 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Puzzling over protein cysteine phosphorylation – assessment of proteomic tools for S-phosphorylation profiling
A. K.
Buchowiecka
*
Institute of Technical Biochemistry, Lodz University of Technology, 90-924 Lodz, Poland. E-mail: alicja.buchowiecka@p.lodz.pl; Fax: +48-42-631-34-02; Tel: +48-42-631-34-95
First published on 10th June 2014
Abstract
Cysteine phosphorylation has recently been discovered in both prokaryotic and eukaryotic systems, and is thought to play crucial roles in signaling and regulation of cellular responses. This article explores the topics of chemical stability of this type of structural modification and the resulting issues regarding affinity enrichment of S-phosphopeptides and their mass spectrometry-based detection in the course of general proteomics studies. Together, this work suggests that the current advances in phosphoproteomic methodologies provide adequate tools for investigating protein cysteine phosphorylation and appear to be immediately available for practical implementation. The article provides useful information necessary for designing experiments in the emerging cysteine phosphoproteomics. The examples of methodological proposals for S-linked phosphorylation detection are included herein in order to stimulate development of new approaches by the phosphoproteomic community.
 A. K. Buchowiecka | Alicja Buchowiecka is an Assistant Professor at the Institute of Technical Biochemistry of the Lodz University of Technology in Poland. She completed her pre-doctoral fellowship at the Pasteur Institute in Paris, where she studied advanced electrophoretic techniques. After receiving her doctoral degree in biotechnology, she joined the Clinical Breast Care Project at the Windber Research Institute in Pennsylvania, working as a Staff Scientist in a proteomic group for two years. Her work in Poland resulted in a three-year government grant to develop a MS-based method for detecting S-linked glycosylation. Dr Buchowiecka is currently working on selected protein cysteine PTMs using newly developed methods, advanced materials, and MS analytics. |
Introduction
The last decade has witnessed dynamic progress in proteomic studies. A global view of the advanced technologies that drive development of new research methodologies and that enable impressive achievements in the field is given in the recent review articles.1–3 A comprehensive evaluation of the current challenges in large-scale phosphoproteomics is presented by Engholm-Keller and Larsen.4 To date, the majority of phosphoproteomic studies are focused on various aspects of serine, threonine and tyrosine O-phosphorylation that dominate signaling and biological regulation in eukaryotic organisms. In vivo phosphorylation of side chains of six other amino acids (His, Lys, Arg, Asp, Glu and Cys) is much less explored due to the chemical lability of the respective peptidyl phosphoramidates,5–7 peptidyl acyl phosphates and peptidyl phosphorothiolates.Cysteine is one of the least abundant amino acids incorporated into proteins. Studies done on single-residue compositions of proteomes revealed an average cysteine frequency of around 2% for eukaryotic organisms, while for Eubacteria and Archea frequency of ca. 1%.8 It was also shown that on average, approximately 70% of the reduced thiol equivalents within cells are those originating from proteins.9 It is thought that the abundance of cysteine is governed by its function in proteins and that the high reactivity of thiol groups shapes distribution and topology of cysteine residues in protein structures. Isolated cysteine accounts for 55–60% of all protein cysteine residues, but due to their anomalous hydrophobic behavior, cysteine residues are infrequently found on protein surfaces.10 Exposed protein thiols are potentially accessible for redox reactions9,11,12 and, presumably, only a certain fraction of them can experience a reversible, enzymatic S-phosphorylation.
Cysteine phosphorylation is important in the context of cysteine-dependent protein phosphatases (CDPs) which belong to a subfamily of protein tyrosine phosphatases (PTPs) and catalyze the hydrolysis of phosphoester bonds via the formation of a phospho-cysteine intermediate. Unlike the so-called dual specificity phosphatases, which catalyze dephosphorylation of both Tyr and Ser/Thr residues in proteins, CDPs exhibit specificity exclusively for phosphotyrosine-containing substrates. The sulfur atom of the reactive cysteine present in the CDP catalytic center plays the role of the phosphate group acceptor, thus a cysteinyl-phosphate intermediate is formed. In the second step of catalysis, the cysteinyl-phosphate intermediate undergoes nucleophilic attack by a water molecule. Due to the enzyme active site architecture, the phosphate is prevented from being transferred to acceptors other than water. More information on a fundamental role of Cys-dependent PTPs in the regulation of cell signaling is accessible in the extensive review by Nicholas K. Tonks.13 The author presents substantial body of data emphasizing the contribution of phosphatases to signal transduction networks. The work explains that kinases and phosphatases are specific and competing regulators of signaling that are coordinated to determine the response to a physiological stimulus.13
In 2012 Sun et al.14 detected phosphorylation that occurs on a reactive cysteine residue conserved in several global transcriptional regulators that control a broad spectrum of genes in the human pathogen Staphylococcus aureus. The authors have shown that this reversible posttranslational modification (PTM) is critical in regulating expression of virulence determinants and bacterial resistance to antibiotics. Moreover, these studies also suggest that cysteine phosphorylation, previously considered a rare modification, could be more prevalent in nature and may play significant previously unappreciated roles in biological regulation in various organisms. Another group of researchers, reexamining PTMs of recombinantly expressed human cystic fibrosis transmembrane conductance regulator, detected in its sequence S-phosphocysteine.15
These new findings extend a short list of known S-phosphorylated proteins (RESID ID AA0034) and provoke questions regarding the possibility of concurrent detection of S- and O-phosphorylation in large-scale phosphoproteomic studies. What factors interfere with the identification of S-linked phosphorylation, and which ones are favorable? This article suggests that through current advances in phosphoproteomic methodologies, adequate tools for detecting protein cysteine phosphorylation appear to be accessible and ready for implementation.
Stability
O-phosphorylated peptides are quite durable under a broad range of pH values, and their stability can be attributed to the resistance of the phosphate group to hydrolysis or beta-elimination reactions. However, under specific conditions, O-phosphopeptides are susceptible to both of these transformations.16–19 In turn, some chemical properties of S-phosphorylated peptides can be inferred from early studies involving several low molecular S-substituted phosphorothioic acids.20 In general, this class of compounds is very stable at pH >7 up to 12,21,22 but is prone to phosphothiolester bond hydrolysis with its maximum rate at a pH of 2–4. At pH <2 hydrolysis slows down according to a mechanism discussed by Åkerfeldt.20 In another study,22 it was observed that S-phosphorylated cystamine and S-phosphorylated mercaptopropionic acids undergo hydrolysis at a maximal rate at a pH of 3–4, reaching a half-life of about 15 min. at 37 °C. Moreover, thiophilic metal ions such as Ag I and Cu II were efficient promoters of phosphothiolester bond hydrolysis.22 Until now, only two examples of kinetic studies on hydrolysis of S-phosphorylated peptides have been described. In 1988 Pas and Robillard23 demonstrated that at pH 4 the KIIVApCDAGMGSSAM peptide loses a phosphate group with a maximum rate comparable to the kinetics of phosphoenolthiopyruvate hydrolysis (half-life 30 min. at 25 °C). Later Meins et al.24 applied the same procedure to study chemical dephosphorylation of the peptide ENITNLDApCITR. Both peptides originated from Escherichia coli enzymes involved in carbohydrate transport by the phosphoenolpyruvate-dependent phosphotransferase system.Affinity enrichment
Successfully navigating the experimental steps of phosphopeptide analysis beginning with protein lysate and ending with the identification of phosphopeptides is a technically demanding task. Generally, in a typical large-scale phosphoproteomics complex protein samples are first digested with selected proteinase(s) and are then subjected to an obligatory affinity enrichment.25,26 The sample preparation process usually involves lengthy operations that are performed at both low and high pH and may run as long as two days.27 Most frequently, metal oxide affinity chromatography is used for the specific catch and release of phosphopeptides. The enrichment process occurs by electrostatic interactions between charged peptides and the charged solid support surface.28 However, the electrostatic-mediated deposition does not exclude phosphate–metal cation covalent interactions.29–32 For example, detailed studies on stability of the dodecyl phosphate films on titanium dioxide demonstrated enhanced hydrolysis of phosphoester bonds at an alkaline pH, possibly due to phosphate covalent binding.33 In general, charge transfer from phosphate oxygen atoms to coordinately unsaturated titanium cations on the metal oxide surface29,32 increases the electrophilicity of the phosphorus atom, making it more prone to a nucleophilic attack by a water molecule or by a hydroxide ion.O- and S-phosphopeptides that are covalently linked through the phosphate group to metal cation centers by formation of bridging bidentate complexes can be compared to neutral phosphonothioates of the general formula RP(O)(OR1)(SR2). Detailed studies on alkaline hydrolysis of these compounds show that P–S bond cleavage is favored over P–O bond cleavage by a factor of approximately 5.34,35 Interestingly, substantial hydrolytic instability of some O-phosphopeptides during a quick ammonia elution from titanium dioxide was recently demonstrated by Eickner et al.36 and similar conclusions can be drawn from the article by Cheng et al.37 Moreover, some degree of alkaline hydrolysis of O-phosphopeptides is commonly observed on lanthanide-based affinity materials as exemplified in several studies.38–41 The hydrolysis taking place even in a slightly alkaline or slightly acidic milieu can be connected to catalytic activity of lanthanide ions towards phosphate esters.17,42 However, in the following MALDI experiments36 attention should be paid on distinguishing between the MS-signals from hydrolytically dephosphorylated peptides and the identical signals that may originate from a metastable loss of the phosphate group (−80 Da). Moreover, there is also possibility of partial dephosphorylation of phosphopeptides due to their acid hydrolysis with common MALDI matrices.43,70
Together, these findings suggest that ammonia elution performed under conditions that hydrolytically affect O-phosphopeptides, may cause more extensive, if not complete hydrolysis of S-linked phosphopeptides. Thus, one can expect the co-occurrence of phosphopeptides and cysteine-containing peptides in the eluate from materials that do not display affinity for thiol groups, unlike notably Fe III-IMAC resins.18 Nonetheless, to specify conditions for S-phosphopeptide survival in the recovery step, enrichment experiments with the use of model S-phosphorylated peptides are recommended. Such compounds can be obtained by Michael addition of thiophosphoric acid44 to dehydroalanine-containing proteins and peptides.45–47
Several articles regarding phosphopeptide enrichment on innovative supports offer fast techniques, which should not be harmful to a phosphothiolester link.48–52 Some of these novel affinity materials can work at almost neutral milieu, using nonvolatile buffers for loading and elution processes. This necessitates subsequent removal of salts. Recently, effective methods for desalting phosphopeptides under both neutral and alkaline conditions as well as very fast desalting techniques under acidic conditions have been considerably improved.53–55 It ought to be mentioned that unavoidable steps involving low-pH operations, if limited to very short time duty, could also be incorporated into enrichment technology.
Detection
This brief summary outlines procedural conditions required to make co-enrichment for O- and S-phosphopeptides possible. Presented below is the rudimentary concept of an MS-based approach for detection of presumably infrequent S-linked phosphopeptide among abundant O-phosphopeptides, which is strongly supported by information garnered from real-world studies (Fig. 1).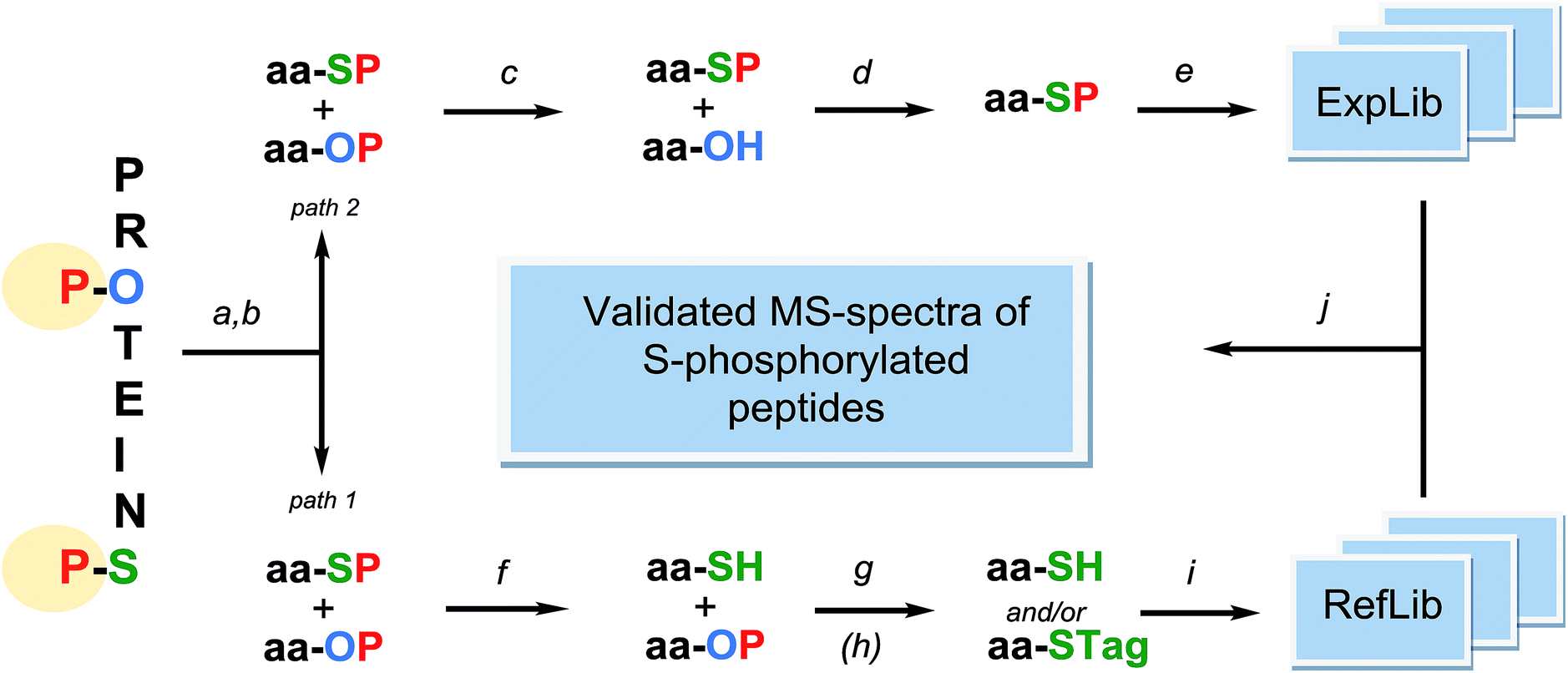 | ||
| Fig. 1 The proposed workflow of mass spectrometry-based detection of S-phosphorylated peptides. PROTEINS – a protein mixture potentially containing O-phosphorylated Ser/Thr/Tyr residues and S-phosphorylated Cys residues; all possibly present labile cysteine PTMs are first reduced/hydrolyzed and then the freed thiol groups are protected by S-alkylation; aa-SP – S-phosphorylated peptides; aa-OP – O-phosphorylated peptides; a – proteolytic digestion under alkaline conditions; b – phosphopeptide affinity co-enrichment; c – hydrolysis by acid phosphatase at pH 7. d – phosphopeptide affinity enrichment (repeated step b); e – tandem MS spectra acquisition; construction of an experimental spectral library ExpLib; f – acid hydrolysis; g – cysteinyl peptides capture on thiol specific materials; h – cysteinyl peptide S-tagging (recommended). i – construction of a reference spectral library RefLib of cysteinyl and S-tagged peptides; calculation of semi-empirical spectra for S-phosphorylated peptides; j – detection of S-phosphorylated peptides by a spectral library approach using ExpLib and RefLib. | ||
It demonstrates selection of currently available research tools to obtain anticipated results, which seem reliable based on certain experimental data that are cited herein.
The mixture of O- and S-phosphopeptides co-enriched from a digest of fully reduced and thoroughly S-protected protein sample56,57 should be split into two equal parts and then subjected to separate dephosphorylation procedures (path 1 and path 2 in Fig. 1). One half of the sample is acid-hydrolyzed in order to dephosphorylate cysteine residues and to generate cysteine-containing peptides, which are further isolated on thiol specific resins56,58 (path 1). The recovered peptides are subjected to LC-MS/MS analysis and the obtained identifiable spectra are then used for building a reference spectral library (RefLib). Optional but recommended S-tagging of the cysteine-containing peptides59,60 can be performed to improve their fragmentation pattern and further sequencing by database searching or de novo sequencing methods can be performed. Alternatively, real MS/MS-spectra of S-tagged and cysteine-containing peptides can be replaced by computationally predicted semi-empirical spectra of respective S-phosphorylated peptides, by analogy to the recently proposed approach of O-phosphopeptide spectra prediction.61,62 The reference library being “extracted” from the analyzed sample covers sequences of peptides potentially bearing S-linked phosphorylation. At the final stage of the analysis, this reference library will serve in the detection and identification of intact S-phosphopeptides from the second half of the analyte, by performing spectrum to spectrum matching.63
The second part of co-enriched O- and S-phosphopeptides (path 2 in Fig. 1) is treated with an appropriate, non-specific acid phosphatase64 at a pH of about 7, which causes a loss of phosphate groups by O-phosphopeptides. Enzymatic dephosphorylation of phosphoproteins by cocktails containing acid phosphatases65 or with acid phosphatases alone66,67 has been described before. Subsequently, most likely intact68 S-phosphorylated peptides are recovered from the mixture by repeating the harmless enrichment procedure. A conceptually similar approach has been implemented in the study of Hu and Tao69 aiming at isolating low abundant phosphotyrosine-containing peptides from a complex mixture of phosphopeptides. Under highly alkaline conditions the beta-elimination reaction removes the phosphate from phosphoserine and phosphothreonine residues while phosphotyrosine residues remain unaffected by hydrolysis.
After enzymatic depletion of interfering O-phosphopeptides, targeted S-phosphopeptides need to be fractionated prior to tandem mass spectrometry. In typical phosphoproteomic experiments, a positive ion ESI-MS/MS is preceded by reversed phase chromatography with low pH elution, which would be destructive for S-phosphorylated structures. Yet, in regard to their predicted half-life, rapid acidic separation14,15 might be acceptable for highly enriched samples. The actual rate of hydrolysis of S-phosphopeptides during their separation on the RP-LC column may actually be much lower than hydrolysis in a homogenous water solution, analogous to the variable context-dependent stability of N-phosphohistidine containing peptides. The latter undergo rapid dephosphorylation at pH <5 (half-life ca. 20 min) but are stable when stored on an acidic C18 RP column can thus be detected by ESI-MS.70 An alternative 2D-LC system involving reversed phase fractionation at alkaline pH71,72 followed by acidic reversed phase HPLC with a short run time appears as a more suitable approach. S-phosphopeptides that survive the LC-ESI step have a chance to be selected for fragmentation in data dependent acquisition (DDA) mode.14,15 However data independent acquisition (DIA), while delivering mixture spectra of co-fragmented peptides, seems to be a method of choice providing higher dynamic range of signal detection. Regardless of the MS/MS acquisition method, further identification of intact S-phosphorylated peptide spectra collected in experimental library (ExpLib) could be facilitated by performing spectrum-to-spectrum matching.61,63 Establishing the reference library originating from the analyzed sample as described above, would enable the use of spectral library search workflow. The outlined approach is fundamentally similar to the methodology of reference-facilitated validation of phosphopeptides, demonstrated by Suni et al.62 and Imanishi et al.73
Final remarks and conclusions
The conceptual discussion outlined above is based heavily on previous empirical accomplishments.74,75 A methodology similar to the one presented herein was applied for the MS-based detection and localization of the S-glycosylation sites in a model mixture of chemically modified hen egg white lysozyme.Briefly, one part of the lysozyme derivatives that contained on average three S-glucosyl residues indeterminately distributed over eight S-protected cysteine residues per molecule was subjected to S-glycosidic bond hydrolysis in a mild alkaline milieu.74,75 This reaction transformed S-glycosylated proteins into respective proteins containing freed thiol groups. After S-tagging and trypsinolysis the resultant peptides were analyzed by complementary MS/MS techniques. The spectra of S-tagged sequences incorporating up to three modified cysteine residues per single peptide were selected for constructing the reference spectral library assigned as “a vicarious library”. The second part of S-glycosylated lysozyme derivatives after trypsinolysis followed by the same tandem MS analyses provided spectra of respective S-glycosylated peptides. The complexity of each reference spectrum was reduced by careful selection of appropriate productions/tag loss ions. This approach allowed detection of the respective spectral pairs of S-tagged/S-glycosylated peptide (of the same sequence) as well as further determination of a glycan size.74,75 In the light of the unambiguously confirmed occurrence of S-glycosylated bacteriocins76,77 the designed and experimentally verified methodology can be eventually tested on native proteomes. It is worthwhile to mention that thoughtful attention to possible co-detection of mono-ADP-S-ribosylation and S-phosphorylation was also given in those studies.
In conclusion, detection of S-linked phosphopeptides by MS-based phosphoproteomics can be successful only if appropriate experimental conditions are used. In the course of routine workflows certain pitfalls jeopardizing the survival of peptidyl phosphorothiolates are to be expected. Prolonged acidic treatments, ammonia elution from commonly used metal oxide materials, and sample contamination with thiophilic metal ions may cause dephosphorylation of S-phosphopeptides. Thus, the possible appearance of cysteinyl peptides in a sample of enriched phosphopeptides may indicate the presence of their S-phosphorylated precursors in the initial protein lysate.
Recognition of mechanisms that lie behind hydrolytic split of the P–S bonding calls for an analytical approach that is relatively harmless for the labile analyte. Detrimental acidic steps should be limited to a few minutes, or rather replaced by operations performed under pH conditions close to neutral. Standard affinity enrichment materials can be exchanged for new ones that insignificantly affect the density of negative charge on the complex-forming phosphate group, therefore minimizing phosphothiolester bond susceptibility to nucleophilic split in an alkaline environment. The final stage of workflow, MS-spectra acquisition in synergy with bioinformatic analysis, can be successfully performed by the use of appropriately selected methods currently applied in phosphoproteomics.
Some concerns in relation to reactivity of S-phosphorylated peptides during isolation or detection processes can be experimentally resolved by the use of model peptides that are easily synthesizable.
Acknowledgements
I gratefully thank Katarzyna M. Skrzypczynska from the University of California, San Francisco, for her critical reading of this manuscript and constructive suggestions.Notes and references
- Y. Zhang, B. R. Fonslow, B. Shan, M.-C. Baek and J. R. Yates III, Chem. Rev., 2013, 113, 2343 CrossRef CAS PubMed.
- M. A. Jünger and R. Aebersold, WIREs Dev. Biol., 2014, 3, 83 CrossRef PubMed.
- P. P. Roux and P. Thibault, Mol. Cell. Proteomics, 2013, 12, 3453 CAS.
- K. Engholm-Keller and M. R. Larsen, Proteomics, 2013, 13, 910 CrossRef CAS PubMed.
- P. V. Attwood, M. J. Piggott, X. L. Zu and P. G. Besant, Amino Acids, 2007, 32, 145 CrossRef CAS PubMed.
- P. G. Besant, P. V. Attwood and M. J. Piggott, Curr. Protein Pept. Sci., 2009, 10, 536 CrossRef CAS.
- J.-M. Kee and T. W. Muir, ACS Chem. Biol., 2012, 7, 44 CrossRef CAS PubMed.
- I. Pe'er, C. E. Felder, O. Man, I. Silman, J. L. Sussman and J. S. Beckmann, Proteins, 2004, 54, 20 CrossRef PubMed.
- R. E. Hansen, D. Roth and J. R. Winther, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 422 CrossRef CAS PubMed.
- S. M. Marino and V. N. Gladyshev, J. Mol. Biol., 2010, 404, 902 CrossRef CAS PubMed.
- R. Requejo, T. R. Hurd, N. J. Costa and M. P. Murphy, FEBS J., 2010, 277, 1465 CrossRef CAS PubMed.
- C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633 CrossRef CAS PubMed.
- N. K. Tonks, FEBS J., 2013, 280, 346 CrossRef CAS PubMed.
- F. Sun, Y. Ding, Q. Ji, Z. Liang, X. Deng, C. C. L. Wong, C. Yi, L. Zhang, S. Xie, S. Alvarez, L. M. Hicks, C. Luo, H. Jiang, L. Lan and C. He, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15461 CrossRef CAS PubMed.
- M. McClure, L. Wilson, M. Ray, L. J. DeLucas, S. Rowe, X. Wu, Q. Dai, J. S. Hong, E. J. Sorscher, J. C. Kappes and S. Barnes, ASMS Conference on Mass Spectrometry and Allied Topics, 2012, TP05 Poster 113, http://app.imswift.com/asms2012/sessions/asms_3385.
- S. Sun, H. Ma, G. Han, R. Wu, H. Zou and Y. Liu, Rapid Commun. Mass Spectrom., 2011, 25, 1862 CrossRef CAS PubMed.
- N. W. Luedtke and A. Schepartz, Chem. Commun., 2005, 43, 5426 RSC.
- J. Thompson, S. R. Hart, C. Franz, K. Barnouin, A. Ridley and R. Cramer, Anal. Chem., 2003, 75, 3232 CrossRef.
- D. T. McLachlin and B. T. Chait, Anal. Chem., 2003, 75, 6826 CrossRef CAS PubMed.
- S. Åkerfeldt, Acta Chem. Scand., 1963, 17, 319 CrossRef PubMed.
- S. Åkerfeldt, Acta Chem. Scand., 1960, 14, 1980 CrossRef PubMed.
- S. Åkerfeldt, Acta Chem. Scand., 1961, 15, 575 CrossRef PubMed.
- H. H. Pas and G. T. Robillard, Biochemistry, 1988, 27, 5835 CrossRef CAS.
- M. Meins, P. Jenö, D. Müller, W. J. Richter, J. P. Rosenbusch and B. Erni, J. Biol. Chem., 1993, 268, 11604 CAS.
- L. Beltran and P. R. Cutillas, Amino Acids, 2012, 43, 1009 CrossRef CAS PubMed.
- J. Fíla and D. Honys, Amino Acids, 2012, 43, 1025 CrossRef PubMed.
- H. Zhou, M. Ye, J. Dong, E. Corradini, A. Cristobal, A. J. R. Heck and H. S. Mohammed, Nat. Protoc., 2013, 8, 461 CrossRef CAS PubMed.
- I. Eriksson, K. Edwards, A. Hagfeldt and V. A. Hernández, J. Phys. Chem. B, 2013, 117, 2019 CrossRef PubMed.
- V. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef.
- C. Jin, Y. Tang, X. Y. Fan, X. T. Ye, X. L. Li, K. Tang, Y. F. Zhang, A. G. Li and Y. J. Yang, Toxicol. Ind. Health, 2013, 29, 235 CrossRef PubMed.
- B. Xu, C. Chen, F. Wang, Y. Bian, K. Cheng, H. Qin, C. Song, J. Zhu, J. Liu, M. Ye and H. Zou, Anal. Methods, 2013, 5, 2939 RSC.
- J. Wan, K. Qian, L. Qiao, Y. Wang, J. Kong, P. Yang, B. Liu and C. Yu, Chem.–Eur. J., 2009, 15, 2504 CrossRef CAS PubMed.
- S. A. Holt, A. P. Le Brun, A. R. J. Nelson and J. H. Lakey, RSC Adv., 2013, 3, 2581 RSC.
- Y.-C. Yang, F. J. Berg, L. L. Szafraniec, W. T. Beaudry, C. A. Bunton and A. Kumar, J. Chem. Soc., Perkin Trans. 2, 1997, 2, 607 RSC.
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345 CrossRef CAS PubMed.
- T. Eickner, S. Mikkat, P. Lorenz, M. Sklorz, R. Zimmermann, H. J. Thiesen and M. O. Glocker, Eur. J. Mass Spectrom., 2011, 17, 507 CrossRef CAS PubMed.
- G. Cheng, Z.-G. Wang, Y.-L. Liu, J.-L. Zhang, D.-H. Sun and J.-Z. Ni, ACS Appl. Mater. Interfaces, 2013, 5, 3182 CAS.
- M. R. Mirza, M. Rainer, C. B. Messner, Y. Güzel, D. Schemeth, T. Stasyk, M. I. Choudhary, L. A. Huber, B. M. Rode and G. K. Bonn, Analyst, 2013, 138, 2995 RSC.
- B. Fatima, M. Najam-ul-Haq, F. Jabeen, S. Majeed, M. N. Ashiq, S. G. Musharraf, M. A. Shad and G. Xu, Analyst, 2013, 138, 5059 RSC.
- Y. Sun and H.-F. Wang, J. Chromatogr. A, 2013, 1316, 62 CrossRef CAS PubMed.
- G. Cheng, Y.-L. Liu, Z.-G. Wang, S.-M. Li, J.-L. Zhang and J.-Z. Ni, J. Mater. Chem. B, 2013, 1, 3661 RSC.
- B. Yoo and M. D. Pagel, Molecules, 2013, 18, 3894 CrossRef CAS PubMed.
- E. Remily-Wood, H. Dirscherl and J. M. Koomen, J. Am. Soc. Mass Spectrom., 2009, 20, 2106 CrossRef CAS PubMed.
- J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Angew. Chem., 2012, 124, 1871 CrossRef CAS; J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2012, 51, 1835 CrossRef PubMed.
- J. T. Andrew, S. R. Hart, C. Franz, K. Barnouin, A. Ridley and R. Cramer, Anal. Chem., 2003, 75, 3232 CrossRef.
- D. T. McLachlin and B. T. Chait, Anal. Chem., 2003, 75, 6826 CrossRef CAS PubMed.
- Y. Kyono, N. Sugiyama, M. Tomita and Y. Ishihama, Rapid Commun. Mass Spectrom., 2010, 24, 2277 CrossRef CAS PubMed.
- B. R. Fonslow, S. M. Niessen, M. Singh, C. C. L. Wong, T. Xu, P. C. Carvalho, J. Choi, S. K. Park and J. R. Yates III, J. Proteome Res., 2012, 11, 2697 CrossRef CAS PubMed.
- B. Iliuk, V. A. Martin, B. M. Alicie, R. L. Geahlen and W. A. Tao, Mol. Cell. Proteomics, 2010, 9, 2162 Search PubMed.
- E. Kinoshita-Kikuta, A. Yamada, C. Inoue, E. Kinoshita and T. Koike, OMICS: J. Integr. Biol., 2011, 1, 157 Search PubMed.
- M. Fischnaller, R. Bakry, R. M. Vallant, L. A. Huber and G. K. Bonn, Anal. Chim. Acta, 2013, 761, 92 CrossRef CAS PubMed.
- Z.-G. Wang, G. Cheng, Y.-L. Liu, J.-L. Zhang, D.-H. Sunc and J.-Z. Niab, J. Mater. Chem. B, 2013, 1, 1491 RSC.
- R. M. Vallant, Z. Szabo, S. Bachmann, R. Bakry, M. Najam-ul-Haq, M. Rainer, N. Heigl, C. Petter, C. W. Huck and G. K. Bonn, Anal. Chem., 2007, 79, 8144 CrossRef CAS PubMed.
- H. Chen, D. Qi, C. Deng, P. Yang and X. Zhang, Proteomics, 2009, 9, 380 CrossRef CAS PubMed.
- S. Zheng, X. Wang, J. Fu, X. Hu, X. Xiao, L. Huang, Y. Zhou and H. Zhong, Anal. Chim. Acta, 2012, 724, 73 CrossRef CAS PubMed.
- J. Paulech, N. Solis and S. J. Cordwell, Biochim. Biophys. Acta, 2013, 1834, 372 CrossRef CAS PubMed.
- J. Paulech, N. Solis, A. V. Edwards, M. Puckeridge, M. Y. White and S. J. Cordwell, Anal. Chem., 2013, 85, 3774 CrossRef CAS PubMed.
- A. Palani, J. S. Lee, J. Huh, M. Kim, Y. J. Lee, J. H. Chang, K. Lee and S. W. Lee, J. Proteome Res., 2008, 7, 3591 CrossRef CAS PubMed.
- V. C. Cotham, Y. Wine and J. S. Brodbelt, Anal. Chem., 2013, 85, 5577 CrossRef CAS PubMed.
- X. Qiao, R. Wang, H. Yan, T. Wang, Q. Zhao, L. Zhang and Y. Zhang, Rapid Commun. Mass Spectrom., 2014, 28, 256 CrossRef CAS PubMed.
- Y. Hu and H. Lam, J. Proteome Res., 2013, 12, 5971 CrossRef CAS PubMed.
- V. Suni, A. Maiolica, R. Aebersold, S. Y. Imanishi and G. L. Corthals, J. Am. Soc. Mass Spectrom., 2012, 23 Search PubMed , Supplement I, the 60th ASMS Conference on Mass Spectrometry and Allied Topics, TP469. http://link.springer.com/journal/13361/23/1/suppl/page/1.
- A. Guthals, J. D. Watrous, P. C. Dorrestein and N. Bandeira, Mol. BioSyst., 2012, 8, 2535 RSC.
- A. Anand and P. K. A. Srivastava, Appl. Biochem. Biotechnol., 2012, 167, 2174 CrossRef CAS PubMed.
- J. R. Smith, M. Olivier and A. S. Greene, Physiol. Genomics, 2007, 31, 357 CrossRef CAS PubMed.
- J. Szczepanowska, X. Zhang, C. J. Herring, J. Qin, E. D. Korn and H. Brzeska, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8503 CrossRef CAS.
- E. W. Bingha and H. M. Farrell Jr, Biochim. Biophys. Acta, 1976, 429, 448 CrossRef.
- H. Neumann, J. Biol. Chem., 1968, 243, 4671 CAS.
- L. Hu and W. A. Tao, Chin. J. Chromatogr., 2011, 29, 869 CAS.
- A. Sickmann and H. E. Meyer, Cold Spring Harb. Protoc., 2007 DOI:10.1101/pdb.prot4628.
- A. Tholey, H. Toll and C. G. Huber, Anal. Chem., 2005, 77, 4618 CrossRef CAS PubMed.
- X. S. Yue and A. B. Hummon, J. Proteome Res., 2013, 12, 4176 CrossRef CAS PubMed.
- S. Y. Imanishi, V. Kochin, S. E. Ferraris, A. de Thonel, H. M. Pallari, G. L. Corthals and J. E. Eriksson, Mol. Cell. Proteomics, 2007, 6, 1380 CAS.
- A. K. Buchowiecka, Polish Patent Application P-397240, 2011-12-05.
- A. K. Buchowiecka and J. Dębski, Acta Biochim. Pol., 2011, 58 Search PubMed Supplement 2, 183; 2nd Congress of Biochemistry and Cell Biology, Cracow, Poland, September 5th–9th, 2011, Oral presentation O18.2. http://www.actabp.pl/pdf/Supl2_11/S18.pdf.
- T. J. Oman, J. M. Boettcher, H. Wang, X. N. Okalibe and W. A. van der Donk, Nat. Chem. Biol., 2011, 7, 78 CrossRef CAS PubMed.
- H. Venugopal, P. J. B. Edwards, M. Schwalbe, J. K. Claridge, D. S. Libich, J. Stepper, T. Loo, M. L. Patchett, G. E. Norris and S. M. Pascal, Biochemistry, 2011, 50, 2748 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |