 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing permanganate chemiluminescence detection for the determination of glutathione and glutathione disulfide in biological matrices†
Zoe M.
Smith
a,
Jessica M.
Terry
a,
Neil W.
Barnett
a,
Laura J.
Gray
b,
Dean J.
Wright
c and
Paul S.
Francis
*a
aCentre for Chemistry and Biotechnology, Faculty of Science, Engineering and Built Environment, Deakin University, Locked Bag 20000, Geelong, Victoria 3220, Australia. E-mail: paul.francis@deakin.edu.au
bSchool of Medicine, Faculty of Health, Deakin University, Locked Bag 20000, Geelong, Victoria 3220, Australia
cFlorey Institute of Neuroscience and Mental Health, The University of Melbourne, 30 Royal Pde, Parkville, Victoria 3052, Australia
First published on 25th March 2014
Abstract
Acidic potassium permanganate chemiluminescence enables direct post-column detection of glutathione, but its application to assess the redox state of a wider range of biological fluids and tissues is limited by its sensitivity. Herein we show that the simple on-line addition of an aqueous formaldehyde solution not only enhances the sensitivity of the procedure by two orders of magnitude, but also provides a remarkable improvement in the selectivity of the reagent towards thiols such as glutathione (compared to phenols and amino acids that do not possess a thiol group). This enhanced mode of detection was applied to the determination of glutathione and its corresponding disulfide species in homogenised striatum samples taken from both wild type mice and the R6/1 transgenic mouse model of Huntington's disease, at both 8 and 12 weeks of age. No significant difference was observed between the GSH/GSSG ratios of wild type mice and R6/1 mice at either age group, suggesting that the early disease progression had not significantly altered the intracellular redox environment.
Introduction
The tripeptide glutathione (GSH) is a critical physiological component endogenous to all biological tissues and fluids.1,2 Existing primarily in its reduced form, an overproduction of reactive oxygen species (ROS) initiates the conversion of GSH to its corresponding disulfide (GSSG).2–4 An alteration in the level of GSH (or the GSH/GSSG ratio) is one of the first indications of cellular oxidative stress, a status which has been implicated in the pathophysiology of conditions such as Alzheimer's, Parkinson's and Huntington's disease.2–5 Analytical methodologies to determine GSH often involve complex sample pre-treatment steps such as derivatisation of the analyte for fluorescence detection.3,6–8 GSSG is normally measured after disulfide bond reduction, in which case it is then treated by the same derivatisation as the native GSH. Subjecting biological samples to these lengthy derivatisation procedures, however, can artificially alter the GSH/GSSG ratio through auto-oxidation. Methods of detection that enable direct measurement (without derivatisation), such as electrochemical9 and chemiluminescence,10–12 are therefore an attractive alternative to fluorescence.6,7,13,14Several new approaches for chemiluminescence detection of GSH have recently emerged.10–12,15 Li et al. reported the determination of thiols such as cysteine, GSH, N-acetylcysteine and captopril based on the sensitised chemiluminescence reaction with acidic potassium permanganate and quinine.15 This system actually contains two light producing pathways: (i) the reduction of permanganate to form an excited state manganese(II) species, and (ii) the oxidation of the thiols to generate an excited intermediate capable of transferring energy to the quinine fluorophore.16 The manganese(II) pathway can be considerably enhanced by addition of sodium polyphosphate rather than quinine, which improves sensitivity by eliminating the background emission resulting from the reaction between the sensitiser and oxidant.17,18 In 2011, McDermott et al. utilised this approach for the post-column chemiluminescence detection of GSH in cultured skeletal muscle cells (C2C12 myotubes) following a rapid isocratic separation.11 The limit of detection was 5 × 10−7 M, compared to 6.5 × 10−6 M reported by Li and co-workers.11,15 McDermott and co-workers also utilised a colloidal manganese(IV) reagent for the direct post-column chemiluminescence detection of thiols and disulfides. The limits of detection of 7 × 10−8 M for GSH and 1 × 10−7 M for GSSG were suitable for their determination in whole blood samples,12 but they are inadequate to measure the relatively low concentration of GSSG in various other clinically relevant physiological fluids, cells and tissues.
Herein we investigate a range of enhancers in order to improve the sensitivity of acidic potassium permanganate chemiluminescence reagent towards thiol compounds. We show that the on-line addition of an aqueous formaldehyde solution can not only improve the sensitivity towards thiol compounds, but also dramatically enhance the selectivity of the reagent. We apply this enhanced chemiluminescence reagent system to the determination of GSH and GSSG in mouse striatum.
Experimental
Chemicals
Deionised water (Continental Water Systems, Victoria, Australia) and analytical grade reagents were used unless otherwise stated. Chemicals were obtained from the following sources: N-acetylcysteine (Nacys), N-cyclohexyl-3-aminopropanesulfonic acid (CAPS), L-cysteine (Cys), L-cystine (CYSS), N-ethylmaleimide (NEM), L-glutathione (GSH), L-glutathione disulfide (GSSG), glyoxal, homocysteine (Hcys), homocystine (HCYSS), L-lysine (Lys), 2-mercaptoethanol, L-methionine (Met), L-phenylalanine (Phe), quinine, serotonin hydrochloride (5-HT), sodium polyphosphate, tris(2-carboxyethyl)phosphine hydrochloride (TCEP), L-tryptophan (Trp) and L-tyrosine (Tyr) from Sigma-Aldrich (New South Wales, Australia); potassium permanganate, and formaldehyde (37%) from Chem-Supply (South Australia, Australia); methanol, sulfuric acid, and tris(hydroxymethyl)methylamine from Merck (Victoria, Australia); morphine from GlaxoSmithKline (Victoria, Australia), formic acid from Hopkin and Williams (Essex, England) and hydrochloric acid (32% w/v) from Ajax Finechem (New South Wales, Australia).Stock solutions (1 × 10−3 M) of N-acetylcysteine, cysteine, cystine, GSH, GSSG, homocysteine and homocystine were prepared and diluted as required in 0.01% formic acid (pH 2.8). Formaldehyde was filtered and diluted to the required concentration with deionised water. The acidic potassium permanganate reagent (2.5 × 10−4 M) was prepared by dissolving solid potassium permanganate in a solution of sodium polyphosphate (1% (m/v)) and adjusting to pH 3 with sulfuric acid.
Flow injection analysis
The manifold was constructed from a Gilson Minipuls 3 peristaltic pump (John Morris Scientific, NSW, Australia) with bridged PVC pump tubing (white/white, 1.02 mm i.d., DKSH, Queensland, Australia), PTFE manifold tubing (0.8 mm i.d., Cole-Parmer Instrument Company, Illinois, USA) and a six-port injection valve (Vici 04W-0192L Valco Instruments, Texas, USA) equipped with a 70 μL sample loop. A custom built flow-cell (a tight coil of 0.8 mm i.d. PTFE tubing) was mounted against an extended range photomultiplier tube (Electron Tubes model 9828SB, ETP, NSW, Australia) and encased in a light-tight housing. The output signal from the detector was obtained using an e-corder 410 data acquisition system (eDAQ, NSW, Australia). Analytes were injected into a carrier stream (deionised water) which merged with a stream of either deionised water or formaldehyde (2 M) at a confluence point located at the entrance of a 1 m reaction coil. This stream was then combined with the acidic potassium permanganate reagent just prior to entry into the flow-cell.High performance liquid chromatography
Chromatographic analysis was carried out on an Agilent Technologies 1260 series liquid chromatography system, equipped with a quaternary pump, solvent degasser system, and autosampler (Agilent Technologies, Forest Hill, Victoria, Australia) using an Alltech Alltima C18 column (250 mm × 4.6 mm i.d., 5 μm) equipped with an Alltima C18 guard column, at room temperature, with an injection volume of 10 μL and a flow rate of 1 mL min−1. Isocratic elution was performed with 97% solvent A (deionised water adjusted to pH 2.8 with formic acid) and 3% solvent B (methanol). An analogue to digital interface box (Agilent Technologies) was used to convert the signal from the chemiluminescence detector. Before use in the HPLC system, all sample solutions and solvents were filtered through a 0.45 μm nylon membrane. For post-column chemiluminescence measurements the column eluate (1 mL min−1) and a formaldehyde enhancer (2 M; 1 mL min−1) were merged at a T-piece located 10 cm from the entrance of the detector. This stream was then combined with the potassium permanganate reagent (2.5 × 10−4 M; 1 mL min−1) just prior to entry into the flow-cell. A GloCel chemiluminescence detector (Global FIA, WA, USA) containing a dual-inlet serpentine flow-cell and photomultiplier tube (model 9828SB, ETP, NSW, Australia) set at a constant voltage of 900 V from a stable power supply (PM20D, ETP) was utilised in this system. The reagent and enhancer solutions were propelled by a 12×6 Dual Piston Pump (Scientific Systems Inc., PA, USA) equipped with a pulse damper and self-flushing pump heads.Chemiluminescence spectra
A Cary Eclipse fluorescence spectrophotometer (Varian, Mulgrave, Victoria, Australia) equipped with R928 photomultiplier tube (Hamamatsu, Japan) was operated in bio/chemiluminescence mode to collect chemiluminescence spectra. A two-line flow manifold was used to continuously merge the analyte (5 × 10−5 M; 3.5 mL min−1) and reagent solutions (2.5 × 10−4 M; 3.5 mL min−1) at a confluence point located just prior to the entrance of a coiled PTFE flow-cell (200 μL, 0.8 mm i.d.) that was mounted against the emission window of the spectrophotometer. A three-line continuous flow manifold was subsequently used to merge the analyte solution (1 × 10−7 M; 3.5 mL min−1) with the formaldehyde enhancer (2.0 M; 3.5 mL min−1) at a confluence point located at the entrance of a short (1 m) reaction coil. This stream was then combined with the acidic potassium permanganate reagent (2.5 × 10−4 M; 3.5 mL min−1) just prior to entry into the flow-cell. Final spectra were an average of 20 scans (1 s gate time; 1 nm data interval; 20 nm band-pass; PMT, 800 V) and corrected as previously described.19Mice
R6/1 transgenic hemizygote males20 were originally obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and bred with CBB6 (CBAÅ × C57/B6) F1 females to establish the R6/1 colony at the Florey Institute of Neuroscience and Mental Health (FINMH). After weaning, animals were group housed (4 mice per cage with 2 of each genotype) and maintained on a 12 h light/dark cycle with access to food and water ad libitum. Mice were handled in accordance with the guidelines of the FINMH Animal Ethics Committee and the National Health and Medical Research Council (NHMRC).Sample collection
Immediately after cervical dislocation, mice brains were dissected on ice and snap frozen in liquid nitrogen, before being stored at −80 °C. Tissue was allowed to defrost on ice prior to homogenization. Tissue homogenates were prepared in 0.1% formic acid, using a motorized pestle. Samples were allowed to rest on ice for 10 min prior to centrifugation at 8000 G for 15 min. Supernatants were removed for analysis.Sample analysis
For GSH determination the sample was initially diluted 10-fold into aqueous formic acid (0.01%). A 50 μL aliquot of the diluted supernatant was then combined with 450 μL of aqueous formic acid (5%), immediately prior to analysis using HPLC with acidic potassium permanganate chemiluminescence detection. For GSSG determination, a second aliquot of the diluted supernatant (100 μL) was combined with a Tris–HCl buffer (0.675 M; 20 μL; pH 8.0) and NEM (6.3 × 10−3 M; 20 μL) and mixed for 30 s. 2-Mercaptoethanol (8 × 10−3 M; 20 μL) was then added and mixed for a further 30 s. Following addition of TCEP (7.8 × 10−4 M; 20 μL) the solution was gently heated at 50 °C for 60 min to allow complete disulfide bond reduction. Finally, aqueous formic acid (5%, 20 μL) was introduced to re-acidify the sample prior to filtration and analysis.Results and discussion
Preliminary investigations
In an attempt to provide a more analytically useful signal from GSH, four compounds previously reported to enhance the emission intensity from the reaction of various organic compounds with acidic potassium permanganate17,18,21–23 were examined: N-cyclohexyl-3-aminopropanesulfonic acid (CAPS), quinine, glyoxal and formaldehyde. Using a three-line flow injection analysis manifold, a GSH solution (1 × 10−6 M) was injected into a deionised water carrier (3.5 mL min−1) which merged with the enhancer solution (3.5 mL min−1) at a confluence point located at the entrance of a short reaction coil. A continuously flowing stream of the reagent (2.5 × 10−4 M; 3.5 mL min−1) then merged with the combined analyte-enhancer solution just prior to entry into the flow-cell. Under these conditions, each of the enhancers resulted in emissions greater than that obtained with only deionised water in the enhancer stream. A small improvement in the chemiluminescence signal was obtained when using solutions of CAPS, quinine and glyoxal at 1.0 mM (Fig. 1). A much larger increase in signal intensity was observed with a more concentrated solution of glyoxal (∼600-fold enhancement), but this response was lower than that obtained from the corresponding blank (i.e. the reaction of permanganate and the enhancer without GSH). The addition of formaldehyde resulted in a significant increase in signal intensity (over three orders of magnitude), with a relatively small increase in the corresponding blank response.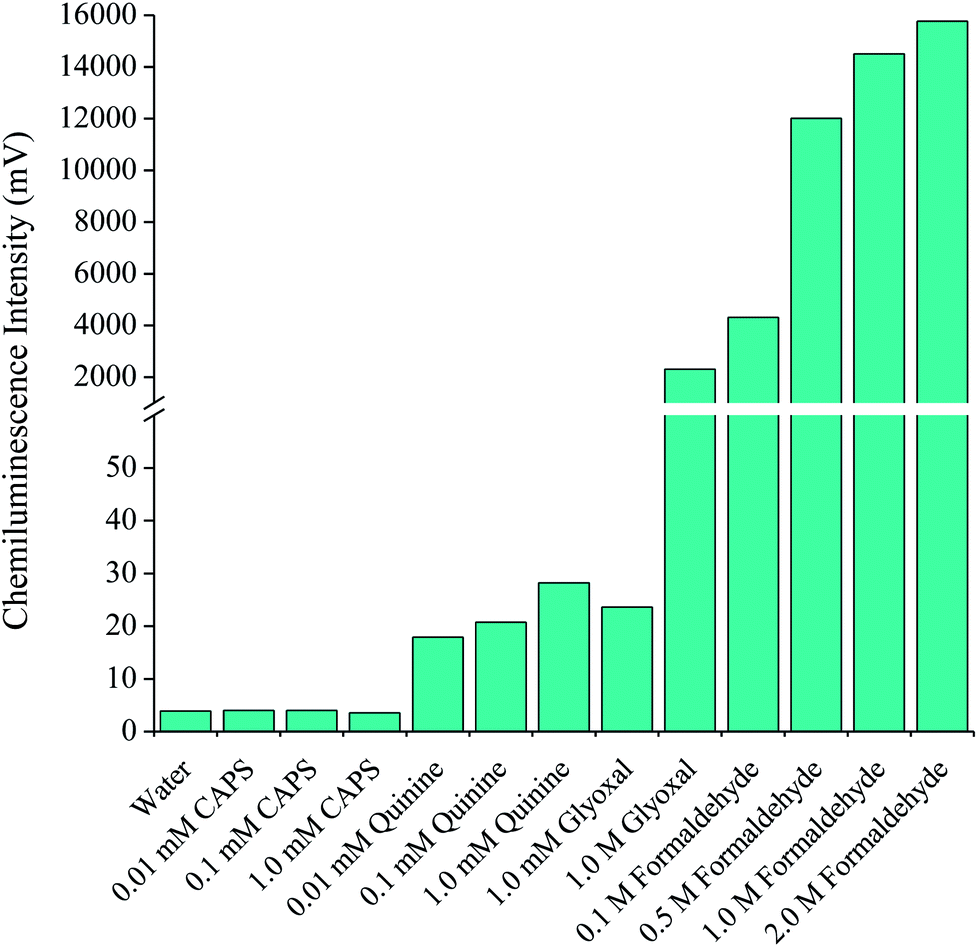 | ||
| Fig. 1 Chemiluminescence intensities for GSH (1 × 10−6 M) upon reaction with acidic potassium permanganate and various enhancer solutions using flow injection analysis. | ||
Confirmation of the emitting species
The mechanism of enhancement of permanganate chemiluminescence systems by formaldehyde has not yet been elucidated.17 It has been postulated that the enhancer is oxidised to form excited singlet oxygen or an unknown intermediate that either emits light or transfers energy to other compounds.24–27For the chemiluminescence reaction of GSH with the acidic potassium permanganate reagent, we observed a broad single band with an apparent emission maximum at approximately 687 nm (Fig. S1a†), which is characteristic of the electronically excited manganese(II) species formed in the oxidation of organic analytes by permanganate in the presence of sodium polyphosphate (λmax = 689 ± 5 nm; 4T1 → 6A1 transition28,29). The incorporation of a formaldehyde enhancer (2 M) into the system, increased the overall emission intensity but did not alter the spectral distribution (Fig. S1b†). It was therefore concluded that the same Mn(II) species was responsible for the emission in both cases.
Analyte screen
Fifteen analytes (5 × 10−6 M) were screened with the acidic potassium permanganate reagent under reaction conditions previously found to afford the greatest chemiluminescence response for GSH.11 These responses (Fig. 2a) were then compared to those obtained when incorporating the formaldehyde enhancer (Fig. 2b). With the exception of glycine and lysine, the addition of formaldehyde increased the chemiluminescence intensity for each analyte. Moderate enhancement (59- to 197-fold) was obtained for species containing a disulfide bond (CYSS, HCYSS, GSSG). However, this improvement in sensitivity was still insufficient to allow direct post-column quantification of GSSG in clinical samples. The greatest improvements in signal intensity were obtained for the thiol containing species: Cys, Hcys, GSH and Nacys, with increases of between 528- and 4417-fold. Conversely, the response from two phenolic analytes that are known to produce very intense signals with the acidic potassium permanganate (morphine and serotonin) were only enhanced 2- to 3-fold. Therefore, the formaldehyde enhancer not only affords much greater sensitivity, but also provided a remarkable improvement in selectivity towards thiol compounds such as GSH, thus reducing potential interferences in complex biological samples.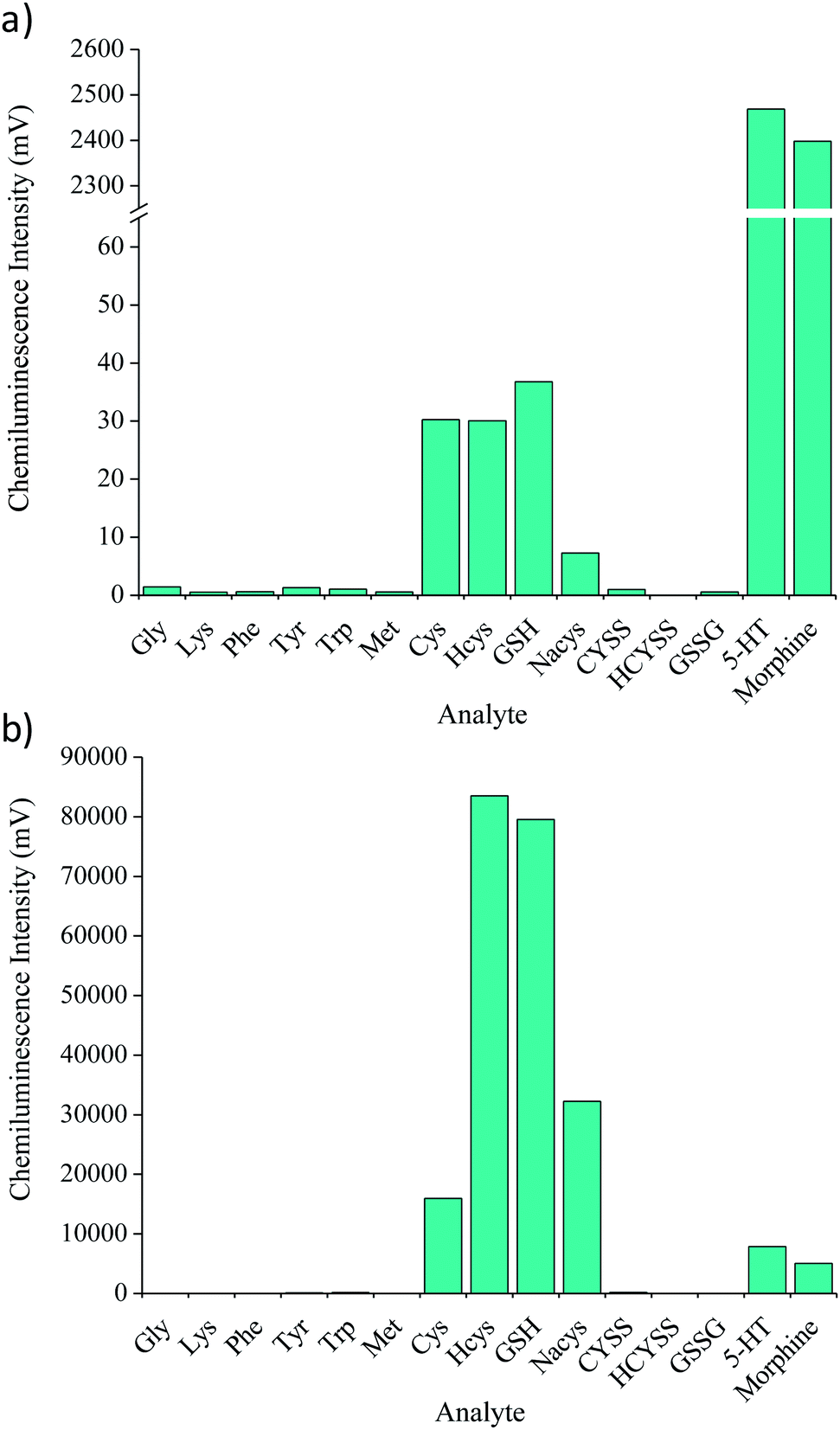 | ||
| Fig. 2 Chemiluminescence intensities for selected analytes (5 × 10−6 M) with (a) acidic potassium permanganate (2.5 × 10−4 M in 1% (m/v) sodium polyphosphate, pH 3) and (b) the permanganate reagent and formaldehyde, using flow injection analysis methodology. | ||
High performance liquid chromatography
A mobile phase consisting of 97% deionised water adjusted to pH 2.8 with formic acid and 3% methanol enabled the separation of four routinely measured thiols in under 15 min with high resolution (Fig. 3). Similar to the observations of the preliminary flow injection analysis experiments, the addition of 2 M formaldehyde to the detection system resulted in an increase in the chemiluminescence response (peak area) for these thiol analytes by approximately two orders of magnitude.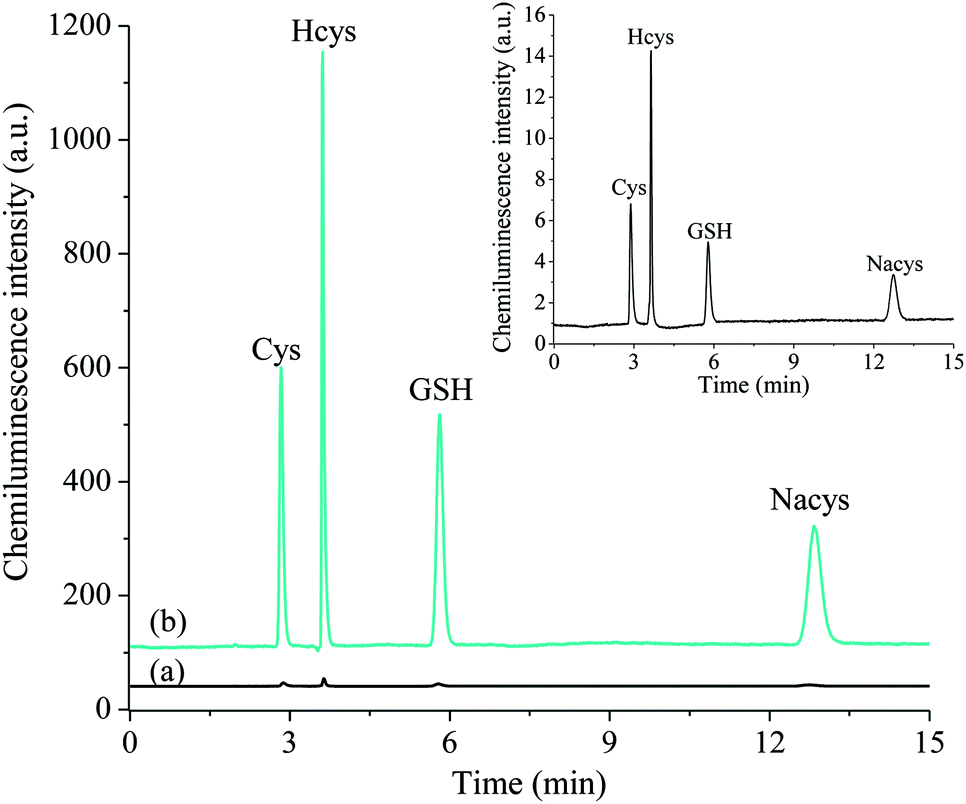 | ||
| Fig. 3 HPLC separation of a mixture of four biologically important thiols; cysteine (Cys), homocysteine (Hcys), glutathione (GSH) and N-acetylcysteine (Nacys) (1 × 10−5 M), using (a) acidic potassium permanganate (2.5 × 10−4 M) chemiluminescence detection (20 μL injection volume) and (b) acidic potassium permanganate and formaldehyde (2.0 M) (10 μL injection). The unenhanced separation is also shown inset. | ||
It should be noted that signal increases obtained when using a formaldehyde enhancer are typically accompanied by a deleterious increase in background emission.17,18 Peristaltic pumps (which are often used to propel reagent and enhancer solutions into the flow-cell for post-column chemiluminescence detection) contribute greatly to baseline noise because of the pulsation of solution flow. To minimise this effect, we employed two dual piston pumps equipped with pulse dampers to deliver reagent and enhancer streams to the detector.
Calibration curves were prepared using eighteen standard solutions between 1 × 10−9 M and 1 × 10−5 M. Each of the thiols exhibited a highly linear relationship between signal (peak area) and analyte concentration (Table 1). The precision of repeated injections (n = 10; 5 × 10−6 M) was excellent, with relative standard deviations of between 0.49% and 1.46%. Limits of detection, defined as a signal to noise ratio of 3, were in the range of 1 × 10−8 M and 5 × 10−8 M. Most importantly, the limit of detection for GSH was 50-fold superior to that of the previously published HPLC procedure utilising permanganate chemiluminescence detection.11
After quantification of the free GSH (Fig. 4a), a second aliquot of sample was used to establish the concentration of GSSG. The free thiol was blocked with the masking agent, N-ethylmaleimide (Fig. 4b) and then the disulfide bond reduction procedure was performed to liberate GSH, which was quantified as described above (Fig. 4c). As such, the gains in sensitivity for GSH were also translated to the detection of GSSG.
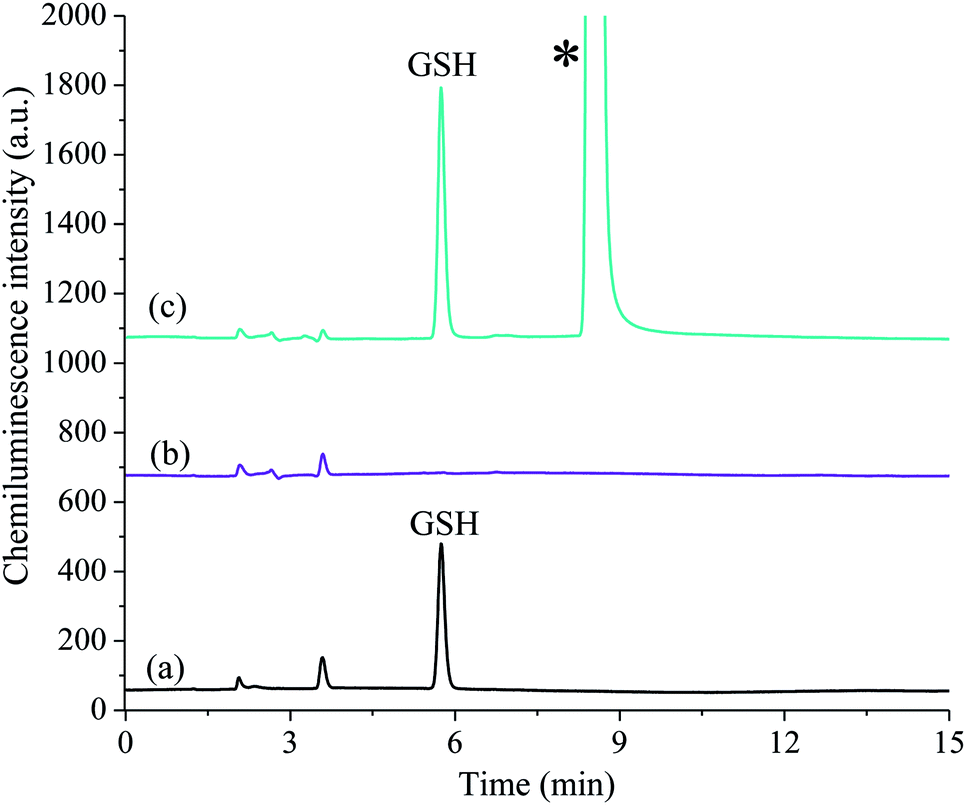 | ||
| Fig. 4 The analysis of a mixture of GSH and GSSG standards (1 × 10−5 M) with post-column chemiluminescence detection using acidic potassium permanganate and a formaldehyde enhancer. (a) Detection of GSH, (b) signal after addition of Tris–HCl buffer and NEM (thiol blocking step), (c) detection of GSSG as GSH after the addition of 2-mercaptoethanol (to react excess NEM) and TCEP (to break the disulfide bond). *This peak corresponds to the excess 2-mercaptoethanol and does not interfere with the analysis. | ||
Determination of GSH/GSSG in mouse striatum
To demonstrate the viability of this method for the analysis of biological tissues, the procedure was applied to the determination of GSH and GSSG in mouse striatum. Progressive degeneration of this brain region is the primary neuropathalogic feature of Huntington's disease.30,31 Several studies have reported signs of oxidative stress in the brain tissue of subjects suffering from the late stages of the disease; however, it is not clear if this begins in early stages or if it is secondary to tissue degeneration.31–33 Quantification of GSH and GSSG in animal models of Huntington's disease is therefore invaluable in determining the stage at which oxidative stress begins to occur.We examined homogenised striatum samples taken from both wild type mice and the R6/1 transgenic mouse model of Huntington's disease, at both 8 weeks of age (when affective-like phenotypes begin to emerge34) and 12 weeks of age (when motor dysfunction occurs20). The acidic supernatant was initially diluted 10-fold into aqueous formic acid (0.01%). A portion of the diluted supernatant was then further diluted 10-fold in 5% formic acid immediately prior to analysis (Fig. 5a). For GSSG determination, a second aliquot of dilute supernatant (100 μL) was subjected to the thiol blocking and disulfide bond reduction procedure as described above. The sample was then re-acidified with 20 μL of 5% formic acid prior to analysis (Fig. 5b).
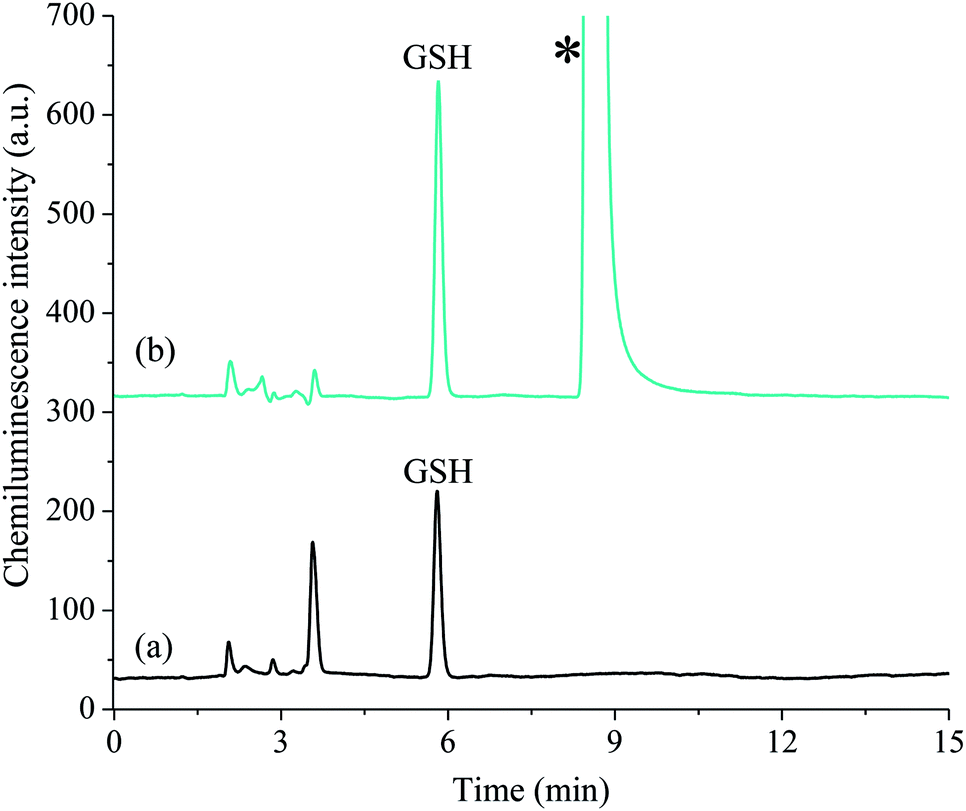 | ||
| Fig. 5 Typical acidic potassium permanganate chemiluminescence chromatograms obtained for the analysis of mouse striatum, (a) detection of GSH (total 1/100 dilution) and (b) detection of GSSG as GSH after performing the disulfide bond reduction procedure (total 1/20 dilution). *This peak corresponds to the excess 2-mercaptoethanol and does not interfere with the analysis. | ||
Using this method, both GSH and GSSG were detected in all samples tested. Despite the complexity of the sample matrix, the selectivity of the reagent combined with the ability to perform large dilutions resulted in only a few observable peaks from other compounds. GSH was present in the range 1.21–3.21 μmol g−1 whilst GSSG was present in the range 0.16–0.44 μmol g−1 (Fig. 6). There was no significant difference in the levels of GSH, GSSG, or the ratio of GSH/GSSG, at either time point (t(8) = 2.89, p = 0.20; see Table S1† for further statistical data). This suggests that the disease progression had not significantly altered the intracellular redox environment. This preliminary result may indicate that significant changes in antioxidant capacity are not occurring in the prodromal stages of the R6/1 Huntington's disease model. However, it may be that such changes in glutathione homeostasis are compartmentalised between different cell types (glia and neurons) and/or subcellular structures (mitochondria and cytoplasm). Thus further analysis is needed to confirm if such changes are occurring. These results do, however, demonstrate the analytical utility of this method for the measurement of GSH/GSSG levels in complex biological matrices.
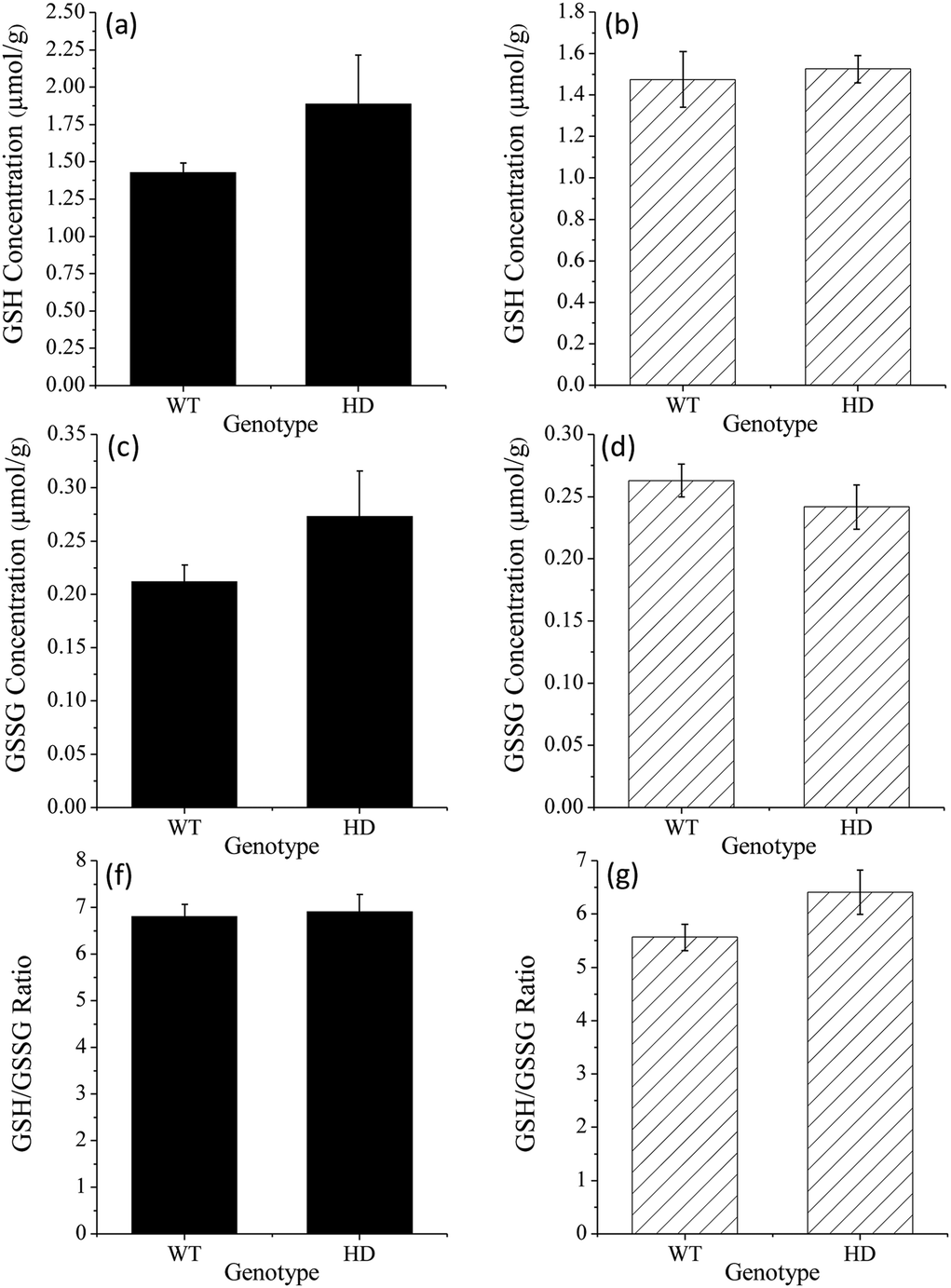 | ||
| Fig. 6 Influence of genotype (wild type (WT) or R6/1 (HD)) on intracellular GSH, GSSG and the GSH/GSSG ratio in moue striatum taken from (a, c, and e) 8 week old and (b, d, and f) 12 week old animals. Values are reported as means of 5 determinations (±SEM). | ||
Conclusions
Applying a formaldehyde enhancer to chemiluminescence reactions with acidic potassium permanganate not only afforded significant increases in signal intensities, but also increased selectivity of the reagent towards thiol compounds. Applying this to the previously developed HPLC method for thiol detection, we were able to improve the detection limits for the important biological thiol GSH by over an order of magnitude whilst also halving the injection volume. The improved method was successfully applied to the detection of GSH/GSSG in mouse striatum.Acknowledgements
The authors thank Deakin University and the Australian Research Council (FT100100646) for funding. The R6/1 mice were generously provided by Dr Anthony Hannan and Dr Thibault Renoir from the Florey Institute of Neuroscience and Mental Health, The University of Melbourne. The R6/1 colony was supported in part by a Future Fellowship (FT100100835) awarded to Dr Hannan. Dean Wright was supported by an Australian Postgraduate Award through the University of Melbourne.Notes and references
- A. Meister and M. E. Anderson, Annu. Rev. Biochem., 1983, 52, 711–760 CrossRef CAS PubMed.
- D. Giustarini, I. Dalle-Donne, D. Tsikas and R. Rossi, Crit. Rev. Clin. Lab. Sci., 2009, 46, 241–281 CrossRef CAS PubMed.
- A. Pastore, G. Federici, E. Bertini and F. Piemonte, Clin. Chim. Acta, 2003, 333, 19–39 CrossRef CAS.
- D. M. Townsend, K. D. Tew and H. Tapiero, Biomed. Pharmacother., 2003, 57, 145–155 CrossRef CAS.
- M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS PubMed.
- T. Toyo'oka, J. Chromatogr. B, 2009, 877, 3318–3330 CrossRef CAS PubMed.
- P. Monostori, G. Wittmann, E. Karg and S. Túri, J. Chromatogr. B, 2009, 877, 3331–3346 CrossRef CAS PubMed.
- R. E. Hansen and J. R. Winther, Anal. Biochem., 2009, 394, 147–158 CrossRef CAS PubMed.
- J. C. Harfield, C. Batchelor-McAuley and R. G. Compton, Analyst, 2012, 137, 2285–2296 RSC.
- S. Bai, Q. Chen, C. Lu and J.-M. Lin, Anal. Chim. Acta, 2013, 768, 96–101 CrossRef CAS PubMed.
- G. P. McDermott, P. S. Francis, K. J. Holt, K. L. Scott, S. D. Martin, N. Stupka, N. W. Barnett and X. A. Conlan, Analyst, 2011, 136, 2578–2585 RSC.
- G. P. McDermott, J. M. Terry, X. A. Conlan, N. W. Barnett and P. S. Francis, Anal. Chem., 2011, 83, 6034–6039 CrossRef CAS PubMed.
- E. Camera and M. Picardo, J. Chromatogr. B, 2002, 781, 181–206 CrossRef CAS.
- F. Carlucci and A. Tabucchi, J. Chromatogr. B, 2009, 877, 3347–3357 CrossRef CAS PubMed.
- Y. Li, A. Zhang, J. Du and J. Lu, Anal. Lett., 2003, 36, 871–879 CrossRef CAS PubMed.
- J. L. Adcock, P. S. Francis and N. W. Barnett, J. Fluoresc., 2009, 19, 867–874 CrossRef CAS PubMed.
- J. L. Adcock, P. S. Francis and N. W. Barnett, Anal. Chim. Acta, 2007, 601, 36–67 CrossRef CAS PubMed.
- J. L. Adcock, N. W. Barnett, C. J. Barrow and P. S. Francis, Anal. Chim. Acta, 2014, 807, 9–28 CrossRef CAS PubMed.
- P. S. Francis, J. L. Adcock and N. W. Barnett, Spectrochim. Acta, Part A, 2006, 65, 708–710 CrossRef PubMed.
- L. Mangiarini, K. Sathasivam, M. Seller, B. Cozens, A. Harper, C. Hetherington, M. Lawton, Y. Trottier and H. Lehrach, et al. , Cell, 1996, 87, 493–506 CrossRef CAS.
- N. T. Deftereos, A. C. Calokerinos and C. E. Efstathiou, Analyst, 1993, 118, 627–632 RSC.
- J. A. Murillo Pulgarín, P. Fernández López and P. Hoyas Nuño, Anal. Bioanal. Chem., 2006, 384, 423–430 CrossRef PubMed.
- J. A. Murillo Pulgarín, L. F. García Bermejo and P. Fernández López, Anal. Chim. Acta, 2005, 546, 60–67 CrossRef PubMed.
- G. N. Chen, F. X. Huang, X. P. Wu, Z. F. Zhao and J. P. Duan, Anal. Bioanal. Chem., 2003, 376, 873–878 CrossRef CAS PubMed.
- S. Liao, X. Wu and Z. Xie, Anal. Chim. Acta, 2005, 537, 189–195 CrossRef CAS PubMed.
- S. Fan, Z. Wu, L. Zhang and C. Lu, Anal. Lett., 2002, 35, 1479–1489 CrossRef CAS PubMed.
- H.-Q. Wei and E.-B. Liu, J. Chin. Chem. Soc., 2005, 52, 1043–1048 CAS.
- N. W. Barnett, B. J. Hindson, P. Jones and T. A. Smith, Anal. Chim. Acta, 2002, 451, 181–188 CrossRef CAS.
- J. L. Adcock, P. S. Francis, T. A. Smith and N. W. Barnett, Analyst, 2008, 133, 49–51 RSC.
- J. P. Vonsattel, R. H. Myers, T. J. Stevens, R. J. Ferrante, E. D. Bird and E. P. Richardson Jr, J. Neuropathol. Exp. Neurol., 1985, 44, 559–577 CrossRef CAS PubMed.
- S. E. Browne, A. C. Bowling, U. MacGarvey, M. J. Baik, S. C. Berger, M. M. K. Muqit, E. D. Bird and M. F. Beal, Ann. Neurol., 1997, 41, 646–653 CrossRef CAS PubMed.
- S. E. Browne, R. J. Ferrante and M. F. Beal, Brain Pathol., 1999, 9, 147–163 CrossRef CAS.
- M. A. Sorolla, G. Reverter-Branchat, J. Tamarit, I. Ferrer, J. Ros and E. Cabiscol, Free Radical Biol. Med., 2008, 45, 667–678 CrossRef CAS PubMed.
- T. Renoir, M. S. Zajac, X. Du, T. Y. Pang, L. Leang, C. Chevarin, L. Lanfumey and A. J. Hannan, PLoS One, 2011, 6, e22133 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Corrected chemiluminescence spectra and statistical data for striatum samples. See DOI: 10.1039/c4an00255e |
| This journal is © The Royal Society of Chemistry 2014 |