 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in sample preparation techniques to overcome difficulties encountered during quantitative analysis of small molecules from biofluids using LC-MS/MS
Caroline
Bylda
ab,
Roland
Thiele
a,
Uwe
Kobold
a and
Dietrich A.
Volmer
*b
aRoche Diagnostics GmbH, Penzberg, Germany
bInstitute of Bioanalytical Chemistry, Saarland University, D-66123 Saarbrücken, Germany. E-mail: Dietrich.Volmer@mx.uni-saarland.de; Fax: +49 681 302 2963; Tel: +49 681 302 3433
First published on 13th February 2014
Abstract
Liquid chromatography-mass spectrometry analysis of small molecules from biofluids requires sensitive and robust assays. Because of the very complex nature of many biological samples, efficient sample preparation protocols to remove unwanted components and to selectively extract the compounds of interest are an essential part of almost every bioanalytical workflow. This review describes the most common problems encountered during sample preparation, ways to optimize established sample preparation techniques and important recent developments to reduce or eliminate major interferents from biofluids.
 Caroline Bylda | Caroline Bylda is a PhD student in Professor Volmer's group at the Institute of Bioanalytical Chemistry, Saarland University, in Saarbrücken, Germany. In 2011, she obtained her Master's degree after completing a research thesis on the LC-MS/MS analysis of Δ9-tetrahydrocannabinol and its metabolites in oral fluid. Her current PhD project is a collaborative effort with Roche Diagnostics in Penzberg, Germany, and focusses on LC-MS/MS methods for quantitative analysis of small molecules in biofluids. |
 Dietrich A. Volmer | Dietrich A. Volmer is Krupp Professor and Chair in Analytical Chemistry, and Director of the Institute of Bioanalytical Chemistry in the Department of Chemistry at Saarland University in Saarbrücken, Germany. He received his PhD in Analytical Chemistry from the University of Hannover, Germany. After postdoctoral research in Jefferson (AR, USA), he held academic appointments in Halifax (NS, Canada) and Cambridge (UK), before moving to Saarbrücken. Dietrich Volmer has also been Editor of Rapid Communications in Mass Spectrometry since 2004. His current research program focuses on bioanalytical chemistry, in particular biological mass spectrometry, quantification techniques, gas phase ion chemistry, chemical separations, imaging mass spectrometry, biomarkers and metabolomics. |
Introduction
The primary goal of sample preparation is to isolate one or several target analytes from the other components of the sample mixture (matrix). Depending on their nature and concentration levels, co-components of the sample matrix can influence the quantitation of target analyte(s) during subsequent liquid chromatography-mass spectrometry (LC-MS) or tandem mass spectrometry (LC-MS/MS) experiments if not removed prior to analysis. The development of new LC-MS/MS methods for small molecules in biological fluids is becoming increasingly more challenging, because of the need to continuously achieve higher sensitivity and better assay robustness in complex biofluids such as serum, plasma, urine, oral fluid or cerebrospinal fluid (CSF). In addition, because of the very low concentration levels of pharmaceutical targets, samples often need to be pre-concentrated before analysis. Unfortunately, this does not only increase the concentration of the desired compound in the sample extract but also often raises the levels of interfering components. As a result, very specific and effective sample clean-up procedures are required for sensitive and selective LC-MS/MS assays today.1This short review presents the main difficulties encountered during sample preparation for analysis of small molecules from biofluids by LC-MS/MS and summarizes several critical factors that particular attention should be paid to, followed by an overview of the latest developments in sample preparation techniques to overcome common difficulties with complex biofluids.
Matrix effects
The general term used today to describe problems encountered during analysis of complex biological samples is “matrix effects”. These effects are usually caused by endogenous (e.g. metabolites of the target analyte, proteins or lipids) or exogenous (all substances introduced during sample processing and analysis) compounds. Depending on their chemical properties, it may or may not be necessary to remove all of these interferents from the sample before injection into the LC-MS system. Also, only matrix compounds coeluting with target analytes during the chromatographic separation prior to MS analysis can cause a change in the response of the analyte, either positive (ion enhancement effect) or negative (ion suppression effect).2Different methods have been presented to examine matrix effects. A common approach is the post-extraction spike method,3–5 where the peak area of the target analyte that has been spiked into the biological matrix prior to the sample preparation is compared to the area of the same analyte spiked post-extraction into the biological fluid extract. The ratio between the two values represents the absolute matrix effect. The relative matrix effect is determined by comparing several lots of the biological matrix.3 Obviously, both absolute and relative matrix effects depend strongly on the target analyte and the ionization technique used for LC-MS/MS.
Another popular method is post-column infusion,6–8 where possible matrix effects are assessed by continuous post-column infusion of the analyte after injection of a processed blank serum sample onto the chromatography column. Any variation of signal intensity at or near the retention times of the analyte would indicate the presence of substances from the matrix interfering with the analysis.
Matrix effects have been shown to be dependent on the ionization methods used for the LC-MS method,3 which are usually either electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) in most modern LC-MS/MS assays. The chemical structures and the concentration levels of both analyte and co-eluting mixture components determine whether they outcompete each other during the ionization process.9 For example, ESI is particularly sensitive to co-eluting phospholipids because ESI is strongly biased towards surfactants,10 which enrich at the surface of the droplets during the liquid/gas-phase ion transfer. That is, phospholipids at the surface of droplets can inhibit ejection of analyte ions trapped inside the droplets. On the other hand, APCI is often less affected by suppression effects, as there is no competition between compounds to enter the gas-phase of the mass spectrometer. Nevertheless, APCI still experiences matrix effects in multicomponent samples. As biofluids contain numerous endogenous molecules, often at high levels, with potentially very high basicities and surface activities, ion suppression effects will almost always be present in any LC-MS/MS assay.
Different strategies are available to eliminate or reduce matrix effects. One approach is to optimize the chromatographic separation to separate the analytes from interfering compounds.1,11,12 This can, however, result in long chromatographic run times. Another approach is to optimize the sample preparation, to obtain clean extracts of the target analytes. With proper sample preparation and the use of isotopically labeled standards, many matrix effects can be eliminated or strongly reduced. Some cases remain, however, where the high variability of the matrix composition makes the use of standard addition calibration necessary.13–16
There are several well-known causes for matrix effects in the analysis of clinically-relevant substances from biological samples. For example, hemolyzed or lipaemic samples have great influence on the analysis of serum and plasma samples.17,18 Cases also have been reported, where buffers used for solid-phase extraction (SPE) triggered matrix effects in LC-MS/MS.19 The most important interferents, however, are phospholipids, which not only affect MS response of many analytes greatly, but which are also very difficult to remove from the samples.
Phospholipids
Phospholipids (PPL) are major constituents of cell membranes and are therefore very abundant in serum and plasma.20 They consist of two functional groups: a hydrophilic head group composed of phosphate and choline units, and a hydrophobic tail, made up of fatty acyl chains. The most abundant phospholipids are glycerophosphocholines (GPChos) (70% of total phospholipids) and lysophosphatidylcholines (10% of total phospholipids) (Fig. 1).11 These two groups are known to cause serious ion suppression effects in LC-MS analysis, caused by competition for space on the surface of droplets formed during the ESI process (vide supra).3,10 Phospholipids are present at different concentration levels in serum and plasma samples, depending on the sampling device used.21 A very simple method to monitor possible ion suppression effects from GPCho was described by Little et al. as in-source multiple reaction monitoring (IS-MRM).22 Using the positive ion mode, a common product ion for the most abundant GPCho is trimethylammonium-ethyl phosphate at m/z 184, which was monitored during analysis of an analyte-free sample. This class-specific product ion was generated using in-source dissociation of the eluting GPCho during the chromatographic run.22 Other methods have been described that allow screening for less abundant phospholipids by adding a precursor ion in the negative mode or by using positive ion neutral loss scans.23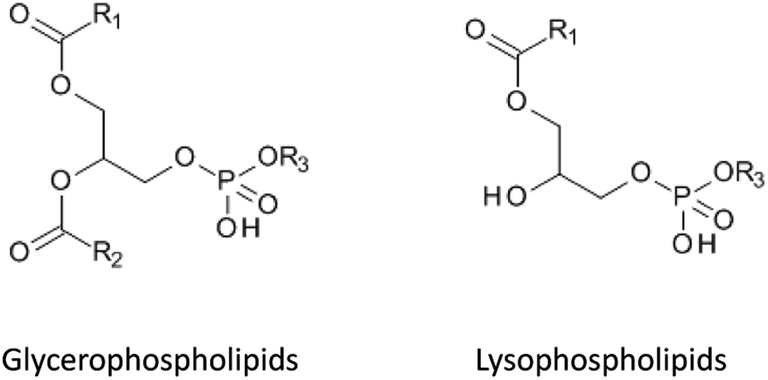 | ||
| Fig. 1 Chemical structures of the two most important groups of phospholipids. | ||
Studies have shown that the use of methanol as a mobile phase for chromatographic separation provided significant advantages over acetonitrile, because elution of all GPCho occurred in a very narrow time window and their retention behavior on reversed-phase columns could be predicted and decreased by increasing the percentage of the organic phase.24 The PPL tended to elute at a high content of the organic mobile phase25 and were completely removed from the system at the end of a run by flushing the analytical column with iso-propanol.26
The behavior of PPL has also been investigated on hydrophobic interaction liquid chromatography (HILIC) columns:27 the compounds were focused into 2 groups of peaks (phosphatidyl cholines and lyso-phosphatidyl cholines) and eluted completely from the column in a one gradient cycle. In comparison, on a reversed-phase material, a strong carry-over was observed from one gradient cycle to another.27
In some cases, where retention times of target analytes and PPL overlapped, elution of the target substance could be shifted after adding mobile phase modifiers.27
Internal standards
The use of isotope-labeled internal standards can help overcome most of the matrix effects during sample preparation and LC-MS/MS analysis. However, in some cases the internal standard cannot completely fulfill its purpose, because of slight differences in the chemical behavior of the target analyte and internal standard. For example, particular attention has to be paid to analytes showing strong protein binding.28 Generally it is necessary to allow enough time for the internal standard to properly equilibrate and bind to the protein before extraction, to ensure identical behavior of the internal standard and target analyte.29 A method has been described to determine the extent of protein binding of corticosteroids.30 In theory, this method could be extended to other substances and be used to compare the protein-affinity of an analyte and its internal standard. It is important that the release of analytes from the protein (e.g. by adding organic solvents for protein precipitation, o-phosphoric acid for breakdown of non-covalent intermolecular interactions31 or dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP) for reduction of disulfide bonds) has the same impact on the analyte and isotope-labeled standard. A case was reported, where the higher susceptibility of the internal standard for matrix effects than the target analyte led to an underestimation of up to 50% in the presence of specific buffers used for SPE sample preparation (Fig. 2).19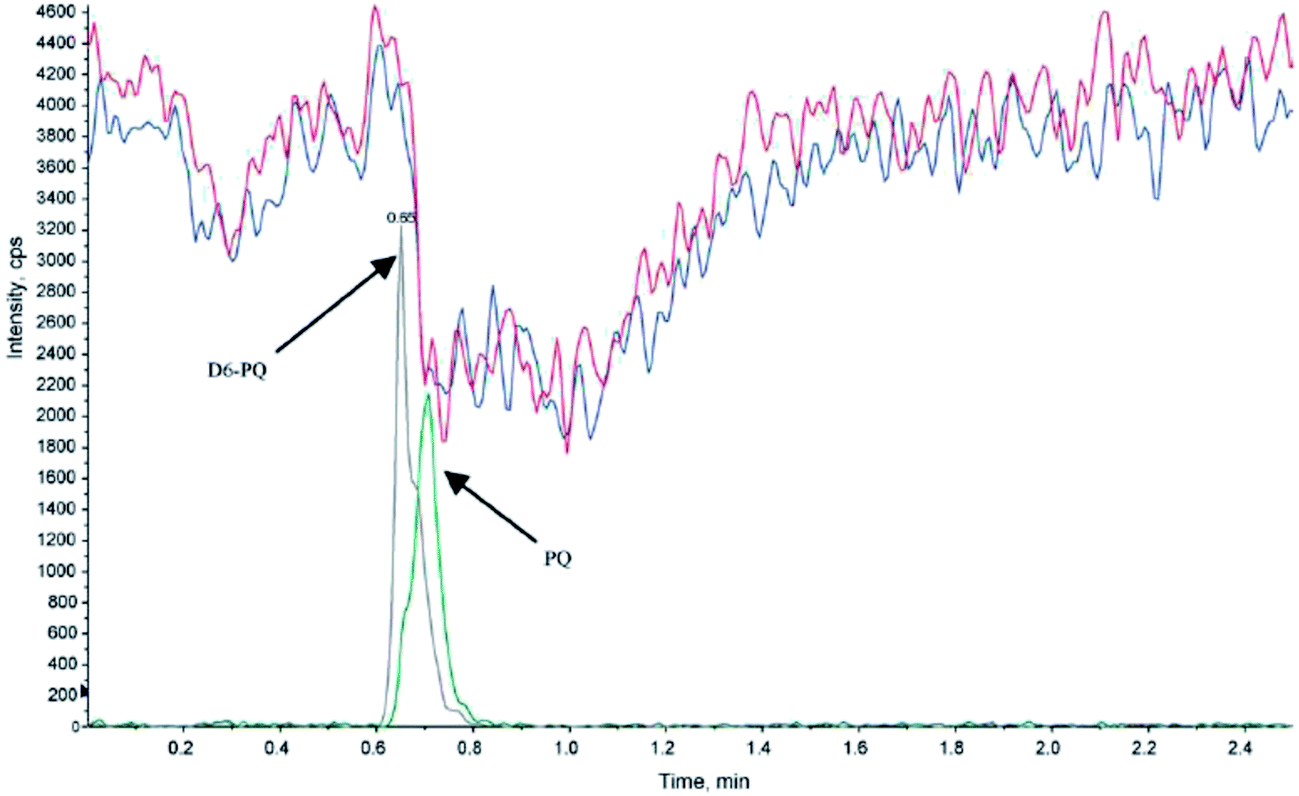 | ||
| Fig. 2 Injection of extracted blank human plasma (+0.2 μL triethylamine, blue and red traces) with an overlay of the control sample (20 ng mL−1, grey and green traces) containing piperaquine (PQ) and internal standard (d6-PQ) during post-column infusion at 10 μL min−1 of PQ and d6-PQ (1.2 ng mL−1). Electrospray ionization of the analytes was performed in positive ion mode; the MRM transitions were m/z 535 → 288 and m/z 541 → 294 for PQ and D6-PQ, respectively (reprinted with permission from ref. 19). | ||
Generally, 13C, 15N or 18O-labeled internal standards are preferable to deuterium labeled analogs,32 because slight differences of physicochemical properties between hydrogen and deuterium can result in small shifts of retention times of the analyte and internal standard. In some cases, this has led to a different degree of ion suppression for the analyte and the internal standard, resulting in changed analyte/internal standard peak area ratios.33,34 Also, deuterium–hydrogen back-exchange can occur, which has led to false positive results.35 Unfortunately, in many cases only deuterated compounds are commercially available, which increases the need to carefully investigate the stability of the reference standards and the influence of matrix effects on the method.
Optimization of established sample preparation methods
Even though there has been some recent interest in quantitative analysis of pharmaceutical compounds from biological samples using ambient, direct mass spectrometry techniques such as desorption electrospray ionization (DESI) or direct analysis in real time (DART), with little or no prior sample preparation or chromatography,36 sample clean-up remains a critical step in most LC-MS analyses of small molecules in biofluids.Protein precipitation
The simplest sample preparation approach for biofluids is protein removal. Proteins can be denatured using acids or heat, or removed by using ultrafiltration cut-off membranes.37 Another possibility is to use organic solvents for protein precipitation (PPT). PPT removes a part of the phospholipid content present in serum and plasma samples, depending on the organic solvent used. Studies have shown that methanol extracts contain 40% more phospholipids compared to acetonitrile,11 and are also less clean than tetrahydrofuran or ethanol extracts.38Solid-phase extraction (SPE)
Silica-based sorbents in SPE cartridges have excellent retention capacity for PPL when eluted with 100% acetonitrile.39 Clean extracts were also obtained by including a washing step with up to 50% methanol, but this strongly affected the recovery of polar analytes.39 Large amounts of methanol eluted significant amounts of phospholipids from silica-based reversed-phase SPE cartridges. Methanol contents of 60, 70 and 80% for elution of samples on phenyl, C8 and C18 phases resulted in a high concentration of phospholipids in the extracts. Acetonitrile appeared to be a stronger eluent for phospholipids on reversed-phase materials when present at levels up to 50%. The same study showed that the recovery of lysophosphatidylcholines decreased with the increasing content of acetonitrile (>50%), reaching its minimum at a 100% organic phase.40 The retention of phospholipids on the sorbent increased by interactions with residual silanol groups, as was shown by comparison of endcapped and non-endcapped materials. Silica-based sorbents were compared to polymeric phases regarding extraction of phospholipids, and the tested materials showed comparable efficiency.40Studies comparing different sample preparation methods in terms of matrix effects and analyte recovery demonstrated that mixed-mode strong anion exchange SPE was more effective than PPT and LLE for polar and non-polar analytes in plasma (Fig. 3).11,41
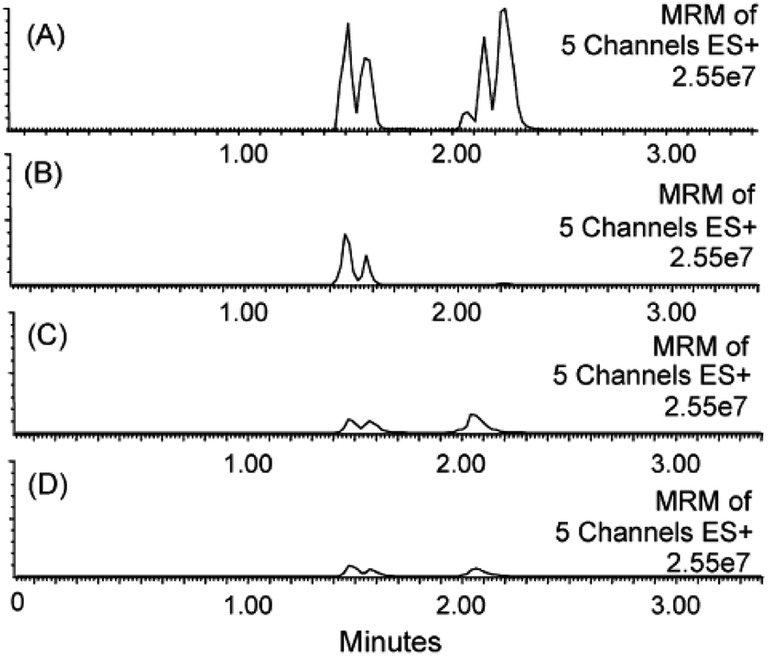 | ||
| Fig. 3 MRM traces for five residual phospholipids in rat plasma extracts after sample preparation by (A) acetonitrile PPT, (B) reversed-phase polymeric SPE, (C) silica-based pure cation exchange, and (D) mixed-mode cation exchange SPE. The phospholipids monitored were 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (m/z 496.35), 1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine (m/z 524.37), 1-hexadecanoyl-2-(9Z,12Z-octadecadienoyl)-sn-glycero-3-phosphocholine (m/z 758.57), 1-(9Z,12Z-octadecadienoyl)-2-(5Z,8Z,11Z,14Z-eicosatetraenoyl)-sn-glycero-3-phosphocholine (m/z 806.57) and a fifth glycerophosphocholine lipid of molecular weight 703.57 Da. MRM analysis was performed on an ESI triple quadrupole LC-MS/MS system using a methanol–water gradient at pH 10 (reprinted with permission from ref. 11). | ||
HILIC-SPE was evaluated as an effective method to remove phospholipids from serum and plasma samples.26 The retention of phospholipids was shown to increase when samples were diluted with acetone. For some applications to urine samples, HILIC materials were more effective than reversed-phase materials.42 The polar metabolites in urine had to be separated from the salts and other polar components present in urine. Orthogonal separation using both HILIC and reversed-phase materials for sample preparation and chromatography improved the effectiveness of sample clean-up.42
Overall, SPE has a very broad range of applications in the LC-MS/MS quantification of small molecules in biofluids.43–47
Liquid–liquid extraction (LLE)
Liquid–liquid extraction has found numerous applications for analysis of pharmaceuticals and their metabolites. The concentration of residual phospholipids in the extract is usually lower compared to other techniques such as mixed-mode SPE; on the other hand, the extraction efficiency for highly polar analytes is also lower.29 The choice of extraction solvent is very important to reduce unspecific extraction of matrix components.41 Halogenated solvents such as chloroform or dichloromethane48–50 are commonly used in combination with hydrophilic solvents (e.g. alcohols) for extraction of polar compounds; they also have high affinity for lipids.38As non-ionized analytes are more efficiently extracted by organic solvents than charged species, particular attention has to be paid to the pH of the sample prior to LLE. As a general rule, the pH should be between pKa and (pKa − 2) for acidic analytes and between pKa and (pKa + 2) for basic analytes,51 to increase the extraction recovery. This obviously applies only if the stability of the main analyte and its potentially labile metabolites is given in this pH range.29
Extraction using methyl-tert-butylether (MTBE) has shown good results,52 but significantly lower analyte recoveries were seen compared to mixed mode SPE and PPT, especially for polar analytes.11 Only traces of phospholipids were found in MTBE and n-butylchloride extracts of serum and plasma samples.53 However, particular attention has to be paid to the process, when several sample preparation steps are combined. The clean extracts obtained with MTBE for untreated serum or plasma can show a high recovery for phospholipids if the samples contain a high percentage of acetonitrile, e.g. after protein precipitation (Fig. 4).53
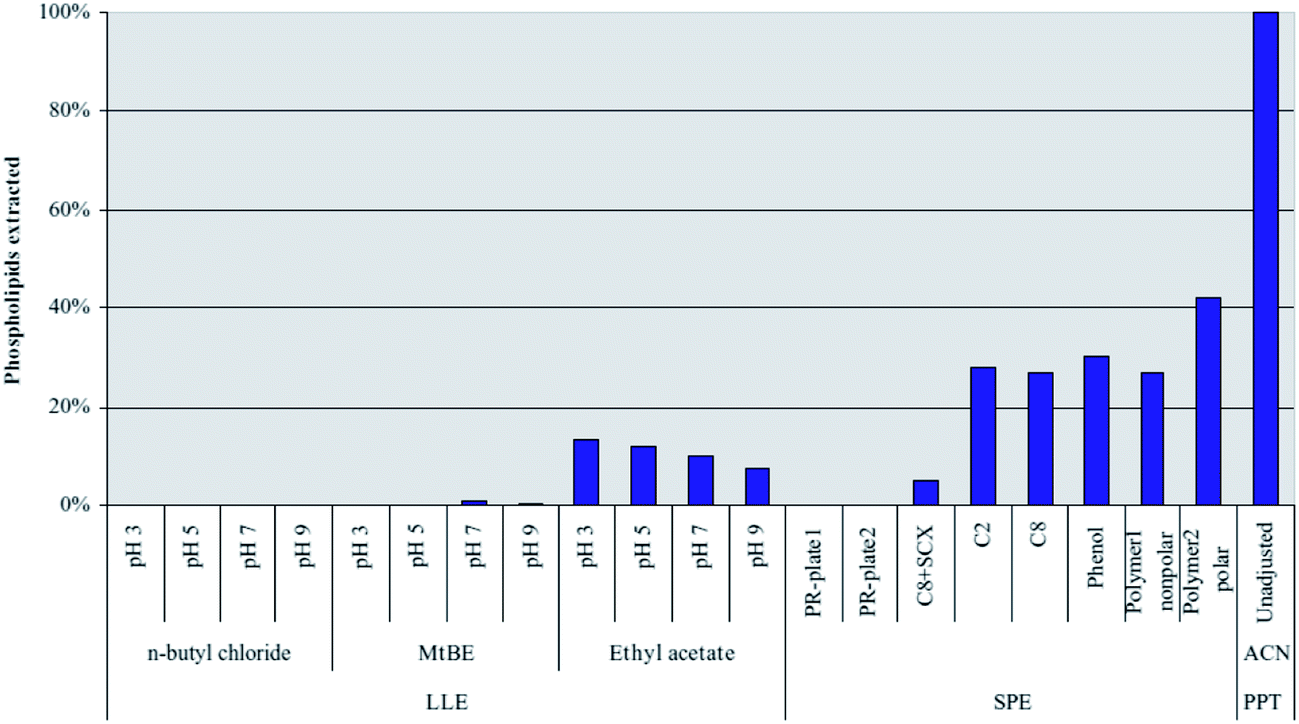 | ||
| Fig. 4 Extraction of C16:0 lysophosphatidylcholine (C16:0 lyso-PC) from human plasma using liquid–liquid extraction with three different solvents at different pH values. Comparison to solid-phase extraction and two commercial phospholipid removal sorbents (PR-plate 1 and PR-plate 2). Lyso-PC was monitored using the following MRM transition: m/z 496 → 184 (reprinted with permission from ref. 53). | ||
Extraction time also plays an important role for the specific extraction of target analytes compared to matrix components. A study showed that a 5 min extraction time yielded a cleaner extract and better recovery for the target compound than 20 min, indicating that matrix compounds diffuse slower into the extraction solvent.52
To improve low recovery rates of LLE for strongly hydrophilic compounds, extraction procedures using water miscible solvents have been considered. Complex methods were reported in the past that use temperatures below 0 °C to achieve phase separation of serum samples and extraction solvent.54 A more convenient way to achieve phase separation between an aqueous sample and a water-miscible solvent is salt-assisted liquid–liquid extraction (SALLE), where the polarity of the aqueous phase is increased by adding high concentration of salt, leading to phase separation.55 This approach has been used for quantitation of pharmaceutical compounds from biofluids using LC56 or LC-MS/MS.57–59
Novel sample preparation methods
Many common interferents can be removed with conventional sample preparation methods (e.g. protein precipitation, SPE, and LLE), but optimization of these techniques for specific applications is often complex, time-consuming and frequently involves multiple steps. Many common interferents can be removed with conventional sample preparation methods (e.g. protein precipitation, SPE, and LLE), but optimization of these techniques for specific applications is often complex, time-consuming and frequently involves multiple steps. Moreover, some challenges involving very small sample volumes and low abundant analytes remain. If repeated analyses are required from the same sample and if no further sampling is possible, sample preparation sometimes has to be performed using a sample volume as low as a few microliters. Similar difficulties apply to assays for metabolites or biomarkers that are present at very low concentration levels in human samples. Here, the method must be able to pre-concentrate the target substance(s), additionally to removing all other components of the matrix. New developments for sample preparation methods are therefore often directed towards simplification and possible automation, miniaturization and specificity enhancements of the clean-up process. New developments for sample preparation methods are therefore often directed towards simplification and possible automation, miniaturization and specificity enhancements of the clean-up process. In the following, the most promising recent developments are briefly summarized.Supported liquid extraction (SLE)
Even though LLE is mostly a very effective sample preparation method, it has limitations, in particular low sample throughput. Several extractions are often required to improve analyte recovery, sample handling is labor-intensive and time-consuming, and emulsions can form at the interface between liquid layers. These limitations can be overcome by using supported liquid extraction (SLE), where aqueous samples are adsorbed on a porous solid support material, e.g. diatomaceous earth. Some studies have shown analyte recovery from SLE that was comparable or higher than LLE.60SLE has been shown to effectively remove the majority of phospholipids when the extraction conditions were carefully optimized.61 The efficiency of several extraction solvents was also compared for SLE:7 ethyl acetate removed about 85%, MTBE removed more than 99% of total phospholipids. Dichloromethane removed 99.5% of the phospholipids when used alone; its removal efficiency decreased to 95% when isopropanol was added. However, addition of water-soluble solvents to the samples (e.g., acetonitrile or methanol) prior to SLE extraction led to higher matrix effects for some analytes.7 Isopropanol combined with dichloromethane also yielded low concentrations of phospholipids in the extract.7
The SLE technique has been implemented in various LC-MS/MS methods recently.62–67 It was particularly powerful for normal phase separation systems, since the high percentage of organic solvent in the eluate did not need to be evaporated prior to injection into the LC-MS/MS system.68
Phospholipid removal plates
The use of hybrid precipitation/SPE plates for selective removal of phospholipids and precipitated proteins has been increasing over the past few years.4,38,69,70 Several types of these plates are now commercially available, e.g. Hybrid SPE™ (Sigma Aldrich), Ostro™ (Waters), Captiva™ ND (Agilent) and Phree™ (Phenomenex). These plates have shown very effective extractions of phospholipids compared to PPT.71 For example, the Hybrid SPE plate specifically retains phospholipids by Lewis acid–base interactions between zirconia ions – which are bonded to the stationary phase – and the phosphate group of the phospholipids. Acetonitrile with 1% formic acid is used as the precipitation agent; formic acid has important influence on the recovery of the analytes.72 Hybrid SPE extracts have shown to contain significantly lower phospholipid concentrations as compared to PPT.4 Ostro uses a combination of protein precipitation and extraction on a C18 sorbent. Several applications using these products have been reported.73–75Other approaches are also possible for removal of phospholipids. A study showed that addition of a colloidal silica suspension together with lanthanum chloride to plasma samples resulted in a reproducible sample clean-up without loss of the analyte of interest.76
Magnetic beads
Magnetic particles and nanoparticles (MNPs) are becoming increasingly interesting for sample preparation. They have been used for extraction and pre-concentration of drugs in complex biological fluids.77–79 They consist of a magnetic core (e.g. Fe3O4) coated with a polymer material, to which specific functionalities can be added (Fig. 5).80 Sample preparation steps are similar to SPE (loading, washing and elution). The magnetic particles suspended in solution can be handled as a liquid. Obviously, the big advantage of magnetic beads is that after sample extraction, the beads are pulled to the tube wall, the supernatant is removed and the wall-bound beads washed with an appropriate solvent. The loaded beads are then re-suspended. The entire procedure is fast and simple, and complete automation is readily possible.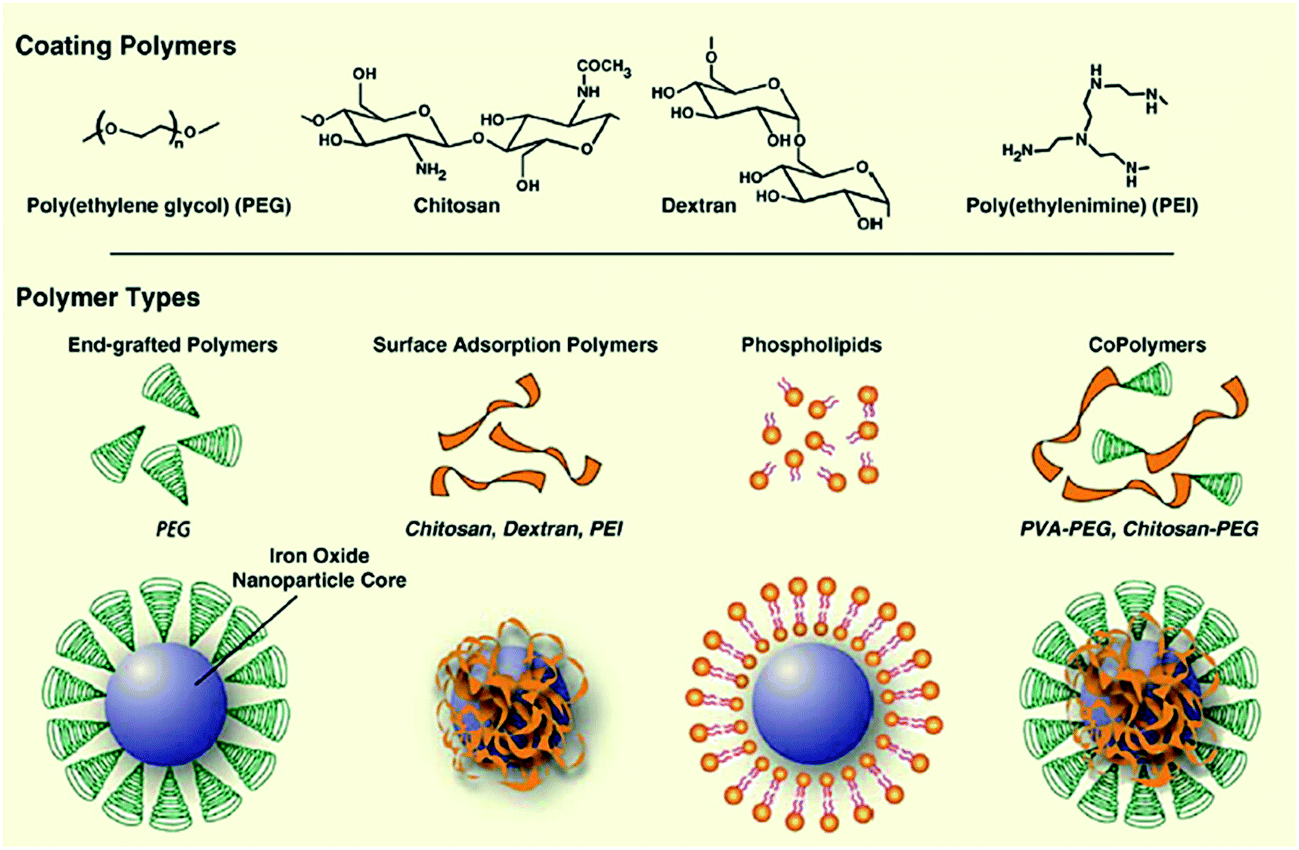 | ||
| Fig. 5 Assembly of polymers onto the surface of magnetic nanoparticle cores (reprinted with permission from ref. 168). | ||
Several applications have been reported, where either analytes are selectively extracted from a complex matrix81–83 or where the matrix components were removed from the sample, leaving a clean extract behind that can be directly injected into the LC-MS system.84 Using matrix-assisted laser desorption/ionization (MALDI), the analytes can also be analyzed without having to be eluted from the magnetic beads first.85 The possible modifications on the surface of the magnetic beads are similar to conventional SPE and involve hydrophobic coatings, ion exchange functionalities, molecular imprinted polymers (MIP),86 restricted access87 or affinity materials.88 Magnetic particles have also been coated with carbon nanotubes and used to extract aromatic compounds.89
Turboflow
Turboflow extraction is usually carried out online before chromatographic separation and uses columns with large particle sizes in conjunction with high flow rates.90 Samples can be directly injected after dilution; sometimes a protein precipitation step is required before injection. The target analytes are retained in the pores of the column, whereas matrix components are flushed through and discarded directly to waste. The analytes are then eluted from the trapping column using organic solvents. This method has the advantage of fast and generic method development but unfortunately it can also show high carry-over effects.91 A study reported that this technique had no significant impact on phospholipid removal from serum and plasma samples, and still needed extensive chromatographic separation after clean-up to avoid matrix effects.92 Other groups reported successful applications for quantification of various substances (drugs, steroids, phenolic compounds, etc.)93–102 in human serum, urine and dried blood spots using reversed-phase, ion exchange or mixed-mode materials.Monolithic spin column extraction
Monolithic spin column extraction is a fast sample preparation method that uses a spin column packed with octadecyl silane-bonded monolithic silica as the extraction device.103 The sample is loaded onto the sorbent by centrifugation; the same procedure is performed for washing and elution steps.104 This technique is fast and easy, requires only small amounts of solvents and allows high sample throughput. Unfortunately, the method can only be applied over a limited pH range because of possible degradation of the monolithic silica phase.104 Several applications have been reported for quantification of various analytes from human samples, using underivatized,105 C18,106–108 ion exchange109 or mixed-mode phases (C–C18, TiO–C18, C18-ion exchange).110–112Microextraction by packed sorbent (MEPS)
This recent sample preparation technique is based on the miniaturization of conventional SPE, using a gas-tight syringe as extraction device. The method is designed for sample volumes from 10 to 1000 μL and can be connected online to LC-MS or GC-MS. Compared to conventional SPE, MEPS is easy to use, faster and needs significantly lower amounts of organic solvents. Additionally, MEPS sorbents can be used for up to 100 extractions.113Packing materials for MEPS are similar to sorbents used for SPE. Essentially, any sorbent material and functionalization can be applied. For example, silica-based materials (C2, C8, C18),114–118 with additional ion exchange functionality119 or even as mixed-mode materials,120 restricted access materials (RAM), HILIC, carbon, polystyrene–divinylbenzene copolymers (PS–DVB) or molecular imprinted polymers113,121 have been utilized for MEPS.
The method has been implemented in several recent reports for quantification of pharmaceutical compounds from human biological samples (urine, plasma, oral fluid and whole blood), including antipsychotic drugs,119 cardiac drugs,114 local anesthetics,115,121 phenolic acid,116 immunosuppressants,117 opioids120 and antidepressants.118 Recent studies have also reported the successful extraction of trazodone from plasma with polymer nano-fibers as the extraction sorbent.122
Carbon nanotubes
Carbon surfaces have the ability to retain substances by strong hydrophobic interactions. These materials are therefore interesting for reversed-phase extractions of hydrophilic substances. Carbon nanotubes (CNTs) are hollow cylinders that consist of one (single-wall carbon nanotubes, SWCNTs) or several (multi-walled carbon nanotubes, MWCNTs) graphene layers.123 Because of their large surface areas, CNTs have a high adsorption capacity. They show high affinity towards aromatic compounds that can be adsorbed via π–π interactions.124 CNTs can be packed into SPE cartridges or used for dispersive solid phase extraction.125–127 Common target analytes are small, hydrophobic molecules extracted from water samples. Very few applications to biofluids have been reported so far. A method for quantitation of diuretics from urine128 has been published as well as plasma peptide analysis.129 The specificity of the extraction can be enhanced by derivatizing the surface of CNTs with functional groups. A method was recently shown for the determination of anti-inflammatory drugs from urine using carboxylated CNTs for sample clean-up.130 To further improve both specificity and handling of the sample clean-up, magnetic CNTs coated with molecular imprinted polymers have been synthetized and used for extraction of BSA from serum samples.131Restricted access materials (RAM)
Restricted access materials allow extractive clean-up of biofluids by utilizing physical and chemical diffusion barriers. RAM consist of a porous material with a restrictive and hydrophilic outer surface that prevents retention of large interfering molecules such as proteins and phospholipids, combined with smaller inner pores with hydrophobic surfaces that only molecules with low molecular weight can reach.132 This technique is commonly used for online sample clean-up, with the advantage that samples dissolved in almost any solvent can be loaded, even MS incompatible solvents, before elution with the mobile phase used for chromatographic separation. There are two types of RAM phases:133 internal surface phase (ISP) materials use size exclusion to prevent the matrix components from reaching the inner layer; semi-permeable surface (SPS) materials chemically exclude matrix components by polymeric- or protein coating of the outer layer. In both cases, the inner layer can be functionalized to enhance the specificity of the method.134 Molecular imprinted polymers are a special form of restricted access materials; they are discussed below.Application of sample clean-up using RAM includes quantification of antimicrobial agents, immunosuppressants etc. from human biological samples prior to LC-MS/MS analysis.135,136 RAM have also been used in combination with magnetic particles to quantify therapeutic drugs and steroids from biofluids.87,137–140 An application was published that reported the synthesis of chiral RAM materials for extraction of enantiomeric drugs from plasma samples.141
Immunosorbents
Immunosorbents use the principle of antigen–antibody affinity for highly specific retention of target substances. The desired antibody is bound to a solid support or gel, which can be used as SPE or micro-SPE sorbent, MEPS or in columns.123 The target analytes can be specifically extracted from complex matrices, which allows thorough sample clean-up prior to instrumental analysis. A study has shown that the capacity of monoclonal antibodies was significantly higher than that of polyclonal antibodies.142 This technique has been used as in-tube SPME to quantify interferon α from plasma samples143 as well as SPE extraction of ProGRP144 and ochratoxin145 from serum. Sample preparation techniques with high specificity towards the target analyte are required if the target analyte is present at very low concentration levels or in cases where structurally similar interferents (e.g. isobars) influence the analysis.146 The immunosorbent extraction usually involves high costs, however, and also requires host animals to grow the required antibodies. Sometimes, the antibodies can be replaced by synthetic alternatives of comparable specificity, such as molecular imprinted polymers or aptamers (see below).Molecular imprinted polymers (MIPs)
MIPs use the principle of affinity chromatography to maximize the specificity for the analyte(s) of interest. The target analyte or a structurally-related compound is used as a template for the synthesis of the MIP by copolymerization of a complex formed by the template and a functional monomer. The template molecule is then removed, leaving a rigid three-dimensional cavity that is complementary to the target analyte.147The synthesis of these adsorbents is often inexpensive and has shown to be fast and reproducible; the materials also have high capacity and can be regenerated and used several times.148 The MIP principle enables highly specific extraction of the target and structurally similar compounds (e.g. a drug and its metabolites) from complex matrices, and pre-concentration of the sample. The specificity of this technique has been shown in several applications. For example, a MIP sorbent developed for tylosin was able to differentiate between tylosin and the closely related narbomycin as well as the remotely similar tylactone. (Fig. 6). Both the target analyte and structurally similar compound were quantitatively extracted, whereas the interfering substance did not show any affinity for the sorbent.149
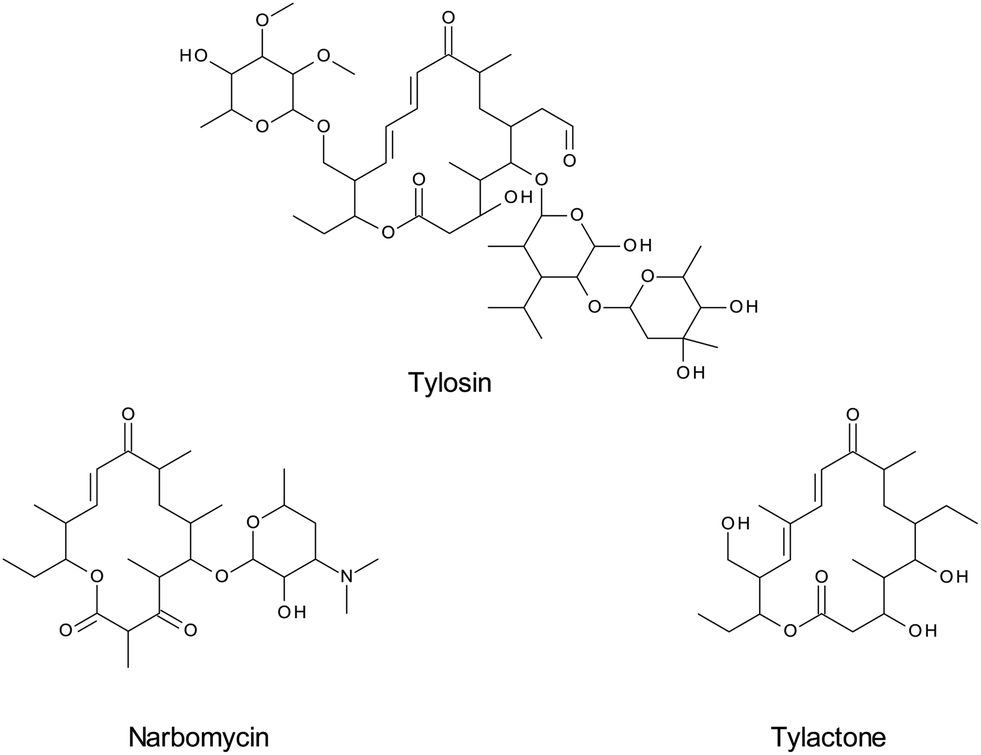 | ||
| Fig. 6 Structures of tylosin and two structurally-related compounds, narbomycin and tylactone. | ||
MIP can be used in various forms, for online or off-line processes such as molecular imprinted solid phase extraction (MISPE),150 magnetic MIP,151,152 solid-phase micro-extraction (SPME), needle/micropipette tip, dynamic liquid–liquid–solid micro-extraction (DLLSME) or molecular imprinted stir-bar sorptive extraction (MI-SBSE).153,154 This concept has been applied to samples with complex matrices, for example, for benzodiazepines in plasma,155 nucleoside reverse transcriptase inhibitors in serum,156 cocaine157 or ketamine158 from hair extract, testosterone159 and tobacco-specific cancer biomarkers160 from urine. MIP-coated fibers for solid phase microextraction (SPME) have also been used for extraction of linezolid from human biofluids.161 This technique has shown to provide much cleaner extracts than other sample preparation methods such as LLE.155 However, this technology still needs some improvement and has several drawbacks, including possible template bleeding, sometimes tedious synthesis procedures, and problematic application to aqueous samples.147,150
Aptamers
Another possibility to increase specificity for the target analyte is the use of aptamers immobilized on a solid sorbent for sample preparation. Aptamers are synthetic single-stranded oligonucleotides capable of binding specific analytes with a high affinity through hydrogen bonding, van der Waals forces and dipole interactions.123,162 They are specifically prepared for each target molecule; that is, several nucleic acids have to be tested in vitro for each target. Selected nucleic acids with high affinity for the analytes are isolated and amplified using a process called systematic evolution of ligands by exponential enrichment (SELEX).163 The major advantage compared to antibodies is that aptamers can be synthetized directly, without the need for laboratory animals. They can be regenerated within minutes and reused several times. The technique has been used for the selective extraction of cocaine from plasma164,165 and for extraction of tetracyclines from biological fluids in combination with ion mobility spectrometry.166 The high affinity of a target substance to an extraction sorbent is clearly shown in these applications as well as the importance of the sequence of the oligonucleotides. The sequence is specific for a particular compound and will become inactive if the oligonucleotides are grafted in a randomized order.163 Recoveries of up to 90% confirm the high specificity of this technique, even in complex samples such as plasma.163 Aptamers have also been immobilized on polymeric nano-fibers and extraction of thrombin from serum was shown.167Conclusion
Common problems encountered during development of an LC-MS/MS assay for the quantification of small molecules from biological samples include loss of sensitivity and specificity due to matrix effects. Sample preparation is therefore an indispensable part of the analytical workflow. The possible influence of matrix effects on LC-MS/MS assays has been extensively studied and several methods have been published to identify and avoid these effects. Considerable progress has been made in the improvement of sample preparation routines in the last few years. New trends are directed towards either increasing the specificity of the extraction for the target analyte or removing as much of the matrix components as possible. Miniaturization and automation of these techniques are on-going efforts, leading to cheaper, more robust and fully automated LC-MS/MS assays that will significantly impact pharmaceutical analyses of biofluids in the future.List of abbreviations
| APCI | Atmospheric pressure chemical ionization |
| BSA | Bovine serum albumin |
| CNT | Carbon nanotubes |
| CSF | Cerebrospinal fluid |
| DART | Direct analysis in real time |
| DESI | Desorption electrospray ionization |
| DLLSME | Dynamic liquid–liquid–solid microextraction |
| DTT | Dithiothreitol |
| ESI | Electrospray ionization |
| GC–MS | Gas chromatography-mass spectrometry |
| GPCho | Glycerophosphocholines |
| HILIC | Hydrophobic interaction liquid chromatography |
| IS-MRM | In-source multiple reaction monitoring |
| ISP | Internal surface phase |
| LC-MS | Liquid chromatography-mass spectrometry |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| LLE | Liquid–liquid extraction |
| MALDI | Matrix-assisted laser desorption/ionization |
| MEPS | Microextraction by packed sorbent |
| MIP | Molecular imprinted polymers |
| MI-SBSE | Molecular imprinted stir-bar sorptive extraction |
| MISPE | Molecular imprinted solid phase extraction |
| MNP | Magnetic nanoparticles |
| MRM | Multiple reaction monitoring |
| MTBE | Methyl-tert-butylether |
| MWCNT | Multi-walled carbon nanotubes |
| PPL | Phospholipids |
| PPT | Protein precipitation |
| PQ | Piperaquine |
| PS–DVB | Polystyrene–divinylbenzene |
| RAM | Restricted access materials |
| SALLE | Salt-assisted liquid–liquid extraction |
| SELEX | Systematic evolution of ligands by exponential enrichment |
| SLE | Supported liquid extraction |
| SPE | Solid-phase extraction |
| SPME | Solid-phase microextraction |
| SPS | Semi-permeable surface |
| SWCNT | Single-wall carbon nanotubes |
| TCEP | Tris(2-carboxyethyl)phosphine |
Acknowledgements
DAV acknowledges research support by the Alfried Krupp von Bohlen und Halbach-Stiftung.References
-
Using Mass Spectrometry for Drug Metabolism Studies, ed. W. A. Korfmacher, 2004 Search PubMed
.
- Y. Modhave, Int. J. Pharm. Phytopharm. Res., 2012, 1, 403–405 CAS
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS
- V. Pucci, S. Di Palma, A. Alfieri, F. Bonelli and E. Monteagudo, J. Pharm. Biomed. Sci., 2009, 50, 867–871 CrossRef CAS PubMed
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 1998, 70, 882–889 CrossRef CAS
- R. Bonfiglio, R. C. King, T. V. Olah and K. Merkle, Rapid Commun. Mass Spectrom., 1999, 13, 1175–1185 CrossRef CAS
- H. Jiang, H. Cao, Y. Zhang and D. M. Fast, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2012, 891, 71–80 CrossRef PubMed
- W. X. Lv, Talanta, 2010, 80, 1406–1412 CrossRef CAS PubMed
- L. L. Jessome and D. A. Volmer, LCGC North Am., 2006, 24, 498–510 CAS
- F. Janusch, L. Kalthoff, G. Hamscher and S. A. Mohring, Bioanalysis, 2013, 5, 2101–2114 CrossRef CAS PubMed
- E. Chambers, D. M. Wagrowski-Diehl, Z. L. Lu and J. R. Mazzeo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2007, 852, 22–34 CrossRef CAS PubMed
- Z. Q. Ye, H. Tsao, H. Gao and C. L. Brummel, Bioanalysis, 2011, 3, 1587–1601 CrossRef CAS PubMed
- M. Ruggieri, C. Tortorella, E. Ceci, D. Paolicelli, V. Solfrizzi, G. Di Bitonto, C. Pica, M. Mastrapasqua, P. Livrea and M. Trojano, Age Ageing, 2011, 40, 391–395 CrossRef PubMed
- S. Kowal, P. Balsaa, F. Werres and T. C. Schmidt, Anal. Bioanal. Chem., 2013, 405, 6337–6351 CrossRef CAS PubMed
- P. Basilicata, N. Miraglia, M. Pieri, A. Acampora, L. Soleo and N. Sannolo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2005, 818, 293–299 CrossRef CAS PubMed
- A. G. Frenich, J. L. M. Vidal, J. L. F. Moreno and R. Romero-Gonzalez, J. Chromatogr. A, 2009, 1216, 4798–4808 CrossRef PubMed
- N. C. Hughes, N. Bajaj, J. A. Fan and E. Y. K. Wong, Bioanalysis, 2009, 1, 1057–1066 CrossRef CAS PubMed
- R. Lucena, P. Moreno, A. Perez-Rico and P. J. Ginel, Vet. J., 1998, 156, 127–131 CrossRef CAS
- N. Lindegardh, A. Annerberg, N. J. White and N. P. Day, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 862, 227–236 CrossRef CAS PubMed
- J. J. Myher, A. Kuksis and S. Pind, Lipids, 1989, 24, 408–418 CrossRef CAS
- V. Gonzalez-Covarrubias, A. Dane, T. Hankemeier and R. J. Vreeken, Metabolomics, 2013, 9, 337–348 CrossRef CAS
- J. L. Little, M. F. Wempe and C. M. Buchanan, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2006, 833, 219–230 CrossRef CAS PubMed
- Y. Q. Xia and M. Jemal, Rapid Commun. Mass Spectrom., 2009, 23, 2125–2138 CrossRef CAS PubMed
- S. Silvester and L. Smith, Bioanalysis, 2012, 4, 879–895 CrossRef CAS PubMed
- Z. M. Liang, Bioanalysis, 2012, 4, 1227–1234 CrossRef CAS PubMed
- T. Van Damme, M. Lachova, F. Lynen, R. Szucs and P. Sandra, Anal. Bioanal. Chem., 2013, 406, 401–407 CrossRef PubMed
- B. J. Tan, A. Negahban, T. McDonald, Y. Z. Zhang and C. Holliman, Bioanalysis, 2012, 4, 2049–2058 CrossRef CAS PubMed
- S. L. Zhou, Q. Song, Y. Tang and N. D. Weng, Curr. Pharm. Anal., 2005, 1, 3–14 CrossRef
- D. E. Mulvana, Bioanalysis, 2010, 2, 1051–1072 CrossRef CAS PubMed
- S. Taylor and A. Harker, J. Pharm. Biomed. Sci., 2006, 41, 299–303 CrossRef CAS PubMed
- S. Tulipani, R. Llorach, M. Urpi-Sarda and C. Andres-Lacueva, Anal. Chem., 2013, 85, 341–348 CrossRef CAS PubMed
- S. Wang, M. Cyronak and E. Yang, J. Pharm. Biomed. Sci., 2007, 43, 701–707 CrossRef CAS PubMed
- C. J. Briscoe, M. R. Stiles and D. S. Hage, J. Pharm. Biomed. Sci., 2007, 44, 484–491 CrossRef CAS PubMed
- M. Jemal, A. Schuster and D. B. Whigan, Rapid Commun. Mass Spectrom., 2003, 17, 1723–1734 CrossRef CAS PubMed
- N. W. Davies, J. A. Smith, P. P. Molesworth and J. J. Ross, Rapid Commun. Mass Spectrom., 2010, 24, 1105–1110 CrossRef CAS PubMed
- S. X. Yu, E. Crawford, J. Tice, B. Musselman and J. T. Wu, Anal. Chem., 2009, 81, 193–202 CrossRef CAS PubMed
- C. Bylda, R. Thiele, U. Kobold and D. A. Volmer, Drug Test. Anal., 2013 DOI:10.1002/dta.1527
- C. Ferreiro-Vera, F. Priego-Capote and M. D. Luque de Castro, J. Chromatogr. A, 2012, 1240, 21–28 CrossRef CAS PubMed
- C. Cote, A. Bergeron, J. N. Mess, M. Furtado and F. Garofolo, Bioanalysis, 2009, 1, 1243–1257 CrossRef CAS PubMed
- M. Lahaie, J. N. Mess, M. Furtado and F. Garofolo, Bioanalysis, 2010, 2, 1011–1021 CrossRef CAS PubMed
- S. G. Chinmoy Ghosh, C. P. Shinde and B. Chakraborty, J. Bioequivalence Bioavailability, 2011, 3, 122–127 Search PubMed
- W. Y. Jian, R. W. Edom, Y. D. Xu and N. D. Weng, J. Sep. Sci., 2010, 33, 681–697 CrossRef CAS PubMed
- C. Bylda, A. Leinenbach, R. Thiele, U. Kobold and D. A. Volmer, Drug Test. Anal., 2012, 4, 668–674 CrossRef CAS PubMed
- D. Gode, M. M. Martin, F. Steiner, C. G. Huber and D. A. Volmer, Anal. Bioanal. Chem., 2012, 404, 433–445 CrossRef CAS PubMed
- R. N. X. Xu, L. M. Fan, M. J. Rieser and T. A. El-Shourbagy, J. Pharm. Biomed. Sci., 2007, 44, 342–355 CrossRef CAS PubMed
- N. Nakov, K. Mladenovska, N. Labacevski, A. Dimovski, R. Petkovska, A. Dimitrovska and Z. Kavrakovski, Biomed. Chromatogr., 2013, 27, 1540–1546 CrossRef CAS PubMed
- Z. Leon, A. Chisvert, I. Tarazona and A. Salvador, Anal. Bioanal. Chem., 2010, 398, 831–843 CrossRef CAS PubMed
- E. L. Oiestad, U. Johansen, M. Stokke Opdal, S. Bergan and A. S. Christophersen, J. Anal. Toxicol., 2009, 33, 372–378 CrossRef PubMed
- J. Ni, H. Ouyang, M. Aiello, C. Seto, L. Borbridge, T. Sakuma, R. Ellis, D. Welty and A. Acheampong, Pharm. Res., 2008, 25, 1572–1582 CrossRef CAS PubMed
- A. Tracqui, P. Kintz, B. Ludes and P. Mangin, J. Chromatogr. B: Biomed. Sci. Appl., 1997, 692, 101–109 CrossRef CAS
- G. Hendriks, D. R. A. Uges and J. P. Franke, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2007, 853, 234–241 CrossRef CAS PubMed
- A. F. Aubry, Bioanalysis, 2011, 3, 1819–1825 CrossRef CAS PubMed
- M. Jemal, Z. Ouyang and Y. Q. Xia, Biomed. Chromatogr., 2010, 24, 2–19 CrossRef CAS PubMed
- M. Yoshida and A. Akane, Anal. Chem., 1999, 71, 1918–1921 CrossRef CAS
- I. M. Valente, L. M. Goncalves and J. A. Rodrigues, J. Chromatogr. A, 2013, 1308, 58–62 CrossRef CAS PubMed
- M. Gupta, A. Jain and K. K. Verma, J. Sep. Sci., 2010, 33, 3774–3780 CrossRef CAS PubMed
- J. Zhang, H. Q. Wu, E. Kim and T. A. El-Shourbagy, Biomed. Chromatogr., 2009, 23, 419–425 CrossRef CAS PubMed
- H. Q. Wu, J. Zhang, K. Norem and T. A. El-Shourbagy, J. Pharm. Biomed. Sci., 2008, 48, 1243–1248 CrossRef CAS PubMed
- P. L. Kole, G. Venkatesh, J. Kotecha and R. Sheshala, Biomed. Chromatogr., 2011, 25, 199–217 CrossRef CAS PubMed
- Y. Zhang, H. Cao and H. Jiang, Bioanalysis, 2013, 5, 285–288 CrossRef CAS PubMed
- O. A. Ismaiel, T. Y. Zhang, R. G. Jenkins and H. T. Karnes, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2010, 878, 3303–3316 CrossRef CAS PubMed
- L. J. Owen, F. C. Wu and B. G. Keevil, Ann. Clin. Biochem., 2013 DOI:10.1177/0004563213501478
- M. T. Furlong, B. He, W. Mylott, S. Zhao, T. Mariannino, J. Shen and B. Stouffer, J. Pharm. Biomed. Sci., 2012, 70, 574–579 CrossRef CAS PubMed
- C. Svanstrom, G. P. Hansson, L. D. Svensson and C. J. Sennbro, J. Pharm. Biomed. Sci., 2012, 58, 71–77 CrossRef PubMed
- A. B. Biotage, LC·GC Eur., 2012, 12 Search PubMed
- F. K. Victor and E. Vandell, LC-GC, 2012, 76 Search PubMed
- K. G. P. R. Nageswara Rao, K. V. Sravan Kumarb and B. Ramesh, Anal. Methods, 2013, 5, 6693–6699 RSC
- X. J. a. Y.-L. C. Jiongwei Pan, Pharmaceutics, 2010, 2, 105–118 CrossRef
- M. Moriarty, A. Lee, B. O'Connell, M. Lehane, H. Keeley and A. Furey, Chromatographia, 2012, 75, 1257–1269 CAS
- H. Jiang, Y. Zhang, M. Ida, A. LaFayette and D. M. Fast, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 2162–2170 CrossRef CAS PubMed
- D. Neville, R. Houghton and S. Garrett, Bioanalysis, 2012, 4, 795–807 CrossRef CAS PubMed
- S. Ahmad, H. Kalra, A. Gupta, B. Raut, A. Hussain and M. A. Rahman, J. Pharm. BioAllied Sci., 2012, 4, 267–275 CrossRef PubMed
- J. M. Cunliffe, J. X. Shen, X. R. Wei, D. P. Dreyer, R. N. Hayes and R. P. Clement, Bioanalysis, 2011, 3, 735–743 CrossRef CAS PubMed
- L. Ye, J. Shi, S. Wan, X. Yang, Y. Wang, J. Zhang, D. Zheng and Z. Liu, Biomed. Chromatogr., 2013, 27, 1532–1539 CrossRef CAS PubMed
- K. Ardjomand-Woelkart, M. Kollroser, L. Li, H. Derendorf, V. Butterweck and R. Bauer, Anal. Bioanal. Chem., 2011, 400, 2371–2381 CrossRef CAS PubMed
- S. T. Wu, D. Schoener and M. Jemal, Rapid Commun. Mass Spectrom., 2008, 22, 2873–2881 CrossRef CAS PubMed
- I. I. Sukhanova, M. A. Dikunets, E. D. Viryus and G. M. Rodchenkov, J. Anal. Chem., 2011, 66, 807–814 CrossRef CAS
- J. F. Peter and A. M. Otto, Proteomics, 2010, 10, 628–633 CrossRef CAS PubMed
- M. Tsunehiro, Y. Meki, K. Matsuoka, E. Kinoshita-Kikuta, E. Kinoshita and T. Koike, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 925, 86–94 CrossRef CAS PubMed
- Z. Karimi, L. Karimi and H. Shokrollahi, Mater. Sci. Eng., C, 2013, 33, 2465–2475 CrossRef CAS PubMed
- A. A. Rajabi, Y. Yamini, M. Faraji and S. Seidi, Med. Chem. Res., 2013, 22, 1570–1577 CrossRef CAS
- K. Konig and M. Vogeser, Eur. J. Mass Spectrom., 2012, 18, 413–417 CrossRef PubMed
- M. Vogeser, A. Geiger, R. Herrmann and U. Kobold, Clin. Biochem., 2009, 42, 915–918 CrossRef CAS PubMed
- K. Konig, S. F. Goethel, V. M. Rusu and M. Vogeser, Clin. Biochem., 2013, 46, 652–655 CrossRef PubMed
- D. Gode and D. A. Volmer, Rapid Commun. Mass Spectrom., 2013, 27, 1011–1018 CrossRef CAS PubMed
- X. Liu, J. Liu, Y. Huang, R. Zhao, G. Liu and Y. Chen, J. Chromatogr. A, 2009, 1216, 7533–7538 CrossRef CAS PubMed
- Y. Wang, Y. X. Wang, L. Chen and Q. H. Wan, J. Magn. Magn. Mater., 2012, 324, 410–417 CrossRef CAS PubMed
- Y. Tao, Z. Chen, Y. Zhang, Y. Wang and Y. Cheng, J. Pharm. Biomed. Sci., 2013, 78–79, 190–201 CrossRef CAS PubMed
- N. Rastkari and R. Ahmadkhaniha, J. Chromatogr. A, 2013, 1286, 22–28 CrossRef CAS PubMed
- L. Couchman, Biomed. Chromatogr., 2012, 26, 892–905 CAS
- W. M. Mullett, J. Biochem. Biophys. Methods, 2007, 70, 263–273 CrossRef CAS PubMed
- L. H. Du and R. L. White, Rapid Commun. Mass Spectrom., 2008, 22, 3362–3370 CrossRef CAS PubMed
- E. Abe, F. Ricard, F. Darrouzain and J. C. Alvarez, Anal. Bioanal. Chem., 2013, 405, 239–245 CrossRef CAS PubMed
- H. Frederiksen, L. Aksglaede, K. Sorensen, O. Nielsen, K. M. Main, N. E. Skakkebaek, A. Juul and A. M. Andersson, Int. J. Hyg. Environ. Health, 2013, 216, 710–720 CrossRef CAS PubMed
- T. Soeborg, H. Frederiksen, P. Fruekilde, T. H. Johannsen, A. Juul and A. M. Andersson, Clin. Chim. Acta, 2013, 419, 95–101 CrossRef CAS PubMed
- T. H. Lassen, H. Frederiksen, T. K. Jensen, J. H. Petersen, K. M. Main, N. E. Skakkebaek, N. Jorgensen, S. K. Kranich and A. M. Andersson, Environ. Res., 2013, 126, 164–170 CrossRef CAS PubMed
- L. Couchman, M. Birch, R. Ireland, A. Corrigan, S. Wickramasinghe, D. Josephs, J. Spicer and R. J. Flanagan, Anal. Bioanal. Chem., 2012, 403, 1685–1695 CrossRef CAS PubMed
- L. Couchman, S. L. Buckner, P. E. Morgan, M. M. Ceesay, A. Pagliuca and R. J. Flanagan, Anal. Bioanal. Chem., 2012, 404, 513–523 CrossRef CAS PubMed
- X. He and M. Kozak, Anal. Bioanal. Chem., 2012, 402, 3003–3010 CrossRef CAS PubMed
- T. Bernsmann, P. Furst and M. Godula, Food Addit. Contam., Part A, 2011, 28, 1352–1363 CrossRef CAS PubMed
- T. F. Metz, T. P. Mechtler, J. J. Orsini, M. Martin, B. Shushan, J. L. Herman, R. Ratschmann, C. B. Item, B. Streubel, K. R. Herkner and D. C. Kasper, Clin. Chem., 2011, 57, 1286–1294 CAS
- R. Harlan, W. Clarke, J. M. Di Bussolo, M. Kozak, J. Straseski and D. L. Meany, Clin. Chim. Acta, 2010, 411, 1728–1734 CrossRef CAS PubMed
- A. Namera, M. Nagao, A. Nakamoto, S. Miyazaki and T. Saito, J. AOAC Int., 2011, 94, 765–774 CAS
- X. J. Huang and D. X. Yuan, Crit. Rev. Anal. Chem., 2012, 42, 38–49 CrossRef CAS
- A. Nakamoto, M. Nishida, T. Saito, I. Kishiyama, S. Miyazaki, K. Murakami, M. Nagao and A. Namura, Anal. Chim. Acta, 2010, 661, 42–46 CrossRef CAS PubMed
- T. Saito, T. Fukushima, Y. Yui, S. Miyazaki, A. Nakamoto, A. Namera and S. Inokuchi, Anal. Bioanal. Chem., 2011, 400, 25–31 CrossRef CAS PubMed
- A. Namera, A. Nakamoto, M. Nishida, T. Saito, I. Kishiyama, S. Miyazaki, M. Yahata, M. Yashiki and M. Nagao, J. Chromatogr. A, 2008, 1208, 71–75 CrossRef CAS PubMed
- T. Saito, S. Morita, I. Kishiyama, S. Miyazaki, A. Nakamoto, M. Nishida, A. Namera, M. Nagao and S. Inokuchi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2008, 872, 186–190 CrossRef CAS PubMed
- A. Namera, T. Saito, S. Miyazaki, S. Ohta, H. Oikawa, A. Torikoshi, H. Shiraishi and M. Nagao, Forensic Toxicol., 2013, 31, 312–321 CrossRef CAS PubMed
- T. Saito, H. Aoki, A. Namera, H. Oikawa, S. Miyazaki, A. Nakamoto and S. Inokuchi, Analytical sciences: the international journal of the Japan Society for Analytical Chemistry, 2011, 27, 999–1005 CrossRef CAS
- T. Saito, N. Miura, A. Namera, S. Miyazaki, S. Ohta, H. Oikawa and S. Inokuchi, Chromatographia, 2013, 76, 781–789 CAS
- A. Namera, S. Yamamoto, T. Saito, S. Miyazaki, H. Oikawa, A. Nakamoto and M. Nagao, J. Sep. Sci., 2011, 34, 2232–2239 CrossRef CAS PubMed
- M. Abdel-Rehim, J. Chromatogr. A, 2010, 1217, 2569–2580 CrossRef CAS PubMed
- S. Magiera, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2013, 938, 86–95 CrossRef CAS PubMed
- A. Abdel-Rehim and M. Abdel-Rehim, Biomed. Chromatogr., 2013, 27, 1188–1191 CrossRef CAS PubMed
- S. Peters, E. Kaal, I. Horsting and H. G. Janssen, J. Chromatogr. A, 2012, 1226, 71–76 CrossRef CAS PubMed
- R. Said, A. Pohanka, M. Abdel-Rehim and O. Beck, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2012, 897, 42–49 CrossRef CAS PubMed
- S. Rani, A. Kumar, A. K. Malik and B. Singh, Chromatographia, 2011, 74, 235–242 CAS
- B. M. da Fonseca, I. E. D. Moreno, M. Barroso, S. Costa, J. A. Queiroz and E. Gallardo, Anal. Bioanal. Chem., 2013, 405, 3953–3963 CrossRef CAS PubMed
- R. Said, A. Pohanka, M. Andersson, O. Beck and M. Abdel-Rehim, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2011, 879, 815–818 CrossRef CAS PubMed
- S. M. Daryanavard, A. Jeppsson-Dadoun, L. I. Andersson, M. Hashemi, A. Colmsjo and M. Abdel-Rehim, Biomed. Chromatogr., 2013, 27, 1481–1488 CrossRef CAS PubMed
- X. J. Kang, C. Pan, Q. Xu, Y. F. Yao, Y. Wang, D. J. Qi and Z. Z. Gu, Anal. Chim. Acta, 2007, 587, 75–81 CrossRef CAS PubMed
- F. Augusto, L. W. Hantao, N. G. S. Mogollon and S. C. G. N. Braga, TrAC, Trends Anal. Chem., 2013, 43, 14–23 CrossRef CAS PubMed
- C. Y. Shi, J. R. Meng and C. H. Deng, J. Mater. Chem., 2012, 22, 20778–20785 RSC
- X. L. Hou, Y. L. Wu, T. Yang and X. D. Du, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2013, 929, 107–115 CrossRef CAS PubMed
- M. M. Li, X. G. Liu, F. S. Dong, J. Xu, Z. Q. Kong, Y. B. Li and Y. Q. Zheng, J. Chromatogr. A, 2013, 1300, 95–103 CrossRef CAS PubMed
- X. D. Du, Y. L. Wu, H. J. Yang and T. Yang, J. Chromatogr. A, 2012, 1260, 25–32 CrossRef CAS PubMed
- T. T. Ho, Z. G. Li, H. Y. Lin and M. R. Lee, J. Chin. Chem. Soc., 2013, 60, 1033–1042 CrossRef CAS
- X. Li, S. Y. Xu, C. S. Pan, H. J. Zhou, X. G. Jiang, Y. Mang, M. L. Ye and H. F. Zou, J. Sep. Sci., 2007, 30, 930–943 CrossRef CAS
- B. Suarez, B. M. Simonet, S. Cardenas and M. Valcarcel, J. Chromatogr. A, 2007, 1159, 203–207 CrossRef CAS PubMed
- Z. H. Zhang, X. Yang, X. Chen, M. L. Zhang, L. J. Luo, M. J. Peng and S. Z. Yao, Anal. Bioanal. Chem., 2011, 401, 2855–2863 CrossRef CAS PubMed
- O. Nunez, H. Gallart-Ayala, C. P. B. Martins, P. Lucci and R. Busquets, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2013, 927, 3–21 CrossRef CAS PubMed
- S. H. Yang, H. Fan, R. J. Classon and K. A. Schug, J. Sep. Sci., 2013, 36, 2922–2938 CrossRef CAS PubMed
- S. Souverain, S. Rudaz and J. -L. Veuthey, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 801, 141–156 CrossRef CAS PubMed
- X. L. Zhou, X. Y. Ye and A. M. Calafat, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2012, 881–82, 27–33 CrossRef PubMed
- V. Neu, N. Delmotte, U. Kobold, T. Dulffer, R. Herrmann, H. von der Eltz and C. G. Huber, Anal. Bioanal. Chem., 2012, 404, 863–874 CrossRef CAS PubMed
- L. Ye, Q. Wang, J. P. Xu, Z. G. Shi and L. Xu, J. Chromatogr. A, 2012, 1244, 46–54 CrossRef CAS PubMed
- Y. Sato, E. Yamamoto, S. Takakuwa, T. Kato and N. Asakawa, J. Chromatogr. A, 2008, 1190, 8–13 CrossRef CAS PubMed
- E. Yamamoto, S. Takakuwa, T. Kato and N. Asakawa, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2007, 846, 132–138 CrossRef CAS PubMed
- K. Bentayeb, R. Batlle, C. Sanchez, C. Nerin and C. Domeno, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2008, 869, 1–8 CrossRef CAS PubMed
- F. Gasparrini, G. Cancelliere, A. Ciogli, I. D'Acquarica, D. Misiti and C. Villani, J. Chromatogr. A, 2008, 1191, 205–213 CrossRef CAS PubMed
- N. Delaunay-Bertoncini, V. Pichon and M. C. Hennion, Chromatographia, 2001, 53, S224–S230 CAS
- A. R. Chaves and M. E. Queiroz, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 928, 37–43 CrossRef CAS PubMed
- B. Winther, M. Nordlund, E. Paus, L. Reubsaet and T. G. Halvorsen, J. Sep. Sci., 2009, 32, 2937–2943 CrossRef CAS PubMed
- N. W. Turner, S. Subrahmanyam and S. A. Piletsky, Anal. Chim. Acta, 2009, 632, 168–180 CrossRef CAS PubMed
- C. Yuan, J. Kosewick, X. He, M. Kozak and S. H. Wang, Rapid Commun. Mass Spectrom., 2011, 25, 1241–1249 CrossRef CAS PubMed
- Y. L. Hu, J. L. Pan, K. G. Zhang, H. X. Lian and G. K. Li, TrAC, Trends Anal. Chem., 2013, 43, 37–52 CrossRef CAS PubMed
- E. V. Piletska, R. Burns, L. A. Terry and S. A. Piletsky, J. Agric. Food Chem., 2012, 60, 95–99 CrossRef CAS PubMed
- S. Piletsky, E. Piletska, K. Karim, G. Foster, C. Legge and A. Turner, Anal. Chim. Acta, 2004, 504, 123–130 CrossRef CAS
- A. Martin-Esteban, TrAC, Trends Anal. Chem., 2013, 45, 169–181 CrossRef CAS PubMed
- L. G. Chen and B. Li, Food Chem., 2013, 141, 23–28 CrossRef CAS PubMed
- Z. M. Zhang, W. Tan, Y. L. Hu and G. K. Li, J. Chromatogr. A, 2011, 1218, 4275–4283 CrossRef CAS PubMed
- Z. G. Xu, C. Y. Song, Y. L. Hu and G. K. Li, Talanta, 2011, 85, 97–103 CrossRef CAS PubMed
- R. Jackson, I. Petrikovics, E. P. C. Lai and J. C. C. Yu, Anal. Methods, 2010, 2, 552–557 RSC
- E. C. Figueiredo, R. Sparrapan, G. B. Sanvido, M. G. Santos, M. A. Z. Arruda and M. N. Eberlin, Analyst, 2011, 136, 3753–3757 RSC
- S. V. Duy, I. Lefebvre-Tournier, V. Pichon, F. Hugon-Chapuis, J. Y. Puy and C. Perigaud, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci, 2009, 877, 1101–1108 CrossRef CAS PubMed
- V. Thibert, P. Legeay, F. Chapuis-Hugon and V. Pichon, Talanta, 2012, 88, 412–419 CrossRef CAS PubMed
- N. Harun, R. A. Anderson and P. A. G. Cormack, Anal. Bioanal. Chem., 2010, 396, 2449–2459 CrossRef CAS PubMed
- B. T. S. Bui, F. Merlier and K. Haupt, Anal. Chem., 2010, 82, 4420–4427 CrossRef CAS PubMed
- H. Hou, X. Zhang, Y. Tian, G. Tang, Y. Liu and Q. Hu, J. Pharm. Biomed. Sci., 2012, 63, 17–22 CrossRef CAS PubMed
- M. Szultka, J. Szeliga, M. Jackowski and B. Buszewski, Anal. Bioanal. Chem., 2012, 403, 785–796 CrossRef CAS PubMed
- M. McKeague, C. R. Bradley, A. De Girolamo, A. Visconti, J. D. Miller and M. C. Derosa, Int. J. Mol. Sci., 2010, 11, 4864–4881 CrossRef CAS PubMed
- E. Luzi, M. Minunni, S. Tombelli and M. Mascini, TrAC, Trends Anal. Chem., 2003, 22, 810–818 CrossRef CAS
- B. Madru, F. Chapuis-Hugon, E. Peyrin and V. Pichon, Anal. Chem., 2009, 81, 7081–7086 CrossRef CAS PubMed
- B. Madru, F. Chapuis-Hugon and V. Pichon, Talanta, 2011, 85, 616–624 CrossRef CAS PubMed
- S. N. Aslipashaki, T. Khayamian and Z. Hashemian, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 925, 26–32 CrossRef CAS PubMed
- J. H. Kim, E. T. Hwang, K. K. Kang, R. Tatavarty and M. B. Gu, J. Mater. Chem., 2011, 21, 19203–19206 RSC
- O. Veiseh, J. W. Gunn and M. Q. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2014 |