 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApoptotic pathways of U937 leukemic monocytes investigated by infrared microspectroscopy and flow cytometry
Giovanni
Birarda
ab,
Diana E.
Bedolla
a,
Elisa
Mitri
cd,
Sabrina
Pacor
c,
Gianluca
Grenci
de and
Lisa
Vaccari
*a
aElettra-Sincrotrone Trieste, SISSI Beamline, S.S. 14 Km 163.5, 34149 Basovizza, Trieste, Italy. E-mail: lisa.vaccari@elettra.eu
bLawrence Berkeley National Laboratory, 1 Cyclotron Rd, Berkeley, CA 94720, USA
cUniversità degli studi di Trieste, Piazzale Europa 1, 34100, Trieste, Italy
dCNR-IOM, TASC Laboratory, S.S. 14 km 163.5 Basovizza, 34149 Trieste, Italy
eMechanobiology Institute (MBI), National University of Singapore, T-Lab 5A Engineering Drive 1, Singapore
First published on 28th March 2014
Abstract
Apoptosis is a strictly regulated cell death mechanism that plays a pivotal role in the normal evolution of multicellular organisms. Its misregulation has been associated with many diseases, making its early and reliable detection a key point for modern cellular biology. In this paper, we propose the use of infrared microspectroscopy (IRMS) as a label-free methodology for the detection of apoptotic-related biochemical processes induced on U937 leukemic monocytes by serum starvation and CCCP-exposure. The spectroscopic results are in agreement with parallel Flow Cytometry (FC) experiments, where plasma membrane integrity and mitochondrial activity were assessed. Spectroscopic outcomes complement FC data and allow drawing a more complete picture of the apoptotic pathways. In particular, we established that the two apoptosis-inducing treatments, cell starvation and CCCP exposure, affect the cell cycle in a different way. With the former, cell death is preceded by a cell cycle arrest, whereas the latter causes an increased cell cycle progression. Spectral data demonstrate that for both conditions apoptosis proceeds through the accumulation of lipid droplets within cells. Moreover, we were able to establish a spectral marker for DNA condensation/fragmentation: the enhancement of the PhI band component centred at ∼1206 cm−1, which is more sensitive than the relative intensity of the PhII band to which phospholipids and carbohydrates also contribute significantly. In conclusion, we demonstrate that the intrinsic multi-parametric nature of IRMS and its application on cells under physiological conditions can be well exploited for the investigation of apoptotic pathways.
Introduction
Apoptosis is a naturally occurring event through which a cell ends its life by Programmed Cell Death (PCD). From the discovery of the phenomenon, first described as PCD in 1964 by Lockshin1 and later in 1972 as apoptosis by Kerr,2 many questions have found an answer but some remain still open.It has been established that apoptosis is the natural route followed for the elimination of cells that are aged, redundant, damaged or infected. It is a fundamental process for the control of embryonic development and for the maintenance of tissue homeostasis, so it is essential for the normal evolution of an organism.3 It is therefore not surprising that the shortcoming of this cellular function is implicated in a myriad of diseases.4 For example, an increased apoptosis has been related to neurodegenerative diseases,5 ischemic damage,6 and to autoimmune diseases7 while, its reduction or suppression leads to uncontrolled cell proliferation, associated with cancer8 and viral infection.9 The development of new drugs for modulating cellular apoptosis can open encouraging perspectives in the treatment of many diseases, and therefore apoptosis related research has increased substantially since the early 1990s.
The apoptotic process is characterised by a series of common morphological features,10 such as cell shrinkage, chromatin condensation and segregation at the nuclear membrane, plasma membrane blebbing and finally cellular breakdown into apoptotic bodies (APs). APs are small cellular fragments, delimited by plasma membranes expressing phosphatidylserine residues, where the potentially toxic or immunogenic cellular content of dying cells is encapsulated. The ultimate fate of APs is to be engulfed and destroyed by macrophages. The AP content (cytosol, condensed chromatin and cellular organelles) may induce an inflammatory response, as happens in necrotic cell death, where the cell is dismembered in an uncontrolled way.11
Morphological changes associated with PCD are related to the biochemical events occurring within cells. Even if signalling pathways may differ from each other depending on the triggering stimuli, two major apoptotic routes have been identified: extrinsic and intrinsic.12 The extrinsic pathway begins outside the cell: it can be initiated by different stimuli that involve the triggering of death receptors of the Tumour Necrosis Factor (TNF) superfamily. In the intrinsic pathway, stress-inducing intracellular stimuli injure the cell, which in turn undergoes PCD. However, both extrinsic and intrinsic pathways involve the activation of cysteine–aspartate-specific proteases, known as caspases, which initiate the proteolytic cascade and execute the cleavage of both DNA, into oligonucleosomal fragments, and specific proteins, which guarantee the integrity of cytoplasm and organelles.13–15
Microscopy techniques, such as light and electron microscopy, have been largely employed for the qualitative determination of apoptosis.16,17 However, they are not useful for a quantitative analysis of the phenomenon. Flow Cytometry (FC) can better accomplish this task. Morphological changes, DNA fragmentation and loss, plasma membrane modification and mitochondrial dysfunction can all be monitored by FC.18 Moreover, to monitor the sequence of events in apoptosis, to distinguish between apoptosis and necrosis, to complement apoptosis-related information with the cell cycle phase distribution or marker expression are all goals achievable by carrying out multi-colour FC experiments.19 However, the multi-parametric advantage offered by FC always implies the use of dyes and often of permeabilizing agents and fixatives that can tune the cellular response and bias the results. Moreover, only circulating or detached cells, which have the ability to flow in the system, can be analysed.
To avoid any possible interference due to fixation processes or probe molecules, label-free techniques, like impedance sensing (IS) or infrared microspectroscopy (IRMS) could be a valid option. Electric cell-substrate impedance sensing (ECIS) is a technique that correlates the electrode impedance variations to changes in shape, motion and, in general, to all those events that involve a cytoskeleton rearrangement in cells.20 This method has been demonstrated to be particularly suitable for the study of biological processes like cell cycle and apoptosis.21–23 Although very useful, the data obtained are mainly related to physical properties (e.g. shape, cell coverage, etc.) and do not provide any chemical information on the system. IRMS on the contrary, is intrinsically a multi-parametric and label-free analytical tool, which can provide a quick and comprehensive overview of the chemical changes happening within the sample. Specifically, it highlights differences in the cellular biochemistry, in terms of composition and structure of the fundamental cell constituents, without the need for any staining. However, the strong absorbance features of water in the mid IR regime24 and the difficulties in handling IR-transparent materials have limited to date the possibility of working under physiological conditions. The use of microfabrication techniques for the production of Visible-Infrared (Vis-IR) transparent devices has recently opened an innovative way for overcoming the current constraints when measuring live cells with this analytical tool. In this decade, our team and others25–29 developed a new generation of microdevices that allow assessing at cellular level the effects of fixatives and dehydration,30,31 determining cell cycle stages,32,33 and accomplishing several other tasks.26
In this paper, we propose IRMS as a tool for investigating the biological events involved in the PCD of U937 leukemic monocytes. We report on the biochemical changes revealed by IRMS of the U937 cell line driven to apoptosis by serum deprivation and mitochondrial membrane depolarization induced by CCCP (carbonyl cyanide m-chloro phenylhydrazone). The spectral shape of unperturbed U937 was compared with those of cells subjected to serum deprivation for 24, 48 and 72 hours and stimulated with CCCP for 2, 6 and 24 hours. The same samples were also tested for membrane damage and mitochondrial dysfunction by FC. A comparison between the IRMS and FC results was done in order to highlight the uniqueness and complementarity of these two techniques.
Experimental
Cell-based assays
Cell line U937 (American Type Culture Collection, Rockville, Md.) was used for the experiments. It is an in vitro established leukemic monocyte cell line that displays many characteristics of human monocytes.34 Control cells were cultured in RPMI 1640 medium supplemented with 10% Fetal Bovine Serum (FBS), 2 mM L-glutamine and penicillin (100 U mL−1)–streptomycin (100 U mL−1). Cells were maintained in an incubator at 37 °C with 5% CO2 and cell passage was routinely done every three days.Apoptosis induction by serum starvation
U937 control cells were cultured 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per /mL in multi-well plates in complete medium supplemented with 10% FBS for 24, 48 and 72 hours without medium exchange (Ctrl_24, Ctrl_48 and Ctrl_72 hereafter). Starved cells were grown under the same conditions without the addition of FBS to RPMI medium (Star_24, Star_48 and Star_72 hereafter).
000 cells per /mL in multi-well plates in complete medium supplemented with 10% FBS for 24, 48 and 72 hours without medium exchange (Ctrl_24, Ctrl_48 and Ctrl_72 hereafter). Starved cells were grown under the same conditions without the addition of FBS to RPMI medium (Star_24, Star_48 and Star_72 hereafter).
Apoptosis induction by CCCP (carbonyl cyanide m-chloro phenylhydrazone)
U937 cells were cultured 500000 cells per /mL in multi-well plates in complete medium supplemented with 10% FBS and let to grow for 24 hours. Then, they were treated with 50 μM CCCP for 2, 6 and 24 hours (CCCP_2 h, CCCP_6 h and CCCP_24 h hereafter).
Flow cytometry determination of cellular apoptosis
Control and treated cells were incubated for 15 minutes with the fluorescent dye DiOC6 (3,3′-dihexyloxacarbocyanine iodide) at 100 nM final concentration. Then, cells were removed from the culture medium, washed with PBS twice and marked with propidium iodide (PI, 10 μg mL−1 final concentration). Double stained cells were immediately run through FC. All cytometry analyses were performed on 10000 events per sample acquired by using a FC500 Beckman–Coulter Instrument and data were submitted to FCS Express Version 3, at the Life Sciences Department of the University of Trieste.
Device microfabrication
All devices were manufactured in LILIT clean rooms, (Laboratory for Interdisciplinary LIThography of IOM-CNR Trieste, Italy).35 CaF2 static devices were employed for the IRMS analysis of control and starved cells, as well as for testing the effects of CCCP at 2, 6 and 24 hours. More details on the device design and fabrication are given elsewhere.36 The biocompatibility of the devices was verified by contact cytotoxicity tests as described elsewhere.30IRMS experiments
All spectroscopic experiments were carried out at the infrared beamline SISSI (Synchrotron Infrared Source for Spectroscopic and Imaging) at Elettra-Sincrotrone Trieste, Italy.37Sample preparation was minimal, in order to the minimize stress level suffered by the cells. Once removed from the incubator, both control and treated cells were washed twice by centrifugation (400 g, 1 min) and re-suspended in a sterile physiological solution of 0.9% NaCl for testing the effects of starvation and complete medium for CCCP-incubation. 1 μL of cells' suspension was dropped into the Vis-IR transparent device and the chip was assembled.
FTIR transmission spectra were acquired using a Bruker Hyperion 3000 Vis-IR microscope equipped with a mid-band HgCdTe detector having a 100 μm sensitive element, coupled with a Bruker Vertex 70 interferometer. Both interferometer and microscope were purged with nitrogen in order to reduce the spectral contributions from the environmental water vapour and carbon dioxide. Spectra were collected in transmission mode using a 15× Schwarzschild condenser and objective and setting the apertures to 20 × 20 μm in order to collect groups of two to three cells. For each spectrum, collected from 900 to 4000 cm−1 in double side, forward/backward acquisition mode with a scanner velocity of 40 kHz, 256 scans were averaged with a spectral resolution of 4 cm−1. Fourier transform was carried out with Mertz phase correction, Blackman–Harris-3 terms apodization function. For each condition 50 to 80 cell groups were collected. An air background was collected with the same acquisition parameters while a buffer spectrum was acquired near to each measured cell group.
Data pre-processing, post-processing and analysis
Raw cell and buffer spectra were corrected for carbon dioxide and water vapour and baseline corrected in the 3730–950 cm−1 spectral region (rubber band method, 32 baseline points) using the OPUS 6.5 routines (Bruker Optics GmbH). An offset correction was then applied. In order to disclose the cellular spectral details hidden by the prominent water features, the buffer spectrum was subtracted from the cell one. The subtraction procedure was carried out using an in-house developed Matlab script. For further details see ref.30 Subtracted live cell spectra do not exhibit appreciable distortions of the spectral features ascribable to Resonant Mie scattering,38 thanks to the close match between the refractive indices of air and cellular components39,40 and water. Therefore, spectra were analysed without any further manipulation.Spectral features driving the discrimination between control and treated cells were highlighted by Hierarchical Cluster Analysis, HCA, performed in an R environment, using HyperSpec41 and stats packages. HCA was performed on vector normalized second derivatives of subtracted spectra (9 smoothing point, Savitsky–Golay algorithm), based on Euclidean distances and Ward's algorithm. Both vector normalization and clustering algorithm were applied in the 3000–2800 & 1760–950 cm−1 spectral region. Given that second derivatives of raw and water-subtracted spectra do not exhibit variations in peak positions,28 subtracted spectra were preferred in order to avoid any possible artefact in clustering due the degradation of the cellular medium, related to the accumulation of catabolites produced by monocytes.42 Absorbance profiles related to each cluster were also obtained by averaging the belonging spectra to the 3050–950 cm−1 spectral region, in order to highlight relative differences in the concentration of the most fundamental cell constituents.
The integral area of the following bands has been calculated with OPUS 6.5: methylene asymmetric stretching, 2945–2900 cm−1 (CH2); methyl asymmetric stretching, 2980–2945 cm−1 (CH3); spectral region 3000–2835 cm−1 (lipids); amide II, 1590–1483 (AmII); phosphate I, 1270–1186 cm−1 (PhI); phosphate II, 1146–1004 cm−1 (PhII); spectral region 1760–950 cm−1. The integral area 3000–2835 plus the 1760–950 cm−1 has been considered indicative of the total cell biomass43 (Cell).
Results
Flow cytometric signatures of apoptotic cells
The apoptotic pathways induced by both serum starvation and CCCP-exposure on U937 leukemic monocytes were followed by FC. Specifically, the integrity of the plasma membrane was probed by the exclusion of propidium iodide (PI) while alterations in the mitochondrial transmembrane potential (ΔΨm) were measured by using the dye 3,3′-dihexyloxacarbocyanide iodide (DiOC6). Cell shrinkage typical of early apoptotic events was assessed by measuring the forward scattering (FSC) while nuclear fragmentation that characterises late apoptosis resulted in a reduced side scattering (SSC). Results for all tested conditions are summarized in Table 1.| % | Viable | Early apoptotic | Late apoptotic | Necrotic |
|---|---|---|---|---|
| Ctrl | 94.91 ± 0.65 | 2.15 ± 0.55 | 1.65 ± 0.30 | 1.29 ± 0.16 |
| Ctrl_24 | 94.81 ± 0.50 | 0.92 ± 0.05 | 3.01 ± 0.49 | 1.26 ± 0.05 |
| Ctrl_48 | 98.01 ± 0.49 | 0.32 ± 0.07 | 0.79 ± 0.45 | 0.88 ± 0.15 |
| Ctrl_72 | 97.46 ± 0.30 | 2.01 ± 0.29 | 0.16 ± 0.02 | 0.37 ± 0.03 |
| Star_24 | 97.23 ± 0.28 | 0.42 ± 0.14 | 1.54 ± 0.24 | 0.81 ± 0.02 |
| Star_48 | 91.41 ± 2.05 | 0.16 ± 0.01 | 7.28 ± 2.05 | 1.15 ± 0.05 |
| Star_72 | 33.91 ± 3.55 | 25.83 ± 2.35 | 39.9 ± 2.66 | 0.36 ± 0.02 |
| CCCP_2 h | 0.00 ± 2.62 | 94.92 ± 1.78 | 1.02 ± 0.14 | 4.06 ± 1.92 |
| CCCP_6 h | 0.03 ± 0.60 | 96.38 ± 0.49 | 1.06 ± 0.17 | 2.53 ± 0.31 |
| CCCP_24 h | 6.90 ± 11.00 | 67.43 ± 9.42 | 12.42 ± 3.99 | 13.25 ± 4.04 |
FC data show that U937 leukemic monocytes are quite resistant to Growth Factor (GF) withdrawal by serum deprivation. Differences from the control can be noticed for 0% FBS only after 72 hours. At day three of starvation, a population of smaller and less granulose cells appears (FSC- and SSC-). These cells have a reduced mitochondrial membrane potential (DiOC6<): some of them retain plasma membrane integrity (early apoptotic cells), while others do not (late apoptotic ones). Conversely, CCCP induces mitochondrial dysfunction and cellular shrinkage within the first 2 hours of incubation. Most of the cells persist in the early apoptotic status for several hours while a partial or complete loss of plasma membrane integrity can be seen only after 24 hours of incubation.
IRMS signatures of apoptosis induced by serum starvation
Using HCA, three major classes of spectral similarity, CL1, CL2 and CL3, were identified and are shown in Fig. 1a. The table in panel 1b shows the distribution of each subset of cells within the clusters. Due to high spectral variability of non-synchronized cells and unavoidable bias on the choice of sampled areas, distribution must not be considered quantitative, but roughly representative of cellular progression toward apoptotic status. Control cells, grown in complete medium supplemented with 10% FBS, are entirely described by CL1 and CL2 clusters at each experimental time point. The effects of serum withdrawal are indeed discernable within the first 24 hours of treatment, since Star_24 and Star_48 cells are still represented by CL1 and CL2, but the CL1 population becomes prevalent. By comparison with FC data, it is possible to reasonably conclude that CL1 and CL2 clusters represent spectral profiles of viable cells, whilst the CL3 cluster represents spectral profiles of apoptotic ones.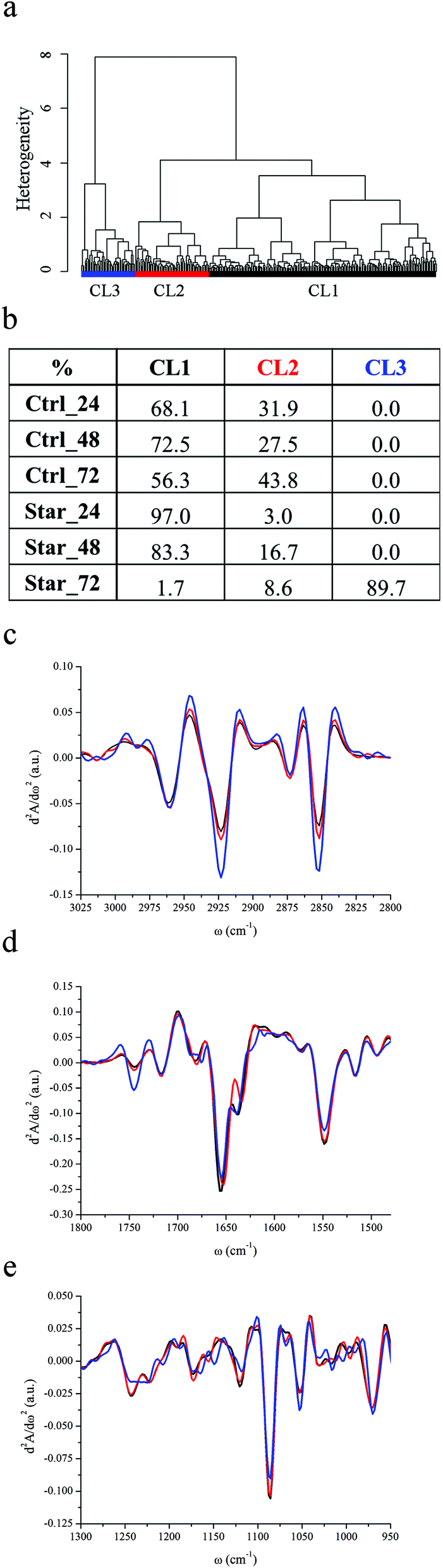 | ||
| Fig. 1 IRMS assessment of U937 apoptosis induced by serum starvation. (a) Dendrogram showing the results of HCA analysis spectra of serum-starved U937 monocyte-derived cells as described in the Experimental section. (b) Distribution of Ctrl_24, Ctrl_48, Ctrl_72, Star_24, Star_48 and Star_72 cells among CL1, CL2 and CL3 clusters. (c–e) CL1, CL2 and CL3 cluster centroids in the spectral regions 3025–2800 cm−1, 1800–1480 cm−1 and 1300–950 cm−1 respectively. | ||
Fig. 1c–e display the centroids of each class in the spectral regions of major variability: 3025–2800 cm−1, 1800–1480 cm−1 and 1300–950 cm−1. In Table 2, the relative intensity of the considered spectral bands of average absorbance spectra associated with each cluster (AbsCL1 to AbsCL3 hereafter) are reported.
| Starvation | |||
|---|---|---|---|
| CL1 | CL2 | CL3 | |
| CH2/CH3 | 1.58 ± 0.07 | 1.67 ± 0.11 | 2.08 ± 0.17 |
| Δ*1–2, Δ*1–3, Δ*2–3 | |||
| Lipids/cell | 0.072 ± 0.014 | 0.085 ± 0.016 | 0.106 ± 0.023 |
| Δ*1–2, Δ*1–3, Δ*2–3 | |||
| PhI/AmII | 0.198 ± 0.016 | 0.198 ± 0.017 | 0.191 ± 0.024 |
| Δ†1–2, Δ*1–3, Δ†2–3 | |||
| PhII/AmII | 0.471 ± 0.063 | 0.430 ± 0.046 | 0.365 ± 0.080 |
| Δ*1–2, Δ*1–3, Δ*2–3 | |||
| CCCP | |||
|---|---|---|---|
| CL1 | CL2 | CL3 | |
| CH2/CH3 | 1.67 ± 0.08 | 1.59 ± 0.04 | 1.86 ± 0.25 |
| Δ*1–2, Δ*1–3, Δ*2–3 | |||
| Lipids/cell | 0.073 ± 0.012 | 0.082 ± 0.010 | 0.101 ± 0.033 |
| Δ*1–2, Δ*1–3, Δ*2–3 | |||
| PhI/AmII | 0.208 ± 0.018 | 0.194 ± 0.014 | 0.218 ± 0.039 |
| Δ*1–2, Δ*1–3, Δ*2–3 | |||
| PhII/AmII | 0.526 ± 0.060 | 0.457 ± 0.045 | 0.473 ± 0.126 |
| Δ*1–2, Δ*1–3, Δ*2–3 | |||
The position of the most intense bands in the 3025–2800 cm−1 is quite well preserved in all clusters (see Fig. 1c): methyl and methylene asymmetric and symmetric stretching bands are at 2961 cm−1, 2873 cm−1, 2923 cm−1, and 2852 cm−1 respectively. The position of the stretching band of the methine groups is also preserved (2897 cm−1), while the C–H stretching of vinyl moieties, centred at 3012 cm−1 for CL1 and CL2, splits into two components at 3007 and 3020 cm−1 in CL3. The methylene to methyl asymmetric stretching ratio is markedly higher for AbsCL3 with respect to AbsCL2 and even more with respect to AbsCL1. The relative content of lipids with respect to the overall cellular biomass follows the same trend. Moreover, the carbonyl ester band of phospholipids is centred at 1745 cm−1 for all three cluster centroids, but, from the inspection of Fig. 1d, it is possible to deduce that its relative contribution to the spectral shape is more relevant for CL3 apoptotic cells.
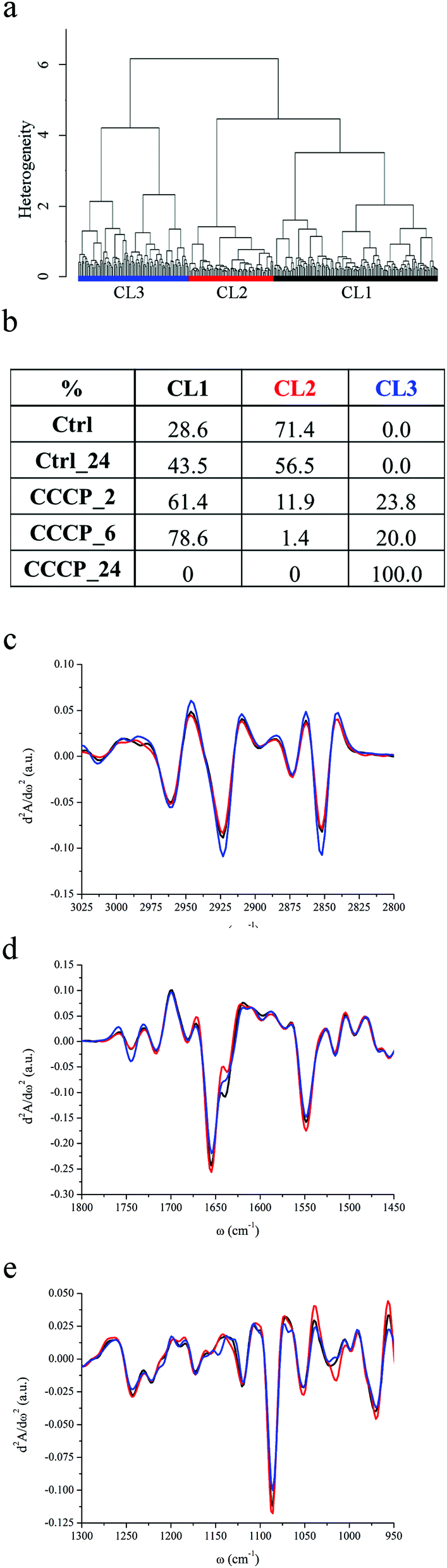 | ||
| Fig. 2 IRMS assessment of U937 apoptosis induced by CCCP exposure. (a) Dendrogram showing the results of HCA analysis on second derivative spectra of U937 monocytes exposed to CCCP as described in the Experimental section. (b) Distribution of Ctrl, Ctrl_24, CCCP_2 h, CCCP_6 h and CCCP_24 h cells among CL1, CL2 and CL3 clusters. (c–e) CL1, CL2 and CL3 cluster centroids in the spectral regions 3025–2800 cm−1, 1800–1480 cm−1 and 1300–950 cm−1 respectively. | ||
Striking differences among the centroids' cluster profiles can be appreciated in the 1300–950 cm−1 region, in which stretching bands of PO2− moieties of the phosphodiester bonds of nucleic acids and phospholipids as well as several bands related to carbohydrates are present. Two major contributions to the PhI band of CL1 and CL2 can be seen in Fig. 1e, centred at ∼1220 and ∼1241 cm−1, and arising respectively from the β-helical form of DNA & RNA and from the A-helical form of double stranded RNA, as well as DNA–RNA hybrid helices.44,45 In contrast, the PhI band for the CL3 cluster is almost completely de-structured, while a shoulder centred at 1206 cm−1 becomes prominent. Similar to PhI, the PhII band shares common spectral features among CL1 and CL2 clusters. It results from the superimposition of three major contributions, centred at 1114, 1086 and 1054 cm−1, and other minor contributions at 1068, 1030 and 1016 cm−1. The same second derivative minima characterise leukemic monocytes belonging to CL3 clusters, but the band shape clearly highlights that the relative intensity of the components at 1114 and 1053 cm−1 changes with respect to the spectral profiles of viable monocytes. The relative intensities of both PhI and PhII bands with respect to cellular proteins were considered, using the area of the amide II band, which is less affected than amide I by uncertainties due to water subtraction. As can be seen from Table 2, the PhI/AmII ratio is comparable between AbsCL1 and AbsCL2, while it slightly but significantly decreases for AbsCL3. Conversely, the PhII/AmII ratio exhibits a higher variability within all three clusters. The spectral region between the PhI and PhII band is also distinctive for CL3. From the cluster centroids in Fig. 1e it is possible to deduce that the intensity of the non-hydrogen bonded stretching mode of C–OH groups at 1172 cm−1 decreases,46 while the hydrogen bonded component at 1164 cm−1 and the band centred at 1150 cm−1, still related to the C–O stretching of protein residues or glycol-materials,47 increases.
The spectral region of 1720–1480 cm−1 is dominated by the protein bands amide I (1710–1590 cm−1) and amide II (1590–1484 cm−1). All clusters are characterised by three major components of amide I, centred at 1680, 1657 and 1640 cm−1 (see Fig. 1d). For CL3, an extra component at 1620 cm−1 is discernible. The amide II band shows the same components for all the families, centred at 1548 and 1515 cm−1, accounting for the α-helix contribution48 and tyrosine amino acid,49 respectively.
IRMS signatures of apoptosis induced by CCCP
HCA of untreated U937 monocytes (Ctrl and Ctrl_24) and of U937 monocytes incubated with a final CCCP concentration of 100 nM for 2 hours (CCCP_2), 6 hours (CCCP_6) and 24 hours (CCCP_24), grouped the data into three major classes of spectral similarity, CL1 to CL3, as shown in Fig. 2a. The table in Fig. 2b panel shows the distribution of each dataset within the clusters, while Fig. 2c–e display the centroids of each class in different spectral ranges. Ctrl and Ctrl_24 cells are entirely grouped in CL1 and CL2 clusters. After 2 and 6 hours of CCCP incubation, CL2 becomes gradually less populated while the remaining cells' subsets distribute prevalently between CL1 and CL3. Upon 24 hours of CCCP exposure, none of the cells is represented by CL1 and CL2, while the entire population belongs to CL3. The global trend deduced by HCA analysis is that CCCP-exposed U937 monocytes preserve most of the characteristics of viable control cells for short and medium-term exposure to CCCP (2 and 6 hours). After 24 hours of exposure, the entire cell population is described by CL3. In parallel, FC reveals that almost the entire cell population is in an early apoptotic status within few minutes after incubation with the protonophore CCCP. As can be seen from Fig. 2c, C–H stretching vibrations of methyl, methylene, methine and vinyl moieties have the same positions detected for the starvation experiment and there are no relevant band shifts in the CCCP treated samples. The relative contents of lipids with respect to the total cellular content as well as the methylene to methyl stretching ratio are higher for AbsCL3 and lower for AbsCL1 and AbsCL2. From Fig. 2d, it is possible to deduce that the peak height of the carbonyl ester band of phospholipids, centred at ∼1745 cm−1 is comparable for AbsCL1 and AbsCL2 while it is higher for AbsCL3.The PhI band of CL1 and CL2 clearly shows the usual components centred at 1240 and 1220 cm−1, while the shoulder at 1206 cm−1 is barely visible (see Fig. 2e). The latter became more pronounced for CL3. The PhII band is again composed of three major contributions, centred at 1114, 1086 and 1054 cm−1, and minor contributions at 1068, 1103 and 1016 cm−1. Differently from what was highlighted for cluster CL3 in starvation experiments, there are no major variations in the relative weight of the three major components. The PhI/AmII ratio has a minimum for AbsCL2 and it is the highest for AbsCL3, while the PhII/AmII ratio has a minimum for AbsCL2 and the highest for AbsCL1 (see Table 2).
Amide I and amide II bands have the same components already identified for the starvation experiments, and major changes have not been detected among the three clusters.
Discussion
Apoptosis is a key event in the control of healthy monocyte population, which during inflammation processes can undergo dramatic variations,50 as well as in pathological conditions of transformed cells. Therefore, the understanding of the sequence of events in monocyte PCD is fundamental in order to develop new strategies to enhance/inhibit the immune response and to control neoplastic proliferation. For this purpose, the U937 monocyte-derived cell line was chosen for the purpose of this study, which aims to complement the outcomes of label-free IRMS on live cells with the ones of a probe-based technique, FC.IRMS experiments on live monocytes have been made possible, thanks to the use of biocompatible microfabricated devices. They have a height of 8.5 microns, that permits to avoid the saturation of the water bending band and, consequently, to disclose the amide I profile by subtracting the water contribution as described elsewhere.30 U937 cells have an average diameter of 8–10 μm and therefore they fit within the devices without suffering any deformation stress that could alter the cellular response as already demonstrated by the authors.51
FC data revealed that U937 leukemic monocytes were quite resistant to GF withdrawal by serum deprivation.52 No appreciable differences were noticed after 24–48 hours of starvation in both cell size and granularity from FSC vs. SSC scattering plots as well as in plasma membrane integrity and mitochondrial activity. Only at day three of starvation, the percentage of both early and late apoptotic cells increased with respect to the control grown with 10% FBS. IRMS results agree perfectly with FC: after 72 hours of starvation, the cell population showed peculiar cellular features, as the ones of cluster CL3.
The mechanism by which growth factor removal may lead to apoptosis is both GF- and cell-dependent, but all possible apoptotic pathways are characterised by a decrease of basic cellular functions, such as the metabolic rate.53,54 This reduction activates endonucleases via cytochrome c (cyt c) release, determining chromatin condensation/fragmentation, and ending with cell death.55 Specifically, Caspase Activated DNase (CAD) cleaves DNA at the linker space between nucleosomes, giving rise to DNA internucleosomal fragments that are roughly a multiple of 180 base pairs (bp).13,56 DNA condensation and fragmentation during apoptosis, responsible for the nuclear morphological changes associated with PCD, is a phenomenon that has been extensively investigated but still under study. Three distinct stages of apoptotic DNA condensation have been recently discovered.57 At stage 1, ring condensation, chromatin condenses as a continuous ring at the interior surface of the nuclear envelope without fragmentation. For the completion of stage 2, necklace condensation, DNase activity is needed: the chromatin ring appears discontinuous and the nucleus shrinks. In stage 3, nuclear collapse/disassembly, the nuclear content splits into individual fragments.
The relative decrease of the integral intensities of both PhI and PhII bands of Star_72 cells reveals that CADs are active upon 72 h of GF withdrawal and that most of the starved cells are likely undergoing stages 1 and 2 of apoptotic DNA condensation. Some of them possibly have a nuclear envelope (NE) almost intact, while others discontinuous, but still present. Spectroscopic evidence of the nuclear condensation was reported also by other authors.58,59 In particular, Gasparri and Muzio demonstrated that the PhII/amideII ratio is inversely correlated to the apoptotic index, as confirmed also in our experiments. However, little information is available on the conformational rearrangements undergone by DNA during PCD. Therefore, the structural changes of nucleic acids, which can be deduced from the de-structuration of the PhI band of Star_72, are probably related to the degradation of higher order DNA but they are hardly accountable in a punctual manner. Whether the spectral components of the Star_72 PhI band other than those at 1240 cm−1 and 1220 cm−1 should be attributed to a partial transition from B to A-like DNA structures or to the stacking of nucleosomal DNA fragments is impossible to establish at the actual state of knowledge. Particularly interesting is the prominence of the band shoulder centred at 1206 cm−1. Whether this component could be assigned to asymmetric stretching of the PO2− group and possibly to Z-DNA,45 to the amide III band47 or to C–O and C–O–C vibrations of polysaccharides60 is hard to tell, but for sure it became prominent in apoptotic cells.
The modulation of PhII band shape during apoptosis also matches with the cleavage of cellular nucleic acids during PCD, as reported also by Gasparri and Muzio.58 Furthermore it might highlight the alteration of the carbohydrate metabolism, reflected also by the spectral shape variation in the 1190–1140 cm−1 energy range. The ensemble of the biochemical processes responsible for synthesis, lysis and transformation of carbohydrates in living cells is quite complex as well as the triggering of the different pathways. This complexity and the numerous contributions determining the spectral shape between 1300 and 900 cm−1 (mainly the phosphate head of phospholipids, the phosphate backbone of nucleic acids and C–O stretching vibrations of mono-, oligo- and poly-saccharides) make difficult any attempt to describe the spectral changes observed. However, they can be associated with the sharp decline in the glucose metabolism consequent to GF withdrawal: the surface expression of glucose receptors is lost after GF deprivation and the remaining intermediates of glycolysis are consumed. When glucose flux is reduced below the level needed for maintaining ΔΨm, cyt c is released resulting in the activation of initiator caspases and PCD commitment.
Once activated, initiator caspases in turn activate execution caspase enzymes, responsible for the deliberate disassembly of the cell into apoptotic bodies during PCD. Caspases are protease enzymes that promote the cleavage of several different proteins; their direct action and the cascade of events promoted permits the ordered dismantling of the dying cell, that requires major modification of the cellular cytoskeleton.61 These events may be responsible for the structural variations of proteins detected in Star_72, as reported also by other authors.58
However, the more striking spectral features of Star_72 apoptotic cells are the increased relative intensities of C–H stretching modes of both methyl and methylene moieties, the higher value of the methylene to methyl ratio and the prominence of the phosphodiester band of phospholipids. The extent of these variations is not fully justified by the formation and accumulation of APs, characteristic of the ultimate stages of apoptosis. Indeed, plasma membrane bulges outward as a consequence of the cleavage of several cytoskeleton components61 but blebbing involves mostly pre-existing membrane components. Moreover, several sets of evidence demonstrate that in Star_72 stage 3 of nuclear DNA condensation has not been accomplished yet: i- optical images of the monocytes within the devices showed round shaped cells, characterised by black spots of the condensed material but not by membrane blebbing; ii- PhI and PhII bands are clearly detectable for Star_72 U937 monocytes.
Recently it has been established that the fluidity of the plasma membrane increases during apoptosis.62,63 An increased lipid mobility could justify the detected vinyl C–H shift but the variation of the methylene to methyl ratio would be expected to be directed in the opposite way: long chains fatty acids possess better association properties with respect to shorter ones, determining more rigid membranes.64 Moreover, the increased relative content of lipids with respect to the cellular content suggests the concurrence of other events. Despite the decrease in both level and activities of the enzymes involved in lipogenesis in apoptotic cells, accumulation of lipid droplets has been established in several primary cells and cell lines.65 The mechanism responsible for this phenomenon is still unclear, probably related to de novo synthesis of neutral lipids as a result of inhibition of mitochondrial fatty acid β-oxidation. Lipid droplets are dynamic cellular organelles made of a central core of neutral lipids, mostly triacylglycerides (TAGs) and cholesterol esters, and an external shell of phospholipid monolayer.66 Their chemical nature perfectly fits with our spectroscopic results, that overall support the hypothesis that apoptosis proceeds through lipid droplet accumulation. Indeed, a higher CH2/CH3 ratio has been determined by 1H magnetic resonance of intact apoptotic Jurkat T-cells, a leukemic monocyte cell line, and these sets of evidence were directly linked to accumulation of TAGs.67
FC results did not point out dramatic differences between Star_24 and Star_48 with respect to the relative controls, while IRMS analysis revealed that the cellular variability decreased within the first 24 hours of starvation. The absence of GF is known to induce cell cycle arrest and, generally speaking, apoptotic stimuli often arrest growth before inducing cell death.68 Cell survival, progression and death share common pathways that make these events tightly interconnected.69 Specific checkpoints are present at all stages of the cell cycle and at their boundaries, which allow cells to enter the next proliferating stage once the previous one has been fully and properly accomplished. In the case of any adverse event, whether the cellular damage produced can be repaired, progression to the cell cycle resumes while, if not, the PCD event cascade starts. It is known that the withdrawal of specific growth factors can prevent the expression of genes needed for DNA synthesis and duplication and it is therefore reasonable to think that CL1 has most of the characteristics of the low-metabolic, quiescent G0 cell cycle phase.32,33,70
The understanding of the experimental results obtained for apoptosis induction by CCCP-exposure requires in-depth analysis of the mechanism of action of the chemical compound. Carbonyl cyanide m-chlorophenyl hydrazone is a lipid-soluble weak acid that acts as an ionophore: it makes the mitochondrial inner membrane permeable to solutes with molecular weight below 1.5 kDa, due to the opening of the so called Mitochondrial Permeability Transition (MPT) pores. The opening of these mitochondrial megachannels leads to the quick dissipation of ΔΨm, to mitochondria swelling and outer membrane integrity damage. These events end with the release of several proteins normally confined within the intermembrane space into the cytoplasm. Among them, cyt c has been recognized to play a pivotal role in apoptosis. CCCP is also an uncoupling agent, which uncouples the respiratory chain from the phosphorylation system (ATP production), acting at this level as an apoptotic agent. FC data clearly reveal that CCCP induces the immediate loss of the mitochondrial inner plasma membrane potential. However, it is well established that the mitochondrial membrane depolarization can be a reversible process and there are several studies that report that the caspase activation induced by CCCP can take place several hours or even days after the cell exposure to the drug.71 This holds true, for example, for the leukemic monocyte cell line Jurkat-neo, where exposure to CCCP 25 μM induced immediate mitochondrial collapse but caspase 3 activation only 24 hours later. Therefore, even if the DiOC6 probe is often used as a diagnostic tool of early apoptosis, it should be exclusively referred to ΔΨm impairment, without implications on the cellular viability as suggested also by other authors.72
From this perspective, functional data from FC and biochemical ones from IRMS can be complemented and summarized as follows. U937 monocytes respond almost immediately to CCCP insult by losing the ΔΨm. However, the mitochondrial permeability transition (MPT) is a necessary event for apoptosis commitment but not sufficient. Actually, cells survive for several hours in this status characterised by an affected steady-state mitochondrial activity, implementing the adaptation mechanism. One of these mechanisms presumably implies the levelling and accumulation of U937 monocytes into the cellular phase identified by CL1. Some cells, more susceptible to the dissipation of ΔΨm (those belonging to CL2, possibly corresponding to cycling cells into S and G2/M phases) in part evolve from CL2 toward not-viable conditions, described by CL3, and in part accumulate in CL1 within the first incubation hours, a time shorter than the U937 cell cycle, that is almost of 24 hours. From the spectral examination of the CL3 cluster centroid, the prominence of the PhI band shoulder centred at 1206 cm−1 emerges, already identified as characteristic of apoptotic Star_72 cells. However, the PhI band of CL3 of CCCP exposed monocytes is much more structured than the one of CL3 of starved cells. These sets of evidence reveal that nucleosome fragmentation is minimal in CCCP exposed cells, also after 24 hours treatment. Indeed, optical images (not shown here), acquired before IRMS data collection at all-time points, showed cellular morphologies quite unperturbed, without the presence of dark cellular spots diagnostic of chromatin condensation and fragmentation, as revealed for Star_72.
Moreover, we did not observe a significant decrement of relative intensity of neither PhI nor PhII bands with respect to cellular proteins for the cells exposed to CCCP for 24 hours, differently from what observed for Star_72 cells. In case DNA fragmentation started, its effect on both PhI and PhII band intensity is not clearly detectable and may be hidden by the contributions of carbohydrates and phospholipids to these bands. In particular, with respect to the lipidome profile, CCCP exposed cells are characterised by a methylene to methyl ratio higher than the ones of viable cells and it is related to an increased lipid content with respect to the total cell biomass, especially of phospholipids as can be deduced by the prominence of the carbonyl stretching band centred at 1745 cm−1 in CL3. It seems reasonable to attribute this trend to the early and progressive accumulation of mobile TAGs in the form of cytoplasmic lipid droplets after U937 exposure to CCCP, as a consequence of the increased intracellular content of reactive oxygen species and related inhibition of fatty acid oxidation due to the loss of ΔΨm.71 This phenomenon goes along with an increment of plasma membrane fluidity as highlighted by the more pronounced contribution of the vinyl stretching band centred at 3013 cm−1 in the CL3 centroid.
Given that lipid droplet accumulation precedes chromosomal DNA fragmentation, it is reasonable to look at CL3 as a spectral family of cells that are suffering an irreversible apoptotic event that follows the ultimate loss of plasma membrane potential and precedes extensive DNA fragmentation.
Conclusion
The results presented in this paper allow highlighting the diagnostic capabilities of IRMS for the detection of cellular apoptosis, and its sensitivity not only to last steps of PCD, but also to early-reversible stages. There are no direct infrared biomarkers of cellular functionality, but IRMS is able to indirectly reveal the biochemical perturbations that result from cell metabolic impairment. Cell cycle arrest and accelerated cellular progression, consequent to the adaptation mechanism during U937 serum withdrawal or CCCP exposure, as well as carbohydrate metabolism alterations are active cellular processes that have been revealed through IRMS. Moreover, it was possible to determine that cell death induced by both GF withdrawal and CCCP exposure proceeds through the accumulation of lipid droplets, an event that can be related to mitochondrial dysfunction and cyt c release. Finally, we revealed that structural changes of nuclear DNA can be detected before evident extensive DNA fragmentation by the decrease of both PhI and PhII bands and thanks to the prominence of the 1206 cm−1 band component that characterises irreversible apoptotic U937 events.The biological meaning of the latter spectral feature is hardly accountable at the actual state of knowledge, since most of the efforts have been directed in the assessment of the degree of DNA fragmentation during apoptosis more than on structural rearrangements undergone by nuclear DNA and histone proteins. However, our results offer a new perspective on the IRMS signatures of apoptosis and its progression, and we can envision that the complement of IRMS with other approaches could eventually provide a global understanding on a variety of cellular processes.
Notes and references
- R. A. Lockshin and C. M. Williams, J. Insect Physiol., 1964, 10, 643–649 CrossRef CAS.
- J. F. Kerr, A. H. Wyllie and A. R. Currie, Br. J. Cancer, 1972, 26, 239–257 CrossRef CAS.
- D. L. Vaux and S. J. Korsmeyer, Cell, 1999, 96, 245–254 CrossRef CAS.
- A. G. Renehan, C. Booth and C. S. Potten, BMJ, 2001, 322, 1536–1538 CrossRef CAS.
- R. M. Friedlander, N. Engl. J. Med., 2003, 348, 1365–1375 CrossRef CAS PubMed.
- E. M. Johnson, L. J. S. Greenlund, P. T. Akins and C. Y. Hsu, J. Neurotrauma, 1995, 12, 843–852 CrossRef.
- K. Eguchi, Intern. Med., 2001, 40, 275–284 CrossRef CAS.
- L. D. Attardi and J. M. Brown, Nat. Rev. Cancer, 2005, 5, 231–237 Search PubMed.
- Y. Shen and T. E. Shenk, Curr. Opin. Genet. Dev., 1995, 5, 105–111 CrossRef CAS.
- A. Saraste and K. Pulkki, Cardiovasc. Res., 2000, 45, 528–537 CrossRef CAS.
- D. Kanduc, A. Mittelman, R. Serpico, E. Sinigaglia, A. A. Sinha, C. Natale, R. Santacroce, M. G. Di Corcia, A. Lucchese, L. Dini, P. Pani, S. Santacroce, S. Simone, R. Bucci and E. Farber, Int. J. Oncol., 2002, 21, 165–170 CAS.
- A. Gewies, ApoReview – Introduction to Apoptosis, 2003, 1–26 Search PubMed.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- N. N. Danial and S. J. Korsmeyer, Cell, 2004, 116, 205–219 CrossRef CAS.
- J. M. Adams, Genes Dev., 2003, 17, 2481–2495 CrossRef CAS PubMed.
- R. Sgonc and J. Gruber, Exp. Gerontol., 1998, 33, 525–533 CrossRef CAS.
- D. Wlodkowic, J. Skommer and Z. Darzynkiewicz, Methods Mol. Biol., 2009, 559, 19–32 CAS.
- Z. Darzynkiewicz, S. Bruno, G. Del Bino, W. Gorczyca, M. A. Hotz, P. Lassota and F. Traganos, Cytometry, 1992, 13, 795–808 CrossRef CAS PubMed.
- W. G. Telford, A. Komoriya, B. Z. Packard and C. B. Bagwell, Methods Mol Biol., 2011, 699, 203–227 CAS.
- I. Giaever and C. R. Keese, Nature, 1993, 366, 591–592 CrossRef CAS PubMed.
- S. Arndt, J. Seebach, K. Psathaki, H.-J. Galla and J. Wegener, Biosens. Bioelectron., 2004, 19, 583–594 CrossRef CAS.
- C. E. Campbell, M. M. Laane, E. Haugarvoll and I. Giaever, Biosens. Bioelectron., 2007, 23, 536–542 CrossRef CAS PubMed.
- C. Xiao, B. Lachance, G. Sunahara and J. H. T. Luong, Anal. Chem., 2002, 74, 5748–5753 CrossRef CAS.
- S. Venyaminov and F. G. Prendergast, Anal. Biochem., 1997, 248, 234–245 CrossRef CAS.
- D. Moss, M. Keese and R. Pepperkok, Vib. Spectrosc., 2005, 38, 185–191 CrossRef CAS PubMed.
- H.-Y. N. Holman, R. Miles, Z. Hao, E. Wozei, L. M. Anderson and H. Yang, Anal. Chem., 2009, 81, 8564–8570 CrossRef CAS PubMed.
- K. L. A. Chan, S. Gulati, J. B. Edel, A. J. de Mello and S. G. Kazarian, Lab Chip, 2009, 9, 2909 RSC.
- E. J. Marcsisin, C. M. Uttero, M. Miljkovic and M. Diem, Analyst, 2010, 135, 3227–3232 RSC.
- E. Mitri, G. Birarda, L. Vaccari, S. Kenig, M. Tormen and G. Grenci, Lab Chip, 2014, 14, 210–218 RSC.
- L. Vaccari, G. Birarda, L. Businaro, S. Pacor and G. Grenci, Anal. Chem., 2012, 84, 4768–4775 CrossRef CAS PubMed.
- D. R. Whelan, K. R. Bambery, P. Heraud, M. J. Tobin, M. Diem, D. McNaughton and B. R. Wood, Nucleic Acids Res., 2011, 39, 5439–5448 CrossRef CAS PubMed.
- D. E. Bedolla, S. Kenig, E. Mitri, P. Ferraris, A. Marcello, G. Grenci and L. Vaccari, Analyst, 2013, 138, 4015–4021 RSC.
- D. R. Whelan, K. R. Bambery, L. Puskar, D. McNaughton and B. R. Wood, Analyst, 2013, 138, 3891–3899 RSC.
- C. Sundström and K. Nilsson, Int. J. Cancer, 1976, 17, 565–577 CrossRef.
- F. Romanato, E. Di Fabrizio, L. Vaccari, M. Altissimo, D. Cojoc, L. Businaro and S. Cabrini, Microelectron. Eng., 2001, 57–58, 101–107 CrossRef CAS.
- G. Birarda, G. Grenci, L. Businaro, B. Marmiroli, S. Pacor and L. Vaccari, Microelectron. Eng., 2010, 87, 806–809 CrossRef CAS PubMed.
- S. Lupi, A. Nucara, A. Perucchi, P. Calvani, M. Ortolani, L. Quaroni and M. Kiskinova, America, 2007, 24, 959–964 CAS.
- P. Bassan, H. J. Byrne, F. Bonnier, J. Lee, P. Dumas and P. Gardner, Analyst, 2009, 134, 1586–1593 RSC.
- B. Mohlenhoff, M. Romeo, M. Diem and B. R. Wood, Biophys. J., 2005, 88, 3635–3640 CrossRef CAS PubMed.
- M. Romeo, B. Mohlenhoff and M. Diem, Vib. Spectrosc., 2006, 42, 9–14 CrossRef CAS PubMed.
- C. Beleites and V. Sergo, hyperSpec: a package to handle hyperspectral data sets in R, http://hyperspec.r-forge.r-project.org/.
- L. Vaccari, G. Birarda, G. Grenci, S. Pacor and L. Businaro, J. Phys.: Conf. Ser., 2012, 359, 012007 CrossRef.
- A. Kretlow, Q. Wang, M. Beekes, D. Naumann and L. M. Miller, Biochim. Biophys. Acta, Mol. Basis Dis., 2008, 1782, 559–565 CrossRef CAS PubMed.
- M. Banyay, M. Sarkar and A. Graslund, Biophys. Chem., 2003, 104, 477–488 CrossRef CAS.
- E. Taillandier and J. Liquier, Methods Enzymol., 1992, 211, 619 Search PubMed.
- H. P. Wang, H. C. Wang and Y. J. Huang, Sci. Total Environ., 1997, 204, 283–287 CrossRef.
- L. Chiriboga, P. Xie, H. Yee, V. Vigorita, D. Zarou, D. Zakim and M. Diem, Biospectroscopy, 1998, 4, 47–53 CrossRef CAS.
- E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Biophys. J., 2006, 90, 2946–2957 CrossRef CAS PubMed.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS.
- D. F. Mangan, S. E. Mergenhagen and S. M. Wahl, J. Periodontol., 1993, 64, 461–466 CAS.
- G. Birarda, G. Grenci, L. Businaro, B. Marmiroli, S. Pacor, F. Piccirilli and L. Vaccari, Vib. Spectrosc., 2010, 53, 6–11 CrossRef CAS PubMed.
- D. R. Plas and C. B. Thompson, Trends Endocrinol. Metab., 2002, 13, 75–78 CrossRef.
- A. Letai, Mol. Cell, 2006, 21, 728–730 CrossRef CAS PubMed.
- M. K. L. Collins, G. R. Perkins, G. Rodriguez-Tarduchy and M. A. Nieto, BioEssays, 1994, 16, 133–138 CrossRef CAS PubMed.
- D. R. Plas and C. B. Thompson, Trends Endocrinol. Metab., 2002, 13, 75–78 CrossRef.
- S. Nagata, Exp. Cell Res., 2000, 256, 12–18 CrossRef CAS PubMed.
- S. Tone, K. Sugimoto, K. Tanda, T. Suda, K. Uehira, H. Kanouchi, K. Samejima, Y. Minatogawa and W. C. Earnshaw, Exp. Cell Res., 2007, 313, 3635–3644 CrossRef CAS PubMed.
- F. Gasparri and M. Muzio, Biochem. J., 2003, 369, 239–248 CrossRef CAS PubMed.
- K. Z. Liu, L. Jia, S. M. Kelsey, A. C. Newland and H. H. Mantsch, Apoptosis, 2001, 6, 269–278 CrossRef CAS.
- Y. Yang, J. Sule-Suso, G. D. Sockalingum, G. Kegelaer, M. Manfait and A. J. El Haj, Biopolymers, 2005, 78, 311–317 CrossRef CAS PubMed.
- O. Ndozangue-Touriguine, J. Hamelin and J. Bréard, Biochem. Pharmacol., 2008, 76, 11–18 CrossRef CAS PubMed.
- D. Jourd'heuil, A. Aspinall, J. D. Reynolds and J. B. Meddings, Can. J. Physiol. Pharmacol., 1996, 74, 706–711 CrossRef CAS.
- E. Gibbons, K. R. Pickett, M. C. Streeter, A. O. Warcup, J. Nelson, A. M. Judd and J. D. Bell, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 887–895 CrossRef CAS PubMed.
- D. A. Los and N. Murata, Biochim. Biophys. Acta, 2004, 1666, 142–157 CrossRef CAS PubMed.
- J. Boren and K. M. Brindle, Cell Death Differ., 2012, 19, 1561–1570 CrossRef CAS PubMed.
- Y. Guo, K. R. Cordes, R. V. Farese Jr and T. C. Walther, J. Cell Sci., 2009, 122, 749–752 CrossRef CAS PubMed.
- N. M. S. Al-Saffar, J. C. Titley, D. Robertson, P. A. Clarke, L. E. Jackson, M. O. Leach and S. M. Ronen, Br. J. Cancer, 2002, 86, 963–970 CrossRef CAS PubMed.
- J. A. King Kl Fau - Cidlowski and J. A. Cidlowski.
- S. Maddika, S. R. Ande, S. Panigrahi, T. Paranjothy, K. Weglarczyk, A. Zuse, M. Eshraghi, K. D. Manda, E. Wiechec and M. Los, Drug Resist. Updates, 2007, 10, 13–29 CrossRef CAS PubMed.
- S. O. Konorov, H. G. Schulze, J. M. Piret, M. W. Blades and R. F. Turner, Anal. Chem., 2013, 85, 8996–9002 CrossRef CAS PubMed.
- A. O. de Graaf, L. P. van den Heuvel, H. B. P. M. Dijkman, R. A. De Abreu, K. U. Birkenkamp, T. de Witte, B. A. van der Reijden, J. A. M. Smeitink and J. H. Jansen, Exp. Cell Res., 2004, 299, 533–540 CrossRef CAS PubMed.
- M. L. Lim, T. Minamikawa and P. Nagley, FEBS Lett., 2001, 503, 69–74 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |