilton
Kobus
b and
Nicolas H.
Voelcker
*ac
aMawson Institute, University of South Australia, Mawson Lakes, SA 5095, Australia. E-mail: nico.voelcker@unisa.edu.au
bSchool of Chemical and Physical Sciences, Flinders University, Bedford Park, SA 5042, Australia
cINM-Leibniz Institute for New Materials, Campus D2 2, Saarbrücken 66123, Germany
First published on 22nd September 2014
Abstract
Surface-assisted laser desorption/ionization mass spectrometry (SALDI-MS) is ideally suited for the high-throughput analysis of small molecules in bodily fluids (e.g. saliva, urine, and blood plasma). A key application for this technique is the testing of drug consumption in the context of workplace, roadside, athlete sports and anti-addictive drug compliance. Here, we show that vertically-aligned ordered silicon nanopillar (SiNP) arrays fabricated using nanosphere lithography followed by metal-assisted chemical etching (MACE) are suitable substrates for the SALDI-MS detection of methadone and small peptides. Porosity, length and diameter are fabrication parameters that we have explored here in order to optimize analytical performance. We demonstrate the quantitative analysis of methadone in MilliQ water down to 32 ng mL−1. Finally, the capability of SiNP arrays to facilitate the detection of methadone in clinical samples is also demonstrated.
Introduction
The quantitative and confirmatory analysis of small molecules is of great interest in forensics and pharmacology, mainly since most synthetic drugs of abuse and medical therapy have molecular weights of less than 500 Da. Similarly, small molecules play a role in cell signalling.1–3 Methadone is a synthetic opioid drug that is used for the treatment of opioid dependency, for example in heroin addicts.4 Patients with opioid dependency experience increased pain sensitivity and intense withdrawal symptoms, which leads to high attrition in withdrawal therapies.5 Methadone maintenance therapy reduces narcotic craving, blocks the euphoric effects associated with opioids, and reduces discomfort in addicts.6 The concentration of methadone in the patient's blood has to be maintained between 150 and 600 ng mL−1 for the treatment to be effective.7 The required daily dosage to maintain this range of methadone blood concentration is very important and has been the subject of several studies.4,8 Therefore, the quantitative detection of methadone in bodily fluids in a non-invasive and high-throughput manner would be highly desirable to assist methadone maintenance programs.Current methods of measuring methadone concentrations involve the use of immunochemical assays from urine samples.9 These assays give only qualitative measurements of the drug, with a positive result generated for concentrations exceeding 300 ng mL−1.10 In some cases, concerns have been raised about spurious results obtained by this method, for example leading to false-positive results in some control patients who have never used methadone.10 This was later attributed to immunochemical cross-reactions with other unrelated drugs present in the patient's urine. A more accurate and quantitative approach was developed for the detection of methadone involving gas chromatography-mass spectrometry (GC-MS) of hair extracts.11 However, this method makes sacrifices on analysis speed and the required multiple sample preparation steps bear the risk of adulteration.11 This limitation can be effectively mitigated when analyzing bodily fluids such as saliva, blood or urine, obtained directly from patients.12
Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) is a soft ionization technique, introduced in the 1980's,13,14 which is commonly used to analyze high molecular weight analytes such as peptides,15 proteins,16 oligonucleotides,17 and polymers.18 Analysis of neat biological fluids is possible and so is quantitation, when using suitable internal standards.19,20 However, the principal weakness of MALDI-MS consists in the analysis of low molecular weight analytes (<700 Da) due to the competitive fragmentation of the matrix molecules, appearing in the lower mass range and producing highly complex spectra that can be tedious if not impossible to interpret.21 Additionally, issues such as matrix-to-analyte ratio and the choice of matrix all affect the analytical performance for analytes across the mass range.22
Matrix-free desorption/ionization approaches have recently been developed, fuelled by the need to obviate the difficulties in small molecule analysis. These approaches rely on nanostructured materials and are collectively termed surface-assisted laser desorption/ionization mass spectrometry (SALDI-MS).23–26
A range of surfaces have been investigated for SALDI-MS including graphite,27,28 carbon nanotubes,29 silica and titania sol–gel systems,27,30,31 and nanostructured silicon.21,23,24 Porous silicon (pSi), for example has successfully been used as a SALDI-MS substrate because of its unique properties, including large surface area, allowing for high loads of analytes to be trapped in the porous layer, and high UV cross-section, facilitating laser energy transfer to the analyte. This technique is commonly referred to as desorption/ionization on silicon (DIOS). Here, interferences in the lower mass range are no longer an issue. However, the infiltration of analytes into the pores in some instances can cause loss in signal intensity due to attenuation of the laser signal.32,33
DIOS has been under extensive investigation as to which factors contributed to efficient desorption/ionization.34,35 Factors such as surface area, optical absorption, thermal conductivity, as well as pH levels and solvent contribution, have all been studied in the context of the mechanisms involved in the desorption and protonation of the analyte molecules.
Submicrometer surface structures and their morphologies have been identified as a key factor in ion generation and therefore, SALDI-MS was successfully performed on other types of nanostructured silicon such as nanocrystalline void-column networks of silicon,36 single-crystalline silicon nanowires fabricated by vapor–liquid–solid (VLS) technique,37 patterned nanocavity arrays,24 microcolumn arrays,38 silicon sub-micrometer grooves and flat silicon roughened with sandpaper,23 and silicon nanowires fabricated using metal-assisted chemical etching (MACE).32
However, since SALDI-MS ion generation depends highly on surface structural dimensions, most of the reports so far fall short on providing nano-scale control over these dimensions. Those that did study the influence of the nanostructure parameters required expensive high-power lasers, photolithography combined with e-beam lithography or reactive ion etching to produce these nanostructures,38,39 which are not suitable for wide and commercial usage as SALDI-MS substrates. MACE on the other hand is a cheap and versatile method that could be implemented under ambient conditions easily, but still suffers from lack of control over the physical dimensions of the structures fabricated. This, however can be solved by combining MACE with a simple lithography technique such as self-assembling nanosphere lithography.40,41 Furthermore, the MACE reaction mechanism allows for the introduction of cracks and pores to the fabricated structures to increase the surface area by manipulating the concentration of the oxidizing agent in the reaction solution as described by Chiappini et al.41 In the investigation described by Wang et al., arrays of hexagonally arranged silicon nanowires fabricated through gold-catalyzed MACE and nanosphere lithography were used for SALDI-MS to detect a group of small peptides.33 The authors used silicon nanowire arrays with 200 nm diameter, 2000 nm length, and an aspect ratio of 10. Furthermore, the inter-nanowire spacing was determined to be in the range of 400 nm. Although the authors were successful in detecting peptides, no systematic study linking analytical performance to structural dimensions was conducted.
In this paper, we adapted the fabrication strategy described by Wang et al.33 but used Ag as a catalyst instead of Au since Ag has been shown to have similar catalytic properties to Au in the context of the MACE reaction and is cheaper.41 We also focused on fabricating and using nanostructures with low aspect ratios and lengths similar to what is typically used in DIOS as opposed to the high aspect ratio structures described by Wang et al.33 Our approach resulted in cylindrical pillars with aspect ratios <10 which we term silicon nanopillars (SiNPs). We explored the flexibility of ordered SiNP formation via MACE and investigated the influence of nanopillar length, aspect ratio and porosity on desorption/ionization performance and efficiency. For this purpose, we elected to use a standard peptide mixture to demonstrate the usability of our substrate to assist in the detection of analytes with masses in the middle range (1–3 kDa). We also chose methadone as a representative of small molecule analytes of <500 Da, in addition to its importance in opioid dependency treatment programs as described previously. The fabrication method involved the use of polystyrene nanosphere (PSNS) lithography as a shadow mask to deposit a thin Ag layer, followed by the MACE process. The SiNPs allowed the detection of peptides and methadone down to a limit of detection (LOD) of 32 ng mL−1. We observed that arrays featuring SiNPs with 450 nm lengths, 450 nm diameters, an aspect ratio of 1.0, and a non-porous morphology afforded the best analytical performance in terms of the detection of methadone and peptides. Furthermore, these SiNP arrays allowed for the detection of methadone in saliva, blood plasma and urine obtained as clinical samples from real methadone-treated patients. Finally, the performance of SiNP arrays was compared with that of conventional DIOS substrates.
Methods and materials
Chemicals and materials
Sulfuric acid (H2SO4, 95–97%) and hydrofluoric acid (HF, 48%) were purchased from Scharlau Chemie (Chem-Supply Pty. Ltd. Australian representation). Hydrogen peroxide (H2O2, 30%) was purchased from Merck (Australia). Nitric acid (HNO3, 70%) was purchased from Sigma-Aldrich (Australia). The PSNS solution was purchased from Polysciences (USA) and diluted (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) with methanol solution containing Triton X-100 (1:400, v/v) before spin-coating. (Tridecafluoro-1,1,2,2-tetrahydrooctyl) dimethylchlorosilane (F13) was purchased from Gelest (USA). A peptide mixture consisting of angiotensin I, angiotensin II, substance P, bombesin, ACTH clip 1–17, ACTH clip 18–39 and somatostatin 28 was purchased from Bruker-Daltonics (Germany). Certified standard solutions of methadone and the internal standard methadone-d3 were kindly provided by Forensic Science South Australia (Australia).
:1, v/v) with methanol solution containing Triton X-100 (1:400, v/v) before spin-coating. (Tridecafluoro-1,1,2,2-tetrahydrooctyl) dimethylchlorosilane (F13) was purchased from Gelest (USA). A peptide mixture consisting of angiotensin I, angiotensin II, substance P, bombesin, ACTH clip 1–17, ACTH clip 18–39 and somatostatin 28 was purchased from Bruker-Daltonics (Germany). Certified standard solutions of methadone and the internal standard methadone-d3 were kindly provided by Forensic Science South Australia (Australia).
Preparation of Si wafers and deposition of PSNS
Prior to the PSNS deposition, flat silicon wafers (P-type, 3–6 Ω cm, 〈100〉, Siltronix, France) were cut to 2 × 2 cm pieces and cleaned by sonication in 1:1 solution of ethanol–acetone for 5 min, followed by further sonication in MilliQ water for 5 min. This was followed by dipping the wafers into boiling piranha solution (2:1 H2SO4:H2O2 v/v) for 1 h to remove organic contaminants, then washed with MilliQ water and dried with N2 jet.
PSNS solution (50 μL) was deposited on cleaned wafers and then spin-coated (Laurell Technologies, WS-650MZ-23NPP spin-coater, USA) at 200 rpm to form a self-assembled, hexagonally arranged monolayer of PSNS. This was directly followed by a 20 s spin at 1500 rpm to remove any excessive solution present around the edges of the wafer.
Preparation of SiNP array
Following PSNS monolayer deposition and self-assembly, the PSNS were tightly packed (or close-packed) alongside each other due to capillary forces driving them together. Upon exposure of this monolayer to O2 plasma treatment the size of the PSNS was reduced uniformly and the close-packed monolayer changed into a non-close-packed arrangement while maintaining the hexagonal long range order. The non-close-packed arrangement of the PSNS was necessary to allow Ag deposition between the PSNS which would subsequently facilitate the MACE process.O2 plasma etching and Ag sputter-coating were both performed using an HHV TF600 sputter-coater fitted with a PLC control system. The O2 plasma was generated with an RF power system using 50 W and oxygen flow rate of 15 cm3 min−1. All plasma etching treatments were conducted for a duration of 7 min at a pressure of 2.00 × 10−2 mbar.
Ag sputter-coating was carried out using a DC power supply set at 100 W with a flow rate of Ar gas set at 10 cm3 min−1 to generate a pressure of 1.00 × 10−2 mbar inside the chamber. All sputter-coating was performed for a total of 4 min to generate a 40 nm layer of Ag.
The PSNS layer was removed by sonication in MilliQ water for 5 min followed by washing with ethanol, acetone, and MilliQ water.
MACE was carried out in a 20 mL polytetrafluoroethylene (PTFE) container by diluting HF (48%, Scharlau, Germany) and H2O2 (30%, Merck, Germany) in MilliQ water. All etching solutions were prepared with HF concentrations of 4.8 M while H2O2 concentrations were varied from 0.1 M to 0.3 M to control the degree of porosity.41 The Teflon container was sealed to prevent dangerous HF vapours from escaping the container, and the reactions were carried out at room temperature for different durations, as specified in the text.
After this, samples were washed with MilliQ water and dipped into concentrated HNO3 (70%, Sigma, USA) for several minutes to remove the Ag layer, followed by washing with MilliQ water and acetone, and drying with a N2 gas jet. The six steps of this fabrication process are shown schematically in Fig. 1.
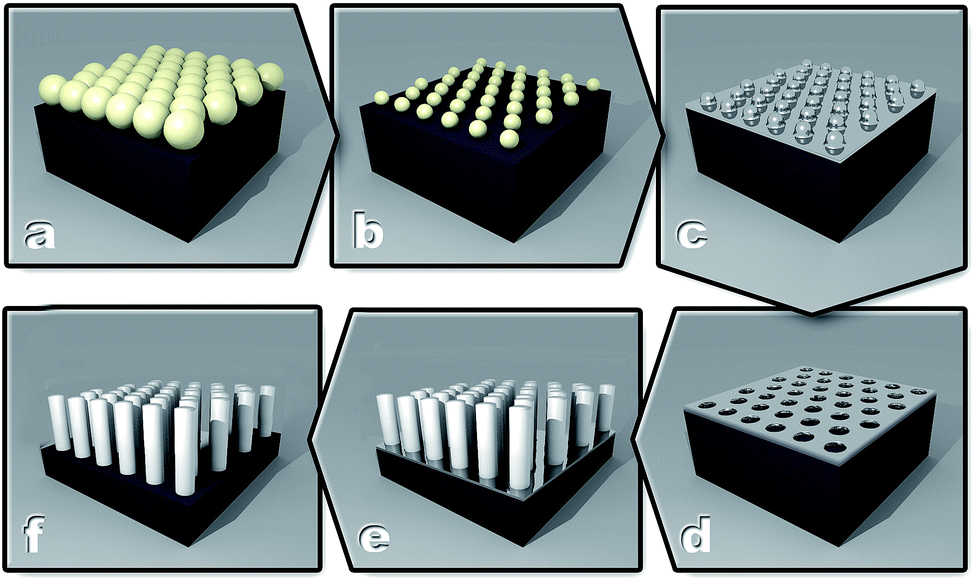 | ||
| Fig. 1 Schematic representation of the SiNP fabrication process. (A) Spin-coating of PSNS on a Si surface, (B) treatment of the PSNS layer with O2 plasma to reduce the size of the PSNS and create a non-close-packed arrangement, (C) sample following Ag deposition, (D) removal of the PSNS layer, (E) etching with HF/H2O2 to produce SiNPs, and (F) SiNPs after the removal of the Ag layer. | ||
Chemical surface modification of SiNP array
SiNPs were oxidized using ozone oxidation treatment at 3.25 g h−1 flow rate from an Ozone Generator 500 (Fischer, Germany) over a 30 min period, yielding a stable thin oxide layer.42 Ozone oxidized SiNP arrays prepared as such were used for all non-clinical samples. Silanization with F13 was carried out on oxidized SiNPs by depositing 100 μL of F13 directly on the surface and allowing the reaction to occur at 95 °C for 30 min, followed by washing with ethanol and drying using a stream of N2 gas. F13 modified SiNP arrays were used with clinical samples.SEM characterization
All SEM imaging and characterizations were done using a Quanta 450 FEG Environmental SEM (FEI, Netherlands) fitted with solid state detector (SSD) and operating at 30 kV in high vacuum mode. Length measurements were performed using the SEM's operating software.Infrared (IR) characterization
IR characterization of the surface chemistry was carried out using a Nicolet Avatar 370 (Thermo Electron Corp., USA) fitted with a mercury–cadmium–telluride (MCT) detector. The collection of spectra was done using a Diffuse Reflectance Infrared Fourier Transform (DRIFT) accessory and recorded using the software OMNIC version 7.3. Scans were recorded in the range of 500–4000 cm−1 at a resolution of 4 cm−1. Background spectra were collected off a clean flat silicon substrate with the same type and resistivity as the SiNP samples in dry air to minimize any background noise that might result from atmospheric water vapor and carbon dioxide.Preparation of methadone and peptide solutions
Stock solutions of methadone at 0.1 mg mL−1 were prepared in methanol from a certified standard ampoule and stored at −20 °C. Working solutions of 4000 ng mL−1 were obtained by diluting the stock solutions with MilliQ water. Working solutions were kept at +4 °C and prepared fresh every 2 weeks. Drug solutions at varying concentrations (100–2000 ng mL−1) containing the corresponding deuterated internal standard at 100 ng mL−1 were prepared from working solutions immediately prior to analysis.Working solutions for the peptide mixture were prepared by dissolving the peptide mixture in 125 μL of 0.1% trifluoroacetic acid (TFA). The working solution was separated into 5 μL aliquots and stored at −20 °C.
Sample deposition method for non-clinical samples
Aliquots of methadone (1 μL) and peptide (2 μL) solutions were deposited onto ozone oxidized SiNP array substrates, and allowed to completely evaporate. Upon evaporation, SiNP substrates were mounted on a modified MALDI target plate (MTP384, Bruker Daltonics, Germany) using double-sided carbon tape.Sample preparation and deposition method for clinical samples
Clinical samples were obtained according to ethics protocol number 0000021954, from known methadone users. Briefly, clinical samples (2 μL) were deposited onto SiNP and DIOS substrates modified with F13, and allowed to interact for 5 min in order to facilitate extraction. For the preparation of DIOS substrates, please see the ESI.† The droplet was removed via pipette and then the resulting droplet area was washed with ammonium citrate buffer (10 mM, 5 μL), pippetting ten times and discarding the solution.SALDI-MS analysis
Mass spectra were collected using an Autoflex Series III MALDI-TOF mass spectrometer (Bruker, Germany) equipped with a SmartBeam (337 nm, Nd:YAG) 200 Hz pulsed laser, operated at 200 Hz frequency and laser attenuator offset of either 30% (for methadone analysis) or 70% (for peptides analysis) in reflectron positive mode. Mass spectra were generated by averaging 100 individual laser shots per spot, while using 5 spots per surface. This was done in order to minimize the effects of surface-to-surface variations during signal measurement. Data acquisition used flexControl 3.3 (build 85) software and data analysis was performed using flexAnalysis version 3.3. Quadratic external calibration of the TOF tube was performed on the monoisotopic masses of α-cyano-4-hydroxycinnamic acid (CHCA) adducts before each analysis.Calculation of limit of detection (LOD)
Measuring the LOD of methadone involved establishing the magnitude of contribution from background noise to the analyte signal that is observed. The background noise was calculated by measuring the average signal intensity ratio between the signal intensity at m/z = 310 (methadone) and m/z = 313 (internal standard, methadone-d3) in the absence of methadone. We averaged eighteen replicates containing methadone-d3 (100 ng mL−1) over 3 different SiNP arrays. The LOD for methadone was then defined as three standard deviations above the average background-to-internal standard ratio that was measured.Results and discussion
Fabrication of SiNP arrays
A PSNS (500 nm) monolayer was first spin-coated onto a flat silicon substrate as a shadow mask. Fig. 2A shows a representative SEM image of non close-packed arrangement of the PSNS monolayer deposited onto the flat silicon surface by means of spin-coating after treatment with O2 plasma to reduce the nanospheres' diameter from the original size of 500 nm to 450 nm, thus creating a 100 nm wide gap between the spheres. In Fig. 2B, the same PSNS layer can be seen after sputter-coating with a 40 nm thick Ag layer. After removal of the PSNS, the remaining Ag layer featured hexagonally arranged holes where the PSNS were originally positioned (Fig. 2C). | ||
| Fig. 2 SEM micrographs of the different stages in the SiNPs fabrication process. (A) Spin-coated monolayer of PSNS after treatment in O2 plasma, (B) PSNS layer after the deposition of a 40 nm Ag layer, (C) the Ag layer after removal of the PSNS layer, and (D) the SiNP array after etching in an aqueous solution of HF/H2O2. | ||
A representative image of a vertical array of SiNPs formed as a result of etching the Ag-covered silicon substrate in HF/H2O2 is shown in Fig. 2D. The length of the SiNPs was determined by controlling the duration of the HF/H2O2 etching.
A set of SiNP arrays was prepared by varying the duration of the etching reactions for each sample in the set as well as the concentration of H2O2 in the etching solution for each duration (concentrations of 0.1 M, 0.2 M and 0.3 M were used), followed by measuring SiNP lengths using SEM imaging of cross-sections of the etched samples. Fig. 3 shows a graph detailing the relationship between the mean SiNP length and the etching duration.
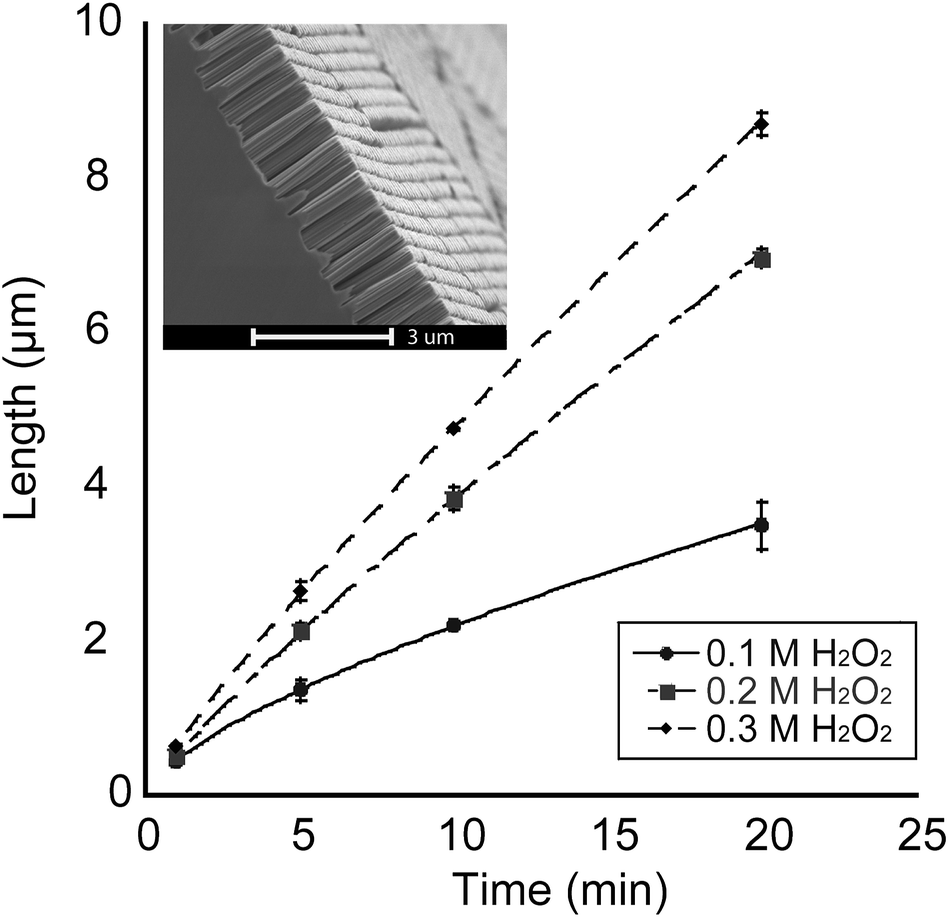 | ||
| Fig. 3 Graph showing the length of the SiNPs as a function of the duration of the HF/H2O2 etching. Etching was carried out at room temperature in a solution of 4.8 M HF with H2O2 concentrations of 0.1, 0.2, and 0.3 M. Inset shows a representative SEM micrograph with a cross-sectional view of a SiNP array. Scale bar is 3 μm. | ||
As Fig. 3 shows, longer etching durations result in an increase in the nanopillar length. Additionally, the rate of this increase was highly dependent on the concentration of the constituents of the etching solution. From Fig. 3, it was evident that increasing the concentration of the oxidizing agent (H2O2) affected the rate of etching and consequently the length of the SiNPs. The variation in the etching rate between the solutions with different H2O2 concentrations started out with negligible difference at lower etching times (∼1 min) and became progressively more prominent as the etching duration increased. Furthermore, the etching rate of the 0.1 M H2O2 solution gradually decreased with increasing etching duration. We postulate that this is a result of diminishing supply of H2O2 molecules as the reaction progresses.
The etching rate became more linear with increasing H2O2 concentration (R2 values of 0.9970 and 0.9975 for 0.2 M H2O2 and 0.3 M H2O2 solutions, respectively). IR spectroscopy characterization of the SiNP arrays revealed the presence of silicon oxide groups at the time when the SALDI-MS experiments were conducted (Fig. S1†).
SALDI-MS detection of peptides
For this section, ozone oxidized SiNP arrays were used. The detection of peptides using different SiNP array morphologies was investigated using a peptide mixture containing angiotensin I and II, substance P, bombesin, ACTH clip 1–17, ACTH clip 18–39, and somatostatin 28. These peptides span a quasimolecular ion weight range from 1047 Da (angiotensin II) to 3147 Da (somatostatin). The peptide mixture was deposited directly onto the SiNP array surface and allowed to dry.Firstly, different SiNP diameters affected signal generation greatly. When nanopillars with 350 nm diameter and inter-nanopillar spacing of 300 nm were used, no signal generation was observed from any of the peptides. However, when the SiNP diameters were set to 450 nm and the inter-nanopillar spacing was 100 nm, signal from the peptides was observed. It appeared that spacing between nanopillars was an important parameter, in analogy to the pores in DIOS platforms where it has been noted that the size of the pores plays an important role in whether or not signal generation can occur.43,44 Additionally, pore sizes in the range of 70–120 nm are commonly used for DIOS based substrates.21,34,45 The inter-nanopillar spacing used here (100 nm) fell within that range. A laser fluence value of 70% of the maximum output was used throughout the analysis of the peptides. It was determined that 70% laser fluence was the optimum setting for peptide detection since increasing the fluence resulted in high background noise, while decreasing the fluence resulted in non-optimum signal intensities. SALDI mass spectra of the peptide mixture on flat silicon and SiNP arrays are depicted in Fig. 4.
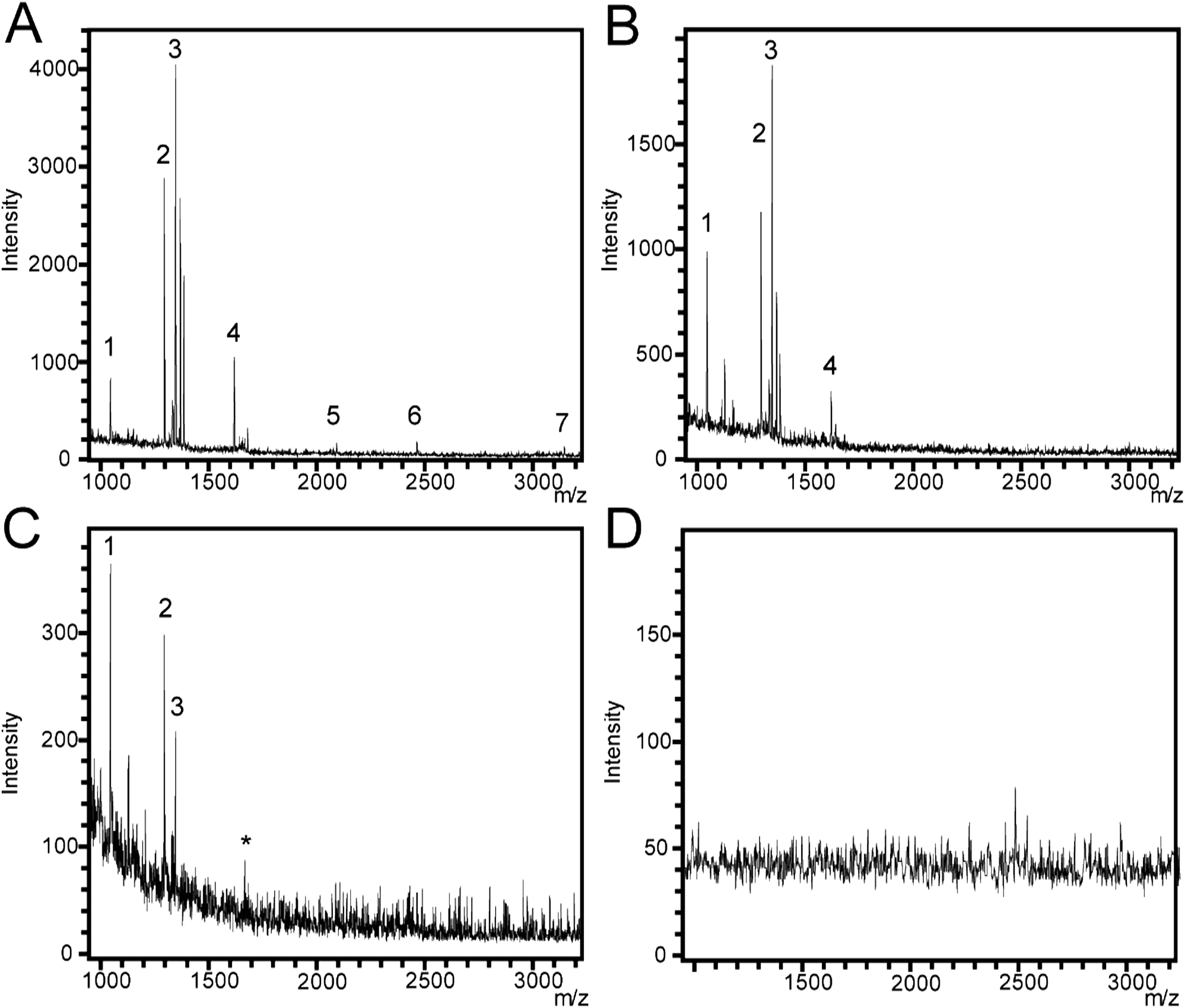 | ||
| Fig. 4 (A–C) Mass spectra collected from SiNP arrays etched for (A) 1 min (450 nm length, aspect ratio of 1.0), (B) 5 min (1360 nm length, aspect ratio of 3.0), and (C) 20 min (3500 nm length, aspect ratio of 7.7). All SiNP arrays had 450 nm diameter and 100 nm spacing and were etched with 4.8 M HF/0.1 M H2O2. Standard peptide mixture spots were deposited on all surfaces. Peak 1 and 2 correspond to the peptides angiotensin II and I, respectively, while peak 3 corresponds to substance P, peak 4 corresponds to bombecin, peak 5 corresponds to ACTH 1–17, peak 6 corresponds to ACTH 18–39 and peak 7 corresponds to somatostatin. The asterisk (*) corresponds to contamination peaks. (D) SALDI mass spectra collected from a flat silicon surface as a negative control. | ||
Mass spectra were collected from SiNP arrays that were etched for 1 min (450 nm), 5 min (1360 nm), and 20 min (3500 nm) (Fig. 4A–C, respectively) in 4.8 M HF/0.1 M H2O2, and on flat silicon (Fig. 4D) as a negative control. We also performed SALDI-MS analysis using MilliQ water on a SiNP array etched for 1 min using the same etching conditions described above as a second experimental control (Fig. S2†) with no significant peaks appearing above background noise. In previous SALDI-MS investigations, signal-to-noise (S/N) ratios of 3, 4, and 5 were specified as acceptable limits for signal detection.46–48 Here, we chose the S/N ratio ≥3 to indicate successful peptide detection. Peptide peaks could not be observed on the flat silicon surface. In contrast, seven peaks were detected for the sample etched for 1 min. The peaks corresponded to angiotensin II and I (labeled 1 and 2, respectively), substance P (3), bombesin (4), the ACTH clips 1–17 and 18–39 (5 and 6, respectively) and somatostatin (7) as well as two small peaks close to peak 3, which correspond to Na+ and K+ adducts to substance P at + 22.98 and + 38.96 Da (with substance P's molecular ion as an offset). For the SiNP array, which was etched for 5 min, peaks 5, 6, and 7 were not detected while Na+ and K+ adducts of peak 3 were still being observed. For the sample etched for 20 min, peaks 4, 5, 6, and 7 were not detected at all. In summary, S/N for all analytes decreased with increasing nanopillar length except for angiotensin II where the highest S/N was detected on the 5 min etched SiNP array. S/N values for the peaks in each spectrum are shown in the ESI (Table S1†). The length of the SiNPs affects the distribution of the analyte and their exposure to the nitrogen laser during the SALDI-MS experiment. Our results suggest that SiNPs with lengths greater than the 1 min etch (450 nm, aspect ratio of 1.0) do not allow effective energy transfer from the laser to the analyte. In longer SiNP arrays, energy transfer from the laser to the analyte may be hindered similar to what has been observed in DIOS for deeper porous layers.23,32,33 The higher molecular weight peptides in the mixture, ACTH clip 1–17, ACTH clip 18–39, and somatostatin 28 were detected only with short SiNP arrays, which suggests that as the SiNP arrays become longer, they become less efficient for ionizing and desorbing molecules with higher molecular weights. Additionally, the 450 nm long SiNPs with an aspect ratio of 1.0 outperformed the reported analyte detection range of commercially available DIOS substrates (MassPREP™ DIOS-target, Waters) where peptides above 2000 Da were not detectable.49 In contrast, we were able to detect ACTH clips 1–17 and 18–39 as well as somatostatin with molecular weights of 2094, 2466, and 3149 Da, respectively.
SALDI-MS of methadone
The encouraging results obtained for the SALDI-MS analysis of peptides using SiNP arrays led us to pursue another analytical application, the detection of methadone. For the detection of methadone in this section, ozone oxidized SiNPs were used. Fig. 5A shows a representative SALDI mass spectrum for methadone (1000 ng mL−1) in water obtained on 450 nm long SiNPs with an aspect ratio of 1.0, which gave the best performance in peptide detection. The quasimolecular ion for methadone (m/z = 310) was clearly observed at an average S/N ratio of 427. In contrast, low signal intensities with no interfering peaks in the mass range for methadone were observed for a background mass spectrum obtained from MilliQ water only (Fig. 5B, note the y-axis scale reduced 200×). A laser fluence of 30% was used in this case since it provided optimum analytical performance and signal intensities.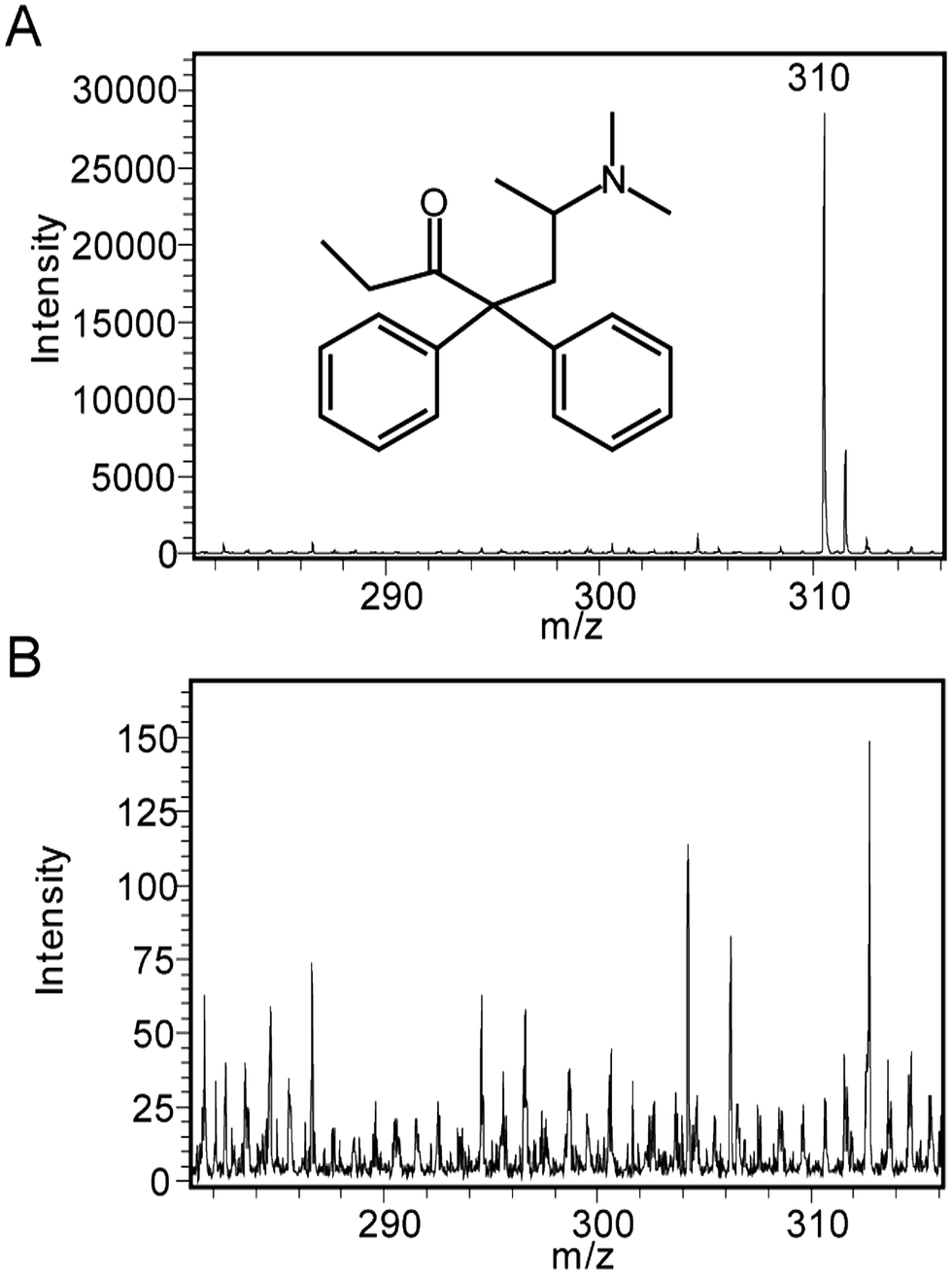 | ||
| Fig. 5 Representative SALDI mass spectra on SiNP arrays etched in a 4.8 M HF/0.1 M H2O2 solution for 1 min (450 nm in length, 450 nm diameter, 100 nm inter-nanopillar spacing, and an aspect ratio of 1.0) for (A) methadone with a MH+ = 310 m/z, and (B) MilliQ water as negative control. | ||
The full mass spectrum is shown in Fig. S3† where, in addition to the presence of the methadone peak at 310 m/z, there was another significant peak appearing at 265 m/z. This has been identified previously as a fragment of methadone.50 Noise levels on the other hand appeared to be significantly low.
The signal intensity of methadone peaks was tested on SiNP arrays with varying lengths below 1200 nm to fine-tune the SiNPs length for highest analytical performance.
Average S/N of the methadone peak were used as a quantitative measure for signal intensity in each case (Fig. 6, n = 3). SiNP arrays with lengths measured as 450 (1 min etch, aspect ratio of 1.0), 730 (2 min etch, aspect ratio of 1.6), 960 nm (3 min etch, aspect ratio of 2.1), and 1170 nm (4 min etch, aspect ratio of 2.6) were used in this case. The SiNP arrays were etched using 0.1 M H2O2, and had diameters of 450 nm according to SEM characterization.
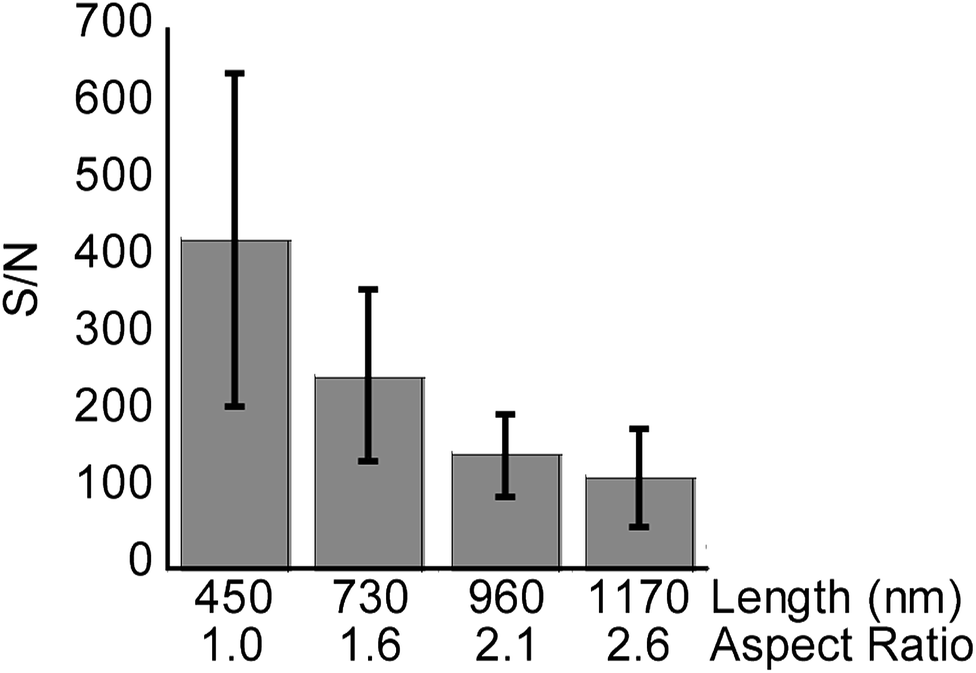 | ||
| Fig. 6 The bars depict the average S/N of methadone (1000 ng mL−1) peaks detected on SiNP arrays, 450 nm in diameter, with different lengths and aspect ratios using SALDI-MS. The lines represent one standard deviation. Number of measurements n = 3. | ||
As in the case with the peptide mixture, the S/N ratio for methadone was seen to decrease with increasing nanopillar length and aspect ratio. This further confirmed the notion that longer SiNPs hinder the process of desorption or attenuate laser irradiation, resulting in non-optimal energy transfer to the analytes.
SALDI-MS studies of nanostructured surfaces are generally in agreement that high surface roughness leads to increased signal intensity of analytes.23,24 We investigated whether further increasing surface roughness of the SiNPs by introducing structural porosity enhances the signal intensity. We introduced this porosity in SiNPs by increasing the concentration of H2O2 in the etching mixture from 0.1 to 0.3 M.41
Fig. 7A shows a SiNP array etched in HF/H2O2 at a concentration of 0.1 M H2O2. The SiNP tips and sidewalls appeared smooth. Increasing the concentration of H2O2 during the etching increases the oxidation rate and encourages localized etching along the SiNP walls.41 At 0.3 M H2O2 concentration, the entire surface of the SiNPs showed increased roughness and formation of pores with sizes between 10 and 200 nm, especially at the tips of the nanopillars and down along the sidewalls (Fig. 7B).
 | ||
| Fig. 7 SEM micrographs detailing the effect of increasing H2O2 concentration on porosity of the SiNPs. The samples were etched for 1 min with a diameter of 450 nm (aspect ratio of 1.0) in a solution containing 4.8 M HF and (A) 0.1 M H2O2, and (B) 0.3 M H2O2. The structural roughness, or porosity increases with increasing H2O2 concentration. | ||
In addition to the signal intensity diminishing as a result of longer etching times and hence longer SiNPs, signal intensity also decreased with increasing H2O2 concentration and hence increasing porosity (Fig. 8, n = 3). However, the latter effect was more pronounced, showing over 10 fold reduction in S/N from samples etched at 0.1 M H2O2 to 0.3 M H2O2. At 1 min etching time, the variation in length and aspect ratios of the SiNPs due to differences in H2O2 concentrations is negligible as is seen in Fig. 3 (450 nm–500 nm). Therefore, the decrease in signal intensity was mostly attributed to the increase in structural porosity. This reduction in signal intensity relates to the adsorption of analyte molecules inside the cracks and pores shown in Fig. 7, which may actually hinder release of those molecules during laser irradiation. Support for this hypothesis comes from the DIOS literature where small pore sizes in the mesopore range perform poorly in SALDI-MS.21,34,43
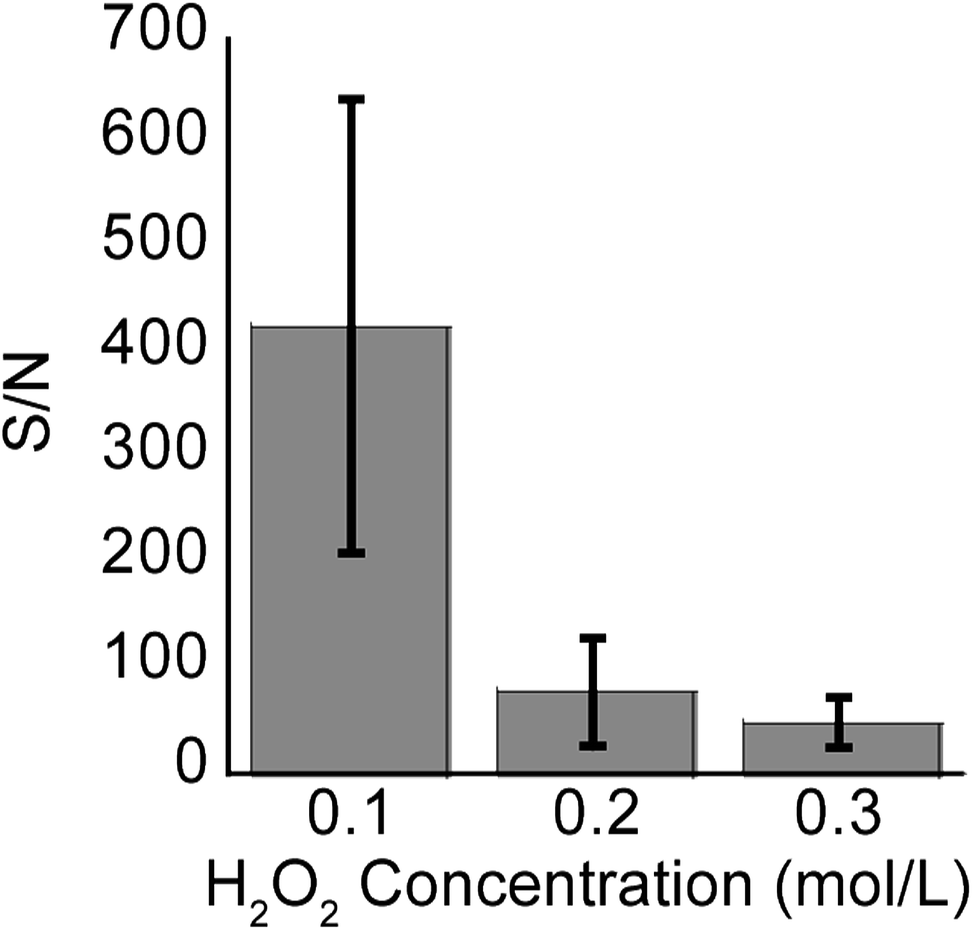 | ||
| Fig. 8 The bars depict the average S/N values for methadone (1000 ng mL−1) analyzed on SiNP arrays etched for 1 min (with an aspect ratio of 1.0) at different peroxide concentrations. The lines represent one standard deviation. Number of measurements n = 3. | ||
The SiNP arrays etched for 1 min were washed with ethanol and MilliQ water, and reused successfully for the detection of methadone without a noticeable decrease in the observed signal intensity. Furthermore, the same SiNP arrays were stored for up to 3 months under ambient conditions, and then reused for SALDI-MS detection of methadone, while showing no decrease in performance (data not shown). The LOD for methadone was evaluated on a SiNP array etched using the conditions which were found to produce the highest S/N, namely 1 min etching in 4.8 M HF/0.1 M H2O2, resulting in SiNPs with 450 nm length, 450 nm diameter, and aspect ratio of 1.0. A deuterated standard of methadone was introduced as an internal standard at a constant concentration of 100 ng mL−1. The LOD for methadone was investigated over the concentration range of 20–2000 ng mL−1. The laser fluence was set to a fixed value of 30% while measuring the signal from each concentration.
Methadone detection showed good linearity (R2 > 0.99) in the signal intensity ratio of drug to internal standard over the concentration range of 20–2000 ng mL−1, demonstrating that quantification of methadone using SiNP arrays can be achieved (Fig. 9A). A LOD value of 32 ng mL−1 was obtained. Fig. 9B shows a close-up of the 0–70 ng mL−1 concentration range, showing that the LOD was determined as three times the noise level. The sensitivity (S) was calculated as the slope of the line of best fit and was defined as the signal ratio of (methadone/methadone-d3) per ng mL−1 where S = 0.013 mL ng−1.
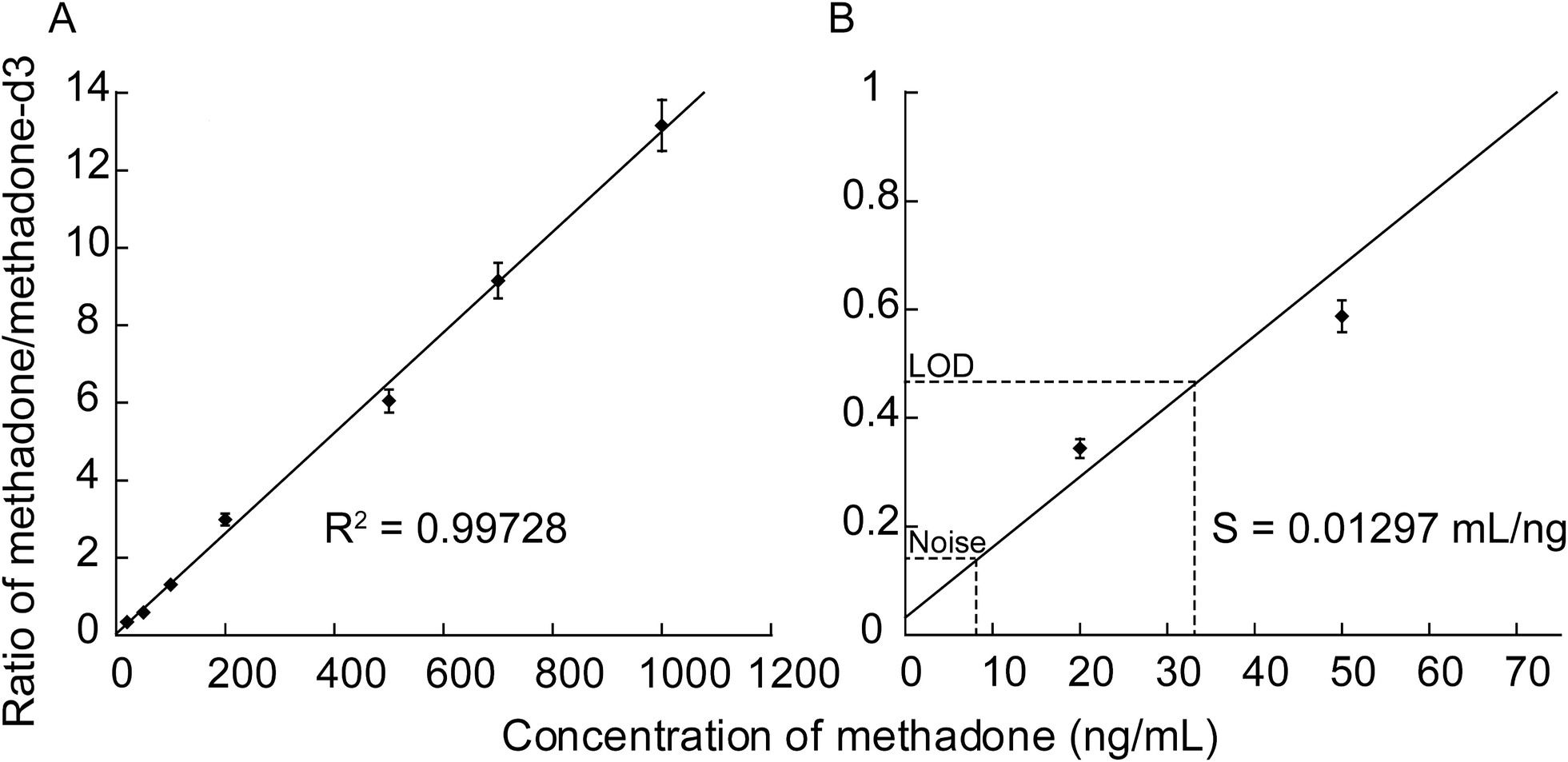 | ||
| Fig. 9 Graph depicting (A) the relationship between the concentration of methadone deposited on a SiNP array etched for 1 min using 4.8 M HF/0.1 M H2O2 solution and the ratio of the signal detected from the methadone to its deuterated standard fixed at a constant concentration of 100 ng mL−1 in each of the solution concentrations tested. (B) shows a close-up of the lower concentration range with the background noise and the LOD displayed graphically, as well as the sensitivity calculated from the slope of the line of best fit (S). | ||
SALDI-MS detection of methadone in real clinical samples
Following the successful determination of LOD of methadone in water, the detection of methadone in real clinical samples was carried out. Several studies have been published so far, detailing the expected methadone concentration found in the urine, blood plasma, and saliva of opioid-dependency patients taking regular doses of methadone.51–53 These concentrations all fall above the LOD established earlier. In order to demonstrate the applicability of SiNP arrays for the detection of methadone in real physiological fluids, real clinical samples of blood plasma, urine, and saliva were obtained from methadone therapy patients and subjected to SALDI-MS for the detection of methadone.Using SiNP arrays with a native oxide surface chemistry resulted in negligible detection from the three fluids. This was attributed to the rinsing protocol used. Rinsing protocols are usually employed for use with DIOS to avoid extraction and derivatization that is required for other MS techniques.43 Due to the relatively non-polar nature of methadone, it is preferentially retained in the biological fluid after the rinsing step rather than being adsorbed on the surface. Alternatively, using dried droplet deposition cannot be implemented with complex biological fluids like saliva and blood since these contain peptides, proteins, and other biomolecules that dry on the surface and interfere with SALDI-MS signals.
For this reason, the surfaces of the SiNP arrays used for clinical samples were F13 modified. This resulted in a significant increase in surface hydrophobicity. Water contact-angle (WCA) measurements are shown in Fig. S4.† Oxidized surfaces showed a WCA of 25° ± 1°, while the F13 modified surfaces showed a WCA of 110° ± 1°. Following this, the clinical samples were tested on the F13 modified SiNP arrays etched with the optimum morphology described previously, namely in 4.8 M HF and 0.1 M H2O2 for 1 min. Furthermore, a standard DIOS surface, fabricated at our lab and modified with the same F13 fluorosilane was used alongside the SiNP arrays in order to compare the performance of both surfaces for the SALDI-MS detection of methadone. The methods and materials for the DIOS substrate fabrication were detailed in the ESI† section and not the main text since it was outside the scope of this work. The results of the comparison are shown in Fig. 10. Three separate measurements were conducted for each clinical sample on both SiNP and DIOS substrates.
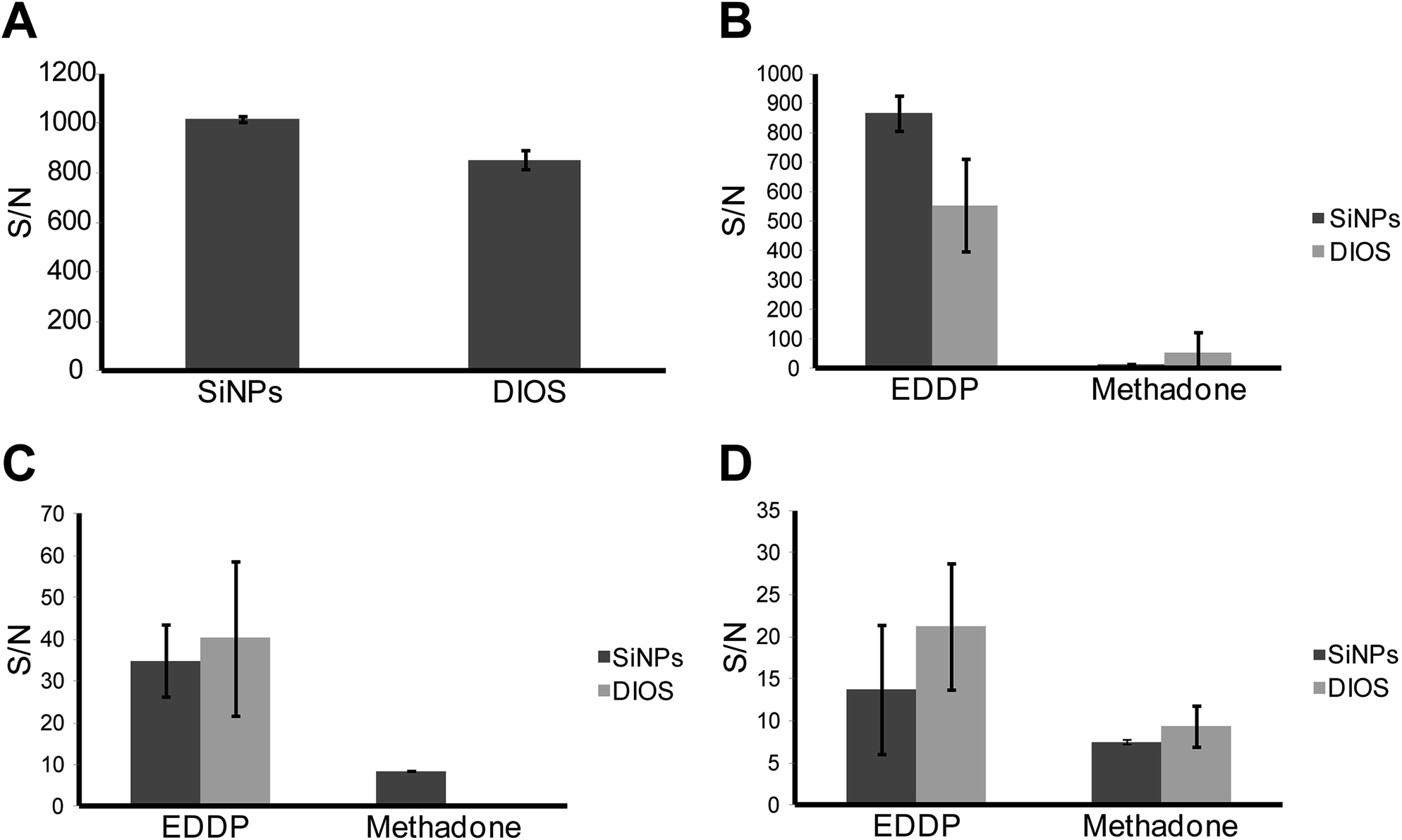 | ||
| Fig. 10 (A) S/N for methadone in MilliQ water using both SiNP arrays and a DIOS surface. (B) S/N of both methadone and its metabolite EDDP in urine detected using SiNPs and DIOS. (C) S/N of methadone and EDDP in blood on both SiNPs and DIOS. (D) S/N of methadone and EDDP in saliva on both SiNPs and DIOS. | ||
Fig. 10A shows a comparison between the analytical performance of both the SiNP array and DIOS surfaces in terms of S/N ratio where the analyte was methadone in MilliQ water (1000 ng mL−1). After functionalization of SiNP arrays with F13, a significant increase in the average S/N from 427, for oxidized SiNP arrays to 1015 was observed (Fig. 6 and 10A). Additionally, the SiNP array performed better than the F13 functionalized DIOS surface. This was confirmed using an unpaired t-test, which showed that the difference in S/N for the detection of methadone was statistically significant (95% confidence) suggesting that SiNP arrays provided better performance. Fig. 10B–D represent S/N values for methadone and its major metabolite detected in urine, blood plasma, and saliva, respectively. The methadone metabolite 2-ethylidene-1,5-dimethyl-3,3-diphenyl-pyrrolidine (EDDP) is formed by N-demethylation and cyclization of methadone in the liver.54 Since the excreted non-metabolized methadone can vary in quantity according to the urine's pH, dose, and the metabolic rate, EDDP content is usually analysed instead since it avoids the variation in metabolism rates between individuals.55
Fig. 10B, demonstrates the superior detection sensitivity of EDDP from urine on SiNP arrays as opposed to DIOS (n = 3, significant with 95% confidence using unpaired t-test). For methadone detection from urine, there was no statistical difference between the two methods (n = 3, unpaired t-test). Fig. 10C depicts the S/N values collected from a clinical blood plasma sample for methadone and EDDP. In this case, both the SiNP array and the DIOS surface performed equally well with no statistical difference in S/N for EDDP (n = 3, unpaired t-test). For methadone however, DIOS provided no signal while the SiNP array provided a S/N of 8.5 (n = 3). It is worth noting that the difference in S/N magnitude between B and C is attributed to the fact that EDDP and methadone accumulate at much higher concentrations in urine compared to blood plasma or saliva.51,53 Finally, Fig. 10D depicts the detection of EDDP and methadone in saliva. In this case, the DIOS surface showed a slightly higher S/N than SiNPs but the difference was not statistically significant (n = 3, unpaired t-test). Furthermore, the signal generated for methadone was equivalent for both surfaces (n = 3, unpaired t-test). The results are summarized and tabulated in Table S2, while the SALDI mass spectra generated from the SiNP array for the clinical samples are listed in Fig. S5.†
Conclusions
We report on the fabrication of ordered SiNP arrays with tuneable length, diameter and porosity. These arrays were successfully used for the detection of peptides with molecular weights of up to 3147 Da in SALDI-MS experiments. Furthermore, methadone was detected without the need for drug extraction and derivatization with a LOD of 32 ng mL−1. A trend between S/N ratio and SiNP length was observed where shorter lengths resulted in increased S/N values for both methadone and peptide analysis. We also showed that the increase in porosity of the SiNPs resulted in a decrease in methadone signal intensity. Additionally, we demonstrated the successful detection of methadone and its metabolite EDDP from real clinical samples of blood plasma, saliva, and urine. We also compared between the analytical performance of the ordered SiNP arrays and a conventional DIOS substrate in terms of S/N. In summary, we believe that ordered SiNP arrays fabricated by MACE are effective SALDI-MS substrates and hold strong potential for high-throughput analysis and quantification of small molecules in applications ranging from drug compliance, to forensics, pharmacology and environmental testing.Acknowledgements
This work was performed (in part) at the South Australian node of the Australian National Fabrication Facility under the National Collaborative Research Infrastructure Strategy to provide nano and microfabrication facilities for Australia's researchers. We would like to thank Marc Cirera for providing us with the schematic representation of the fabrication process shown in Fig. 1. NHV thanks the Alexander von Humboldt Foundation for a Fellowship. Clinical samples were kindly provided by Dr Chris Della Vedova and Mr Declan Neldner from the School of Pharmacy and Medical Sciences, University of South Australia (Ethics protocol number: 0000021954). We would like to acknowledge Forensic Science South Australia (FSSA) and the Australian Research Council (ARC LP110200446) for financial support.Notes and references
- E. J. Want, G. O'Maille, C. A. Smith, T. R. Brandon, W. Uritboonthai, C. Qin, S. A. Trauger and G. Siuzdak, Anal. Chem., 2005, 78, 743–752 CrossRef PubMed.
- M. A. Hashir, G. Stecher, R. Bakry, S. Kasemsook, B. Blassnig, I. Feuerstein, G. Abel, M. Popp, O. Bobleter and G. K. Bonn, Rapid Commun. Mass Spectrom., 2007, 21, 2759–2769 CrossRef CAS PubMed.
- D. S. Smith and S. A. Eremin, Anal. Bioanal. Chem., 2008, 391, 1499–1507 CrossRef CAS PubMed.
- E. Strain, G. E. Bigelow, I. A. Liebson and M. L. Stitzer, JAMA, J. Am. Med. Assoc., 1999, 281, 1000–1005 CrossRef CAS.
- S. H. Schnoll and M. F. Weaver, Am. J. Addict., 2003, 12, S27–S35 CrossRef.
- D. P. Alford, P. Compton and J. H. Samet, Ann. Intern. Med., 2006, 144, 127–134 CrossRef CAS.
- V. P. Dole, JAMA, J. Am. Med. Assoc., 1988, 260, 5 Search PubMed.
- E. C. Strain, M. L. Stitzer, I. A. Liebson and G. E. Bigelow, Ann. Intern. Med., 1993, 119, 23–27 CrossRef CAS.
- S. J. Mulé, M. L. Bastos and D. Jukofsky, Clin. Chem., 1974, 20, 243–248 Search PubMed.
- F. Lancelin, L. Kraoul, N. Flatischler, S. Brovedani-Rousset and M.-L. Piketty, Clin. Chem., 2005, 51, 2176–2177 CAS.
- B. A. Goldberger, A. G. Darraj, Y. H. Caplan and E. J. Cone, J. Anal. Toxicol., 1998, 22, 526–530 CrossRef CAS PubMed.
- O. H. Drummer, Clin. Biochem. Rev., 2006, 27, 147–159 Search PubMed.
- M. Karas and F. Hillenkamp, Anal. Chem., 1988, 60, 2299–2301 CrossRef CAS.
- K. Tanaka, H. Waki, Y. Ido, S. Akita, Y. Yoshida, T. Yoshida and T. Matsuo, Rapid Commun. Mass Spectrom., 1988, 2, 151–153 CrossRef CAS.
- J. Gobom, M. Schuerenberg, M. Mueller, D. Theiss, H. Lehrach and E. Nordhoff, Anal. Chem., 2000, 73, 434–438 CrossRef.
- C. Fenselau, Anal. Chem., 1997, 69, 661A–665A CrossRef CAS.
- J. Koomen, B. Ruotolo, K. Gillig, J. McLean, D. Russell, M. Kang, K. Dunbar, K. Fuhrer, M. Gonin and A. Schultz, Anal. Bioanal. Chem., 2002, 373, 612–617 CrossRef CAS PubMed.
- H. Räder and W. Schrepp, Acta Polym., 1998, 49, 272–293 CrossRef.
- G. Montaudo, F. Samperi and M. S. Montaudo, Prog. Polym. Sci., 2006, 31, 277–357 CrossRef CAS PubMed.
- M. W. Duncan, G. Matanovic and A. Cerpa-Poljak, Rapid Commun. Mass Spectrom., 1993, 7, 1090–1094 CrossRef CAS PubMed.
- Z. Shen, J. J. Thomas, C. Averbuj, K. M. Broo, M. Engelhard, J. E. Crowell, M. G. Finn and G. Siuzdak, Anal. Chem., 2000, 73, 612–619 CrossRef.
- J. Yao, J. R. Scott, M. K. Young and C. L. Wilkins, J. Am. Soc. Mass Spectrom., 1998, 9, 805–813 CrossRef CAS.
- S. Okuno, R. Arakawa, K. Okamoto, Y. Matsui, S. Seki, T. Kozawa, S. Tagawa and Y. Wada, Anal. Chem., 2005, 77, 5364–5369 CrossRef CAS PubMed.
- N. H. Finkel, B. G. Prevo, O. D. Velev and L. He, Anal. Chem., 2005, 77, 1088–1095 CrossRef CAS.
- K. Murray Kermit, K. Boyd Robert, N. Eberlin Marcos, G. J. Langley, L. Li and Y. Naito, in Pure Appl. Chem., 2013, vol. 85, p. 1515 Search PubMed.
- A. M. Dattelbaum and S. Iyer, Expert Rev. Proteomics, 2006, 3, 153–161 CrossRef CAS PubMed.
- T. T. Hoang, Y. Chen, S. W. May and R. F. Browner, Anal. Chem., 2004, 76, 2062–2070 CrossRef CAS PubMed.
- Y. Chen, M. C. Sullards, T. T. Hoang, S. W. May and T. M. Orlando, Anal. Chem., 2006, 78, 8386–8394 CrossRef CAS PubMed.
- S. Xu, Y. Li, H. Zou, J. Qiu, Z. Guo and B. Guo, Anal. Chem., 2003, 75, 6191–6195 CrossRef CAS PubMed.
- Y.-S. Lin and Y.-C. Chen, Anal. Chem., 2002, 74, 5793–5798 CrossRef CAS.
- C.-T. Chen and Y.-C. Chen, Rapid Commun. Mass Spectrom., 2004, 18, 1956–1964 CrossRef CAS PubMed.
- G. l. Piret, H. Drobecq, Y. Coffinier, O. Melnyk and R. Boukherroub, Langmuir, 2009, 26, 1354–1361 CrossRef PubMed.
- C. Wang, J. M. Reed, L. Ma, Y. Qiao, Y. Luo, S. Zou, J. J. Hickman and M. Su, J. Phys. Chem. C, 2012, 116, 15415–15420 CAS.
- Y. Xiao, S. T. Retterer, D. K. Thomas, J.-Y. Tao and L. He, J. Phys. Chem. C, 2009, 113, 3076–3083 CAS.
- R. A. Kruse, X. Li, P. W. Bohn and J. V. Sweedler, Anal. Chem., 2001, 73, 3639–3645 CrossRef CAS.
- A. K. Kalkan and S. Bae, J. Appl. Phys., 2000, 88, 555 CrossRef PubMed.
- E. P. Go, J. V. Apon, G. Luo, A. Saghatelian, R. H. Daniels, V. Sahi, R. Dubrow, B. F. Cravatt, A. Vertes and G. Siuzdak, Anal. Chem., 2005, 77, 1641–1646 CrossRef CAS PubMed.
- Y. Chen and A. Vertes, Anal. Chem., 2006, 78, 5835–5844 CrossRef CAS PubMed.
- B. N. Walker, J. A. Stolee, D. L. Pickel, S. T. Retterer and A. Vertes, J. Phys. Chem. C, 2010, 114, 4835–4840 CAS.
- K. Peng, M. Zhang, A. Lu, N.-B. Wong, R. Zhang and S.-T. Lee, Appl. Phys. Lett., 2007, 90, 163123 CrossRef PubMed.
- C. Chiappini, X. Liu, J. R. Fakhoury and M. Ferrari, Adv. Funct. Mater., 2010, 20, 2231–2239 CrossRef CAS PubMed.
- T. Gao, J. Gao and M. J. Sailor, Langmuir, 2002, 18, 9953–9957 CrossRef CAS.
- T. Guinan, M. Ronci, H. Kobus and N. H. Voelcker, Talanta, 2012, 99, 791–798 CrossRef CAS PubMed.
- M. Ronci, D. Rudd, T. Guinan, K. Benkendorff and N. H. Voelcker, Anal. Chem., 2012, 84, 8996–9001 CAS.
- T. R. Northen, H.-K. Woo, M. T. Northen, A. Nordström, W. Uritboonthail, K. L. Turner and G. Siuzdak, J. Am. Soc. Mass Spectrom., 2007, 18, 1945–1949 CrossRef CAS PubMed.
- S. Baumann, U. Ceglarek, F. Georg Martin and J. Lembcke, et al. , Clin. Chem., 2005, 51, 973–980 CAS.
- H. Wei, K. Nolkrantz, D. H. Powell, J. H. Woods, M.-C. Ko and R. T. Kennedy, Rapid Commun. Mass Spectrom., 2004, 18, 1193–1200 CrossRef CAS PubMed.
- N. Yao, H. Chen, H. Lin, C. Deng and X. Zhang, J. Chromatogr. A, 2008, 1185, 93–101 CrossRef CAS PubMed.
- N. Shenar, S. Cantel, J. Martinez and C. Enjalbal, Rapid Commun. Mass Spectrom., 2009, 23, 2371–2379 CrossRef CAS PubMed.
- L. Sundar and F. Rowell, Analyst, 2014, 139, 633–642 RSC.
- A. M. Bermejo, A. C. S. Lucas and M. J. Tabernero, J. Anal. Toxicol., 2000, 24, 70–72 CrossRef CAS.
- Y.-C. Hsu, B.-G. Chen, S.-C. Yang, Y.-S. Wang, S.-P. Huang, M.-H. Huang, T.-J. Chen, H.-C. Liu, D.-L. Lin, R. Liu and A. W. Jones, Anal. Bioanal. Chem., 2013, 405, 3921–3928 CrossRef CAS PubMed.
- G. Nadia De and F. Nadia, Forensic Toxicol., 2013, 31, 347–350 CrossRef.
- R. A. Totah, P. Sheffels, T. Roberts, D. Whittington, K. Thummel and E. D. Kharasch, Anesthesiology, 2008, 108, 363–374, DOI:10.1097/ALN.0b013e3181642938.
- T. Kelly, P. Doble and M. Dawson, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 814, 315–323 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1, S2, S3, S4, and S5 in addition to Tables S1 and S2. See DOI: 10.1039/c4an01391c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |