 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microfluidic approaches for epithelial cell layer culture and characterisation
Roland
Thuenauer
*ab,
Enrique
Rodriguez-Boulan
c and
Winfried
Römer
ab
aInstitute of Biology II, Albert-Ludwigs-University Freiburg, Schänzlestraße 1, 79104 Freiburg, Germany. E-mail: roland.thuenauer@yahoo.de
bBIOSS – Centre for Biological Signalling Studies, Albert-Ludwigs-University Freiburg, Schänzlestraβe 18, 79104 Freiburg, Germany
cMargaret M. Dyson Vision Research Institute, Weill Cornell Medical College, 1300 York Avenue, New York, New York 10065, USA
First published on 26th February 2014
Abstract
In higher eukaryotes, epithelial cell layers line most body cavities and form selective barriers that regulate the exchange of solutes between compartments. In order to fulfil these functions, the cells assume a polarised architecture and maintain two distinct plasma membrane domains, the apical domain facing the lumen and the basolateral domain facing other cells and the extracellular matrix. Microfluidic biochips offer the unique opportunity to establish novel in vitro models of epithelia in which the in vivo microenvironment of epithelial cells is precisely reconstituted. In addition, analytical tools to monitor biologically relevant parameters can be directly integrated on-chip. In this review we summarise recently developed biochip designs for culturing epithelial cell layers. Since endothelial cell layers, which line blood vessels, have similar barrier functions and polar organisation as epithelial cell layers, we also discuss biochips for culturing endothelial cell layers. Furthermore, we review approaches to integrate tools to analyse and manipulate epithelia and endothelia in microfluidic biochips; including methods to perform electrical impedance spectroscopy; methods to detect substances undergoing trans-epithelial transport via fluorescence, spectrophotometry, and mass spectrometry; techniques to mechanically stimulate cells via stretching and fluid flow-induced shear stress; and methods to carry out high-resolution imaging of vesicular trafficking using light microscopy. Taken together, this versatile microfluidic toolbox enables novel experimental approaches to characterise epithelial monolayers.
 Roland Thuenauer | Roland Thuenauer is a post-doctoral researcher at the Centre of Biological Signalling Studies (BIOSS) at the University of Freiburg, Germany. He studied physics at the Technical University of Graz, Austria. During his Ph.D. studies at the Johannes Kepler University of Linz, Austria, he developed novel microfluidic platforms that allow imaging of vesicular trafficking in live polarised epithelia. Afterwards he joined the research group of Prof. Winfried Römer, where he currently investigates the interaction of bacterial lectins with polarised epithelial cells via traditional and microfluidic approaches. |
 Enrique Rodriguez-Boulan | Enrique Rodriguez-Boulan is a Professor of Cell Biology and Neurosciences at the Dyson Institute of Vision Research, Weill Cornell Medical College, New York. His laboratory interest is in epithelial and retinal cell biology. With M. Cereijido and D. Sabatini, he introduced the MDCK model, which is now the best-studied epithelial cell model. His group has elucidated many fundamental epithelial polarity mechanisms. His retinal research currently focuses on building an in vitro model of the outer retina and on developing drugs to treat Age Related Macular Degeneration, a disease in which photoreceptor cell death is caused by disruption of their main support cell, the retinal pigment epithelium. |
 Winfried Römer | Winfried Römer has been a Professor at the University of Freiburg, Germany, since April 2011 and heads the research group “Synthetic Biology of Signalling Processes”. His research focuses on host–pathogen interactions; in particular, he is interested in understanding the physiological role of bacterial lectins in mammalian cells by using analytical and synthetic approaches. He studied chemistry and biology at the University of Regensburg, Germany. During his Ph.D. studies he developed novel pore-suspending membrane systems and successfully reconstituted membrane proteins. As a post-doctoral fellow and research scientist at the CNRS and Curie Institute in Paris, France, he investigated the cellular uptake of toxins and viruses. |
1 Introduction
Epithelial cells constitute the key functional component of most body organs and organise themselves as selective barriers between the internal medium of the organism and various organ luminal compartments (gut lumen, urinary space, lung air space, lumina of exocrine and endocrine glands, etc.).1In vitro models of epithelia provide well-defined and accessible systems that enable investigation of the basic properties of epithelial cells,1 as well as unraveling of mechanisms of diseases that are caused by malfunctions of the epithelial cell polarity program (e.g. cancer,2 microvillus inclusion disease,3,4 congenital sucrase–isomaltase deficiency,5 cystic fibrosis,6 and ciliopathies7 such as polycystic kidney disease,8,9 retinitis pigmentosa10,11 or Bardet–Biedl syndrome12). Furthermore, in vitro models of epithelia also have important pharmaco-therapeutic applications. As epithelial barriers are a major obstacle that needs to be overcome for targeted drug delivery,13–15in vitro models offer a powerful tool to identify permeable candidate drugs as well as to understand the underlying transport processes.In order to generate well-differentiated epithelial cell layers in vitro, it is necessary to reconstitute their natural microenvironment as closely as possible. This is traditionally achieved by culturing epithelial cells on Transwell filters,1 which allow provision of different culture media to each side of a two-dimensional epithelial cell layer. Another commonly utilised approach is based on placing epithelial cells, such as Madin–Darby canine kidney (MDCK) cells16,17 or Michigan Cancer Foundation-7 (MCF-7) cells,18 in gels resembling the extracellular matrix, where they form self-organised three-dimensional cysts with internal lumina. Although these techniques allow the in vitro generation of epithelial cell layers with some basic features of in vivo epithelia, they do not replicate all features of the in vivo microenvironment of epithelia. Here, microfluidic approaches provide a new perspective, because they enable a much more precise and dynamic control of multiple parameters of the cell's microenvironment. Microfluidic approaches ensure continuous supply of fresh medium while maintaining realistic ratios of cell volume to growth medium volume. Moreover, microfluidic models enable to resemble the challenges faced by epithelia in vivo, such as fluid shear stress or mechanical stretching. In addition, epithelia in microfluidic biochips are easily accessible for analytical tools. Various analytical tools to read out biologically relevant parameters based on fluorescence, mass spectrometry, electrical impedance spectroscopy, and light microscopy have been directly integrated into microfluidic biochips. This review critically discusses recent advances in microfluidic biochip designs that enable novel approaches for culturing and characterising epithelial cell layers. The design and manufacturing of biochips, together with the cell culture and biological characterisation in microfluidic biochips, is a highly interdisciplinary endeavour. Therefore, this review aims to provide an overview for both, biologists interested in novel techniques, and chemists, physicists and engineers interested in finding biologically relevant applications for their innovations, in order to stimulate inter-disciplinary exchange.
2 Background: structure and functions of epithelial cell layers
In order to carry out selective barrier and transport functions, epithelial cells assume a polarised architecture1 (Fig. 1). Tight junctions seal neighbouring cells together, so that the passage of substances along the space between cells, the so-called paracellular pathway, is regulated. The tight junctions also ensure that two distinct plasma membrane domains, the apical and the basolateral plasma membrane domain, can exist without diffusive intermixing.1 This enables polarised epithelial cells to establish and maintain a different lipid and protein composition at their apical and basolateral plasma membrane domains via highly dynamic intracellular trafficking and sorting mechanisms. Such polarised distribution of transporters, carriers and channels between the apical and basolateral plasma membrane domains is the basis for the vectorial transport, secretory, and absorptive functions of epithelial cell layers.1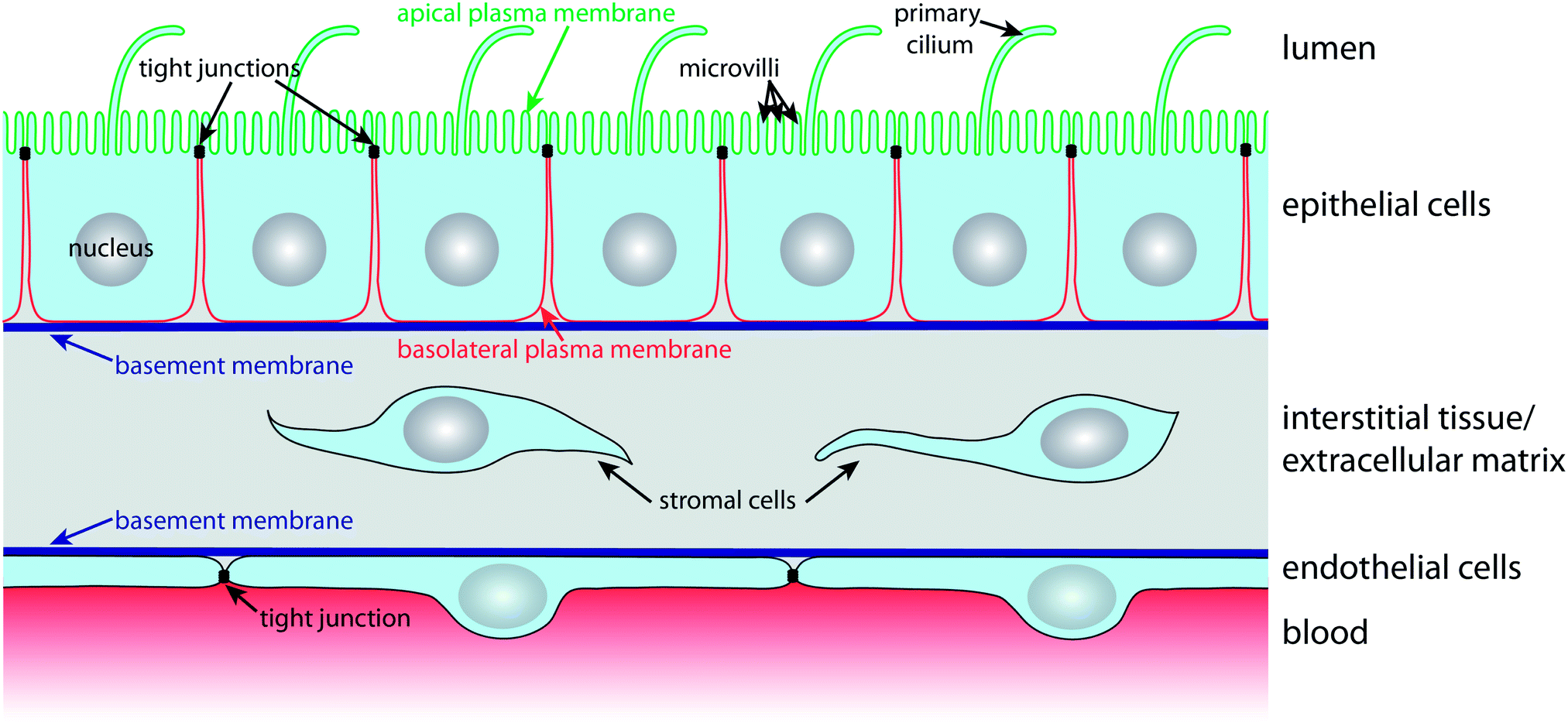 | ||
| Fig. 1 The in vivo environment and architecture of epithelial and endothelial cell monolayers. Polarised epithelial cells possess apical and basolateral plasma membrane domains that are separated by tight junctions. The apical plasma membrane (green) faces the lumen and contains microvilli and the primary cilium. The basolateral plasma membrane (red) contacts other cells and the extracellular matrix. The essential structures of the extracellular matrix are the basement membrane and the interstitial tissue that consists of connective tissue and stromal cells. Endothelial cells line blood vessels and exhibit a similar architecture as epithelial cells in which also tight junctions regulate the passage of substances between neighbouring cells. | ||
The apical plasma membrane faces the luminal space of organs. In many epithelia, such as the small intestinal epithelium or the kidney proximal tubule epithelium, the surface area of the apical plasma membrane is enlarged by actin-filled protrusions, so-called microvilli. In addition, most epithelial cells express a primary cilium, which is a several micrometre long microtubule-supported protrusion from the apical plasma membrane that serves as a multifunctional sensory antenna.19,20
Cell–cell contacts along the lateral part of the basolateral plasma membrane are maintained by intercellular adhesion molecules, such as calcium-dependent cadherins, that contribute to the formation of belt-like adherens junctions and spot desmosomes. The basal domain of the basolateral plasma membrane faces the basement membrane, a condensation of the extracellular matrix (ECM), and expresses a variety of receptors (e.g. integrins) for components of the basement membrane. Nutrients for epithelial cell layers are provided by blood vessels in the underlying interstitial tissue of the ECM. The nutrients traverse the basement membrane, which must be kept appropriately permeable in order to ensure that nutrients reach receptors and transporters in the basolateral plasma membrane that import them into the cell.
Blood vessels are lined on their luminal side by endothelial cells, which are organised according to similar principles as epithelial cells (Fig. 1). Endothelial cells also have tight junctions that confer them with selective barrier functions; importantly, the permeability of their tight junctions varies with the localisation in the vascular system.21 For example, the endothelium that forms the blood–brain barrier (BBB) exhibits very tight, tight junctions in order to maintain and protect the microenvironment of the central nervous system.22 Microfluidic models for culturing and characterising epithelial cell layers are also suitable for endothelial monolayers and we will therefore treat microfluidic biochips for epithelia and endothelia in this review.
It is also noteworthy that epithelial polarity is tightly controlled by a self-organising network of polarity proteins and lipids2,17,23 and a highly organised vesicular trafficking system.1 This is underlined by the fact that malfunctions in epithelial polarity frequently lead to cancer formation.24 In fact, 90% of all human cancers are derived from epithelial cells.2
Furthermore, individual cells can exhibit a uniform directional organisation within the plane of an epithelial monolayer, which is termed planar cell polarity.25 This manifests itself, for example, in a uniform orientation of cilia in the airway epithelium,26 or in the alignment of various protrusions from epithelial cells, such as cuticular hairs in the Drosophila melanogaster wings or pleura.27
3 Microfluidic biochip architectures for culturing epithelial cell layers
Over the last decade increasingly complex microfluidic biochips to culture epithelial cells have been developed. Significant progress has been made to design chips that replicate the microenvironment, 3D-geometries and stimuli faced by epithelial cell layers in vivo.3.1 Basic features of microfluidic biochips for culturing epithelial cells
Virtually all developed biochips for culturing epithelial cell layers are based on polydimethylsiloxane (PDMS). This is mainly due to the fact that for this material soft lithography-based methods, which enable rapid manufacturing of 3D-microstructures, are well established.28,29 Furthermore, PDMS is biologically compatible,30 permeable to gases, which allows oxygen supply for cells, is transparent and exhibits low autofluorescence, which is beneficial for on-chip light microscopy.31 However, PDMS has also some disadvantages, for example its gas permeability makes it also permeable to water vapour, which can cause changes in the osmolarity of the cell culture medium. Furthermore, PMDS has a propensity to absorb small molecules.31Already simple microfluidic biochip designs that consist of channels or chambers in which the cells can grow allow the creation of in vivo-like fluid flow conditions32–35 and/or investigation of the effects of surface topography on epithelial cell layers36 (Fig. 2A). For example, Frohlich et al. demonstrated that a surface topography bearing 0.75 μm wide and deep linear grooves functions in concert with fluid shear stress to align renal epithelial cells and to modulate the formation of tight junctions.36 It is also possible to manufacture channels with circular cross-section in order to replicate cylindrical vessel geometries.37,38
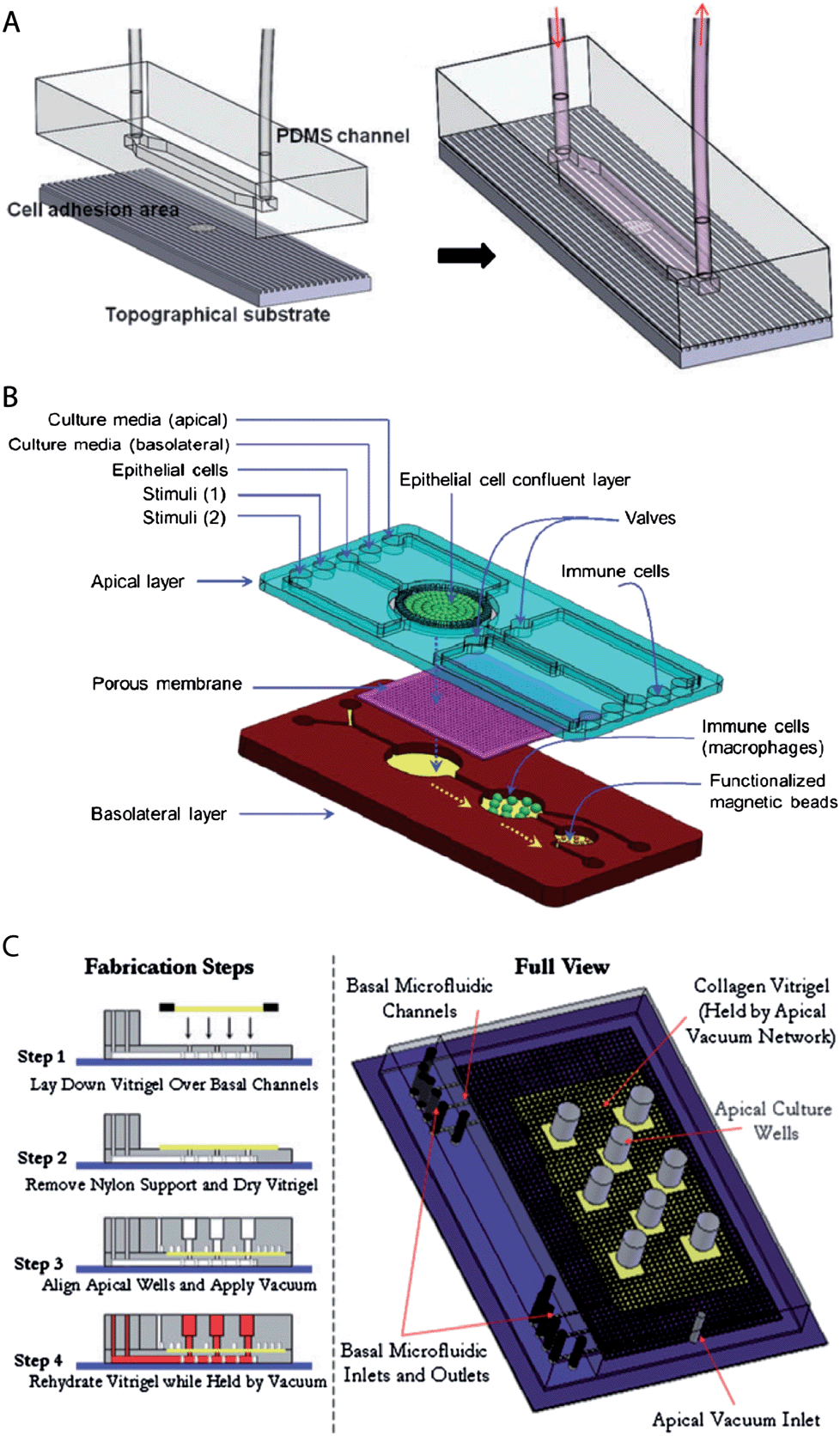 | ||
| Fig. 2 Examples of microfluidic biochip architectures for culturing epithelial cell layers. (A) Simple microfluidic biochip design based on a single channel that additionally incorporates a cell growth substrate with controlled surface topography. Reproduced from ref. 36 with permission. (B) Microfluidic biochip design based on an integrated porous membrane as a growth substrate for the epithelial cell layer. The chip constitutes a miniaturized model of the human gastrointestinal tract, which can be used to monitor immuno-modulatory effects of food.39 To this end, the chip contains additional chambers for the growth of immune cells and performing an immunomagnetic assay. This assay is based on antibody-coated magnetic beads and allows the detection of the secretion of pro-inflammatory cytokines by the immune cells. Reproduced from ref. 39 with permission. (C) Microfluidic biochip design with an integrated collagen vitrigel layer as a growth substrate for an epithelial cell layer. The device enables the generation and culturing of corneal microtissue patches.40 The collagen vitrigel layer is first placed on a PDMS structure containing the basal microfluidic channel system (step 1). After the removal of nylon supports and drying of the collagen vitrigel (step 2), the PDMS structure containing the apical channel system is placed on top and held in place by vacuum (step 3). In order to prepare the chip for cell culture, the collagen vitrigel layer is rehydrated again (step 4). Reproduced from ref. 40 with permission. | ||
However, in vivo, epithelial cells within a monolayer receive their nutrients from the basolateral side (Fig. 1). This can be efficiently mimicked by microfluidic biochip architectures that incorporate a porous membrane (Fig. 2B). The porous membrane carries the epithelial cell layer and separates the channels that allow access from the apical and basolateral side. Pore diameters smaller than 1–2 μm are usually required to prevent the migration of individual cells through the pores. In order to guarantee fast diffusive transport through the pores, the porous membranes should not be thicker than a few ten micrometres.
3.2 Manufacturing and integration of porous membranes
In most chip architectures, commercially available porous membranes made of polyester or polycarbonate with a pore diameter of 0.4 μm, identical to the porous membranes utilised in Transwell filters, are applied. Also polyethersulfone membranes with a very small pore diameter (0.04 μm), which are traditionally used in dialysis devices, have been utilised in microfluidic biochips.41–43The challenge of integrating commercially available porous membranes is their leakage-free bonding to the PDMS-based parts of the chip. Some groups reported that polyester membranes could be bonded to PDMS via oxygen plasma treatment,39,44–46 whereas others combined oxygen plasma with additional treatments, such as sputter-coating the membrane with SiO2 (ref. 47) or functionalisation with (3-aminopropyl)triethoxysilane (APTES).48 As an alternative, fluid PDMS pre-polymer49 or glue50,51 can be utilised to ensure tight bonding of the porous membrane, whereby the PDMS pre-polymer or glue is applied only to non-cell culture areas of the porous membrane. Furthermore, tight clamping of a PDMS/membrane/PDMS sandwich with a microchip holder has been utilised,41–43 which has the advantage that the membrane with an attached cell layer can be extracted after experiments.
Several microstructuring techniques have been developed to manufacture thin PDMS membranes with μm sized pores.52–57 A technique that is easy to use and yields pores down to a few micrometer in diameter is the spin coating of the PDMS pre-polymer on molds that bear thin posts in order to generate the pores.54,56,57 In order to reduce the thickness of the PDMS membrane, the PDMS pre-polymer can be diluted with toluene54 or cyclohexane.57 Another approach is to air-blow pores.55 This is achieved with a substrate that contains tiny holes. During curing of the PDMS pre-polymer, air is blown through the holes, thus locally removing PDMS over the substrate pores. Although this technique is more difficult to use, well-defined pores down to 1 μm in diameter can be produced. Furthermore, methods to manufacture pores in PDMS membranes with sub-micrometer diameter have been described.52 To this end, the PDMS pre-polymer was spin coated on a mold with pyramid-shaped posts. Subsequent etching of PDMS with SF6 plasma allowed the adjustment of the aperture size of the pores on the side of the membrane where the pyramid tips of the mold were located. Porous PDMS membranes have the advantage that the well-established methods to bond PDMS surfaces together58–60 can be applied. In addition, porous PDMS membranes are highly optically transparent with a refractive index (n ∼ 1.4)61 close to water (n = 1.33), which is beneficial for high-resolution microscopy, and are elastic, which enables to perform on-chip mechanical stretching of the epithelial cell layer.62–64
Another alternative are chips that contain adaptors for Transwell filters, which have the advantage that established Transwell filter culturing methods can be combined with microfluidic tools.65 Furthermore, methods for manufacturing and integrating non-flat porous membranes have been developed. This includes techniques to topographically pattern porous membranes in the sub-micrometer scale via hot-embossing without compromising the pores.66 Such surface topography provides spatial cues, similar to physiological cues found in vivo,66 along which cells can align in order to establish planar cell polarity. Esch et al. described a method to manufacture porous membranes made of SU-8 that can be deformed in the μm-scale to provide a non-flat support that resembles the three-dimensional shape of intestinal villi.67 Moreover, a hollow fibre membrane has been successfully integrated in a microfluidic chip in order to mimic the geometry and function of a renal tubule.68 In addition, microfluidic biochips with micro-gap arrays that separate two channels, thus forming horizontal pores between the channels, have been developed.69,70 This allowed manufacturing bifurcations and junctions of micro-gap-spanning BBB-cell layers,69 which is a feature that is often found in vivo.
3.3 Incorporation of extracellular matrix (ECM) coatings and ECM gels
In most biochips for culturing epithelial cell layers, the surfaces on which the cells grow are coated with proteins mimicking the ECM (e.g. collagen,66,71 fibronectin,72 poly-L-lysine,73etc.) in order to facilitate cell adhesion and cell differentiation. Coating is easily achieved by flowing an ECM-protein solution through the cell culture chamber. This approach leads to another interesting set of microfluidic models in which structured ECM gels are utilised as substrates for epithelial cell layers. Puleo et al. manufactured a chip in which a collagen vitrigel layer serves as a separator between apical and basolateral fluid channel networks, thus replacing a porous membrane as a permeable carrier of the epithelial cell layer40 (Fig. 2C). The collagen vitrigel layer has the advantage that, once a corneal epithelial cell layer is grown on top, the gel can be removed by enzymatic degradation, and a supporting stromal cell layer can be grown underneath.40 Furthermore, traditional ECM gel-based culture systems, in which epithelial cells form self-organised three-dimensional cysts, can be equipped with a microfluidic perfusion system.74 Finally, several chips that contain channel sidewalls made of ECM gels or chips that contain entire channel networks within ECM gels have been developed. These approaches, which are described in more detail in excellent recent reviews,75–78 can be utilized to generate endothelial cell-lined vascular networks and provide the means to study angiogenesis and tumour cell invasion under in vivo-like conditions.4 Integration of tools to analyse and manipulate epithelial cell layers in microfluidic biochips
Microfluidic models of epithelia offer the advantage that multiple tools can be integrated on-chip. The following section discusses which tools are available to control and measure the biologically relevant parameters of epithelial cell layers and describes integration techniques. Finally, we highlight microfluidic models of lung epithelium, because there are impressive examples available that illustrate the potential of highly integrated microfluidic biochips to resemble complex epithelial cell layer functions on-chip.4.1 Electrical characterisation
Epithelial cells form electrically tight barrier layers, whereby ‘electrically tight’ means that the tight junctions prevent the passage of ion currents along a paracellular pathway. Although the extent of electrical tightness depends on the cell type (typical TEER values in microfluidic models range from ∼100 Ω cm2 for e.g. Madin–Darby canine kidney II cells50,79 to ∼250 Ω cm2 for e.g. BBB cells80), the measurement of the trans-epithelial electrical resistance (TEER) is a reliable indicator of the development of functional tight junctions and thus the differentiation status of polarised epithelia. TEER measurement is routinely used in Transwell culture systems to validate and monitor the differentiation of epithelial monolayers.81,82Typically, TEER measurements are not performed with DC current, which would have undesirable side effects on the cells and electrodes, but at low AC frequencies (usually 12.5 Hz (ref. 83)). However, more information can be obtained if the electrical resistance is measured over a range of AC frequencies (up to a few MHz), which is known as impedance spectroscopy.65,79,84,85 The impedance of epithelia strongly depends on the AC frequency,85 which indicates that at higher frequencies the influence of the capacitance of the lipid bilayer of the cell membrane becomes dominant. In order to take into account these effects, equivalent circuit models for epithelial barriers have been developed. In their simplest form84,85 (Fig. 3A), a constant phase element (CPh) accounts for the double layer capacitance of the electrodes and a resistor (RM) accounts for the resistance of the medium. The cell monolayer itself is modelled by a resistor (RC) that accounts for the resistance of the paracellular pathway, which corresponds to the TEER, in parallel with a capacitor (CM) that accounts for the capacitance of the cell membrane. An alternative approach to model the frequency-dependence of the electrical resistance of epithelia is finite element modelling.65 Finite element modelling allows the calculation of the electric field for a given geometry of the porous membrane and the cells and thus the direct investigation of the influence of these geometries on the TEER.
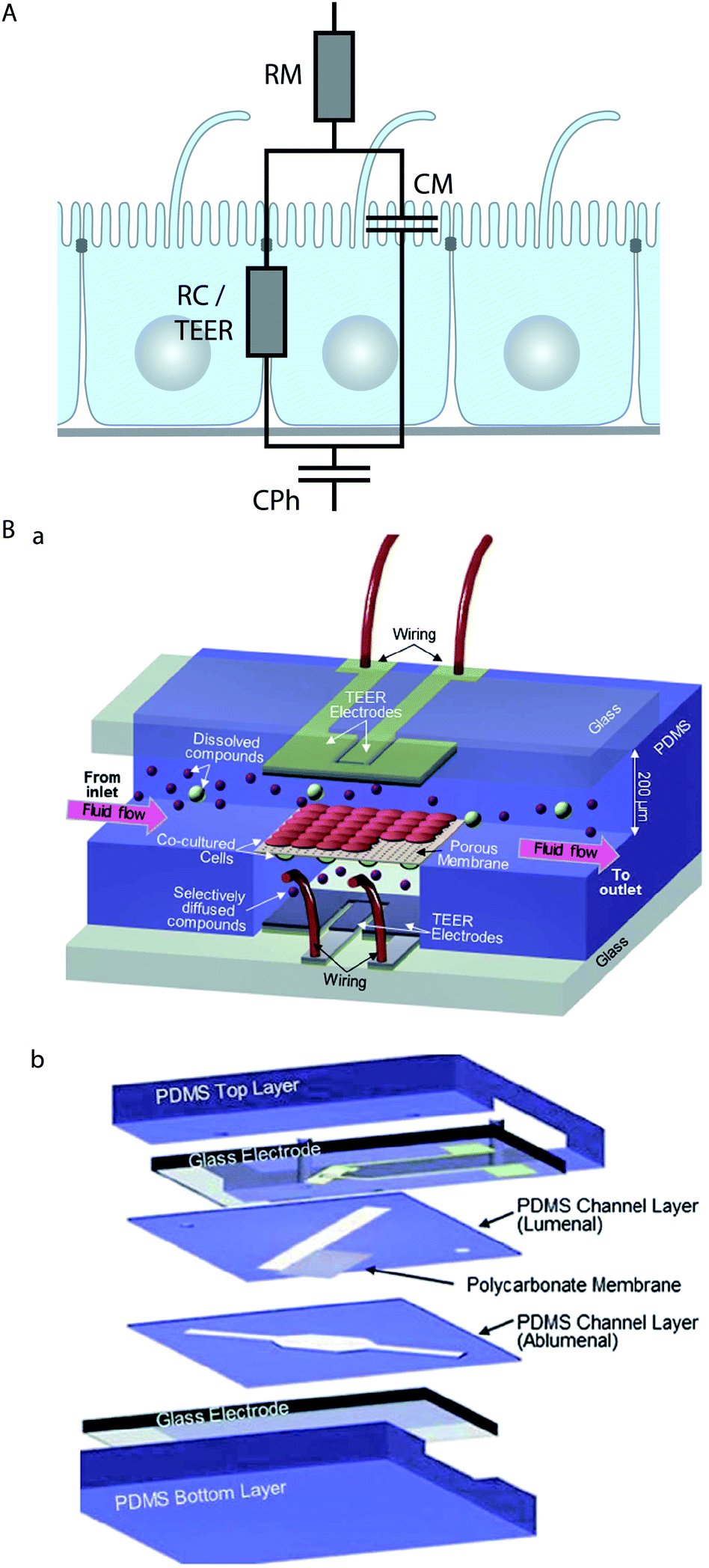 | ||
| Fig. 3 Electrical characterisation of epithelial cell layers in microfluidic biochips. (A) Equivalent circuit diagram of an epithelial monolayer. The constant phase element CPh accounts for the double layer capacitance of the measurement electrodes, the resistor RM accounts for the resistance of the medium, the resistor RC accounts for the resistance of the paracellular route, which is equivalent to the TEER, and the capacitor CM accounts for the capacitance of the plasma membranes. (B) Example for the integration of electrodes to measure the TEER across an endothelial cell layer in a microfluidic biochip. (a) Three-dimensional schematic view of the biochip. (b) Components of the biochip. The biochip contains two perpendicular flow channels that are separated by a porous polycarbonate membrane carrying an endothelial cell layer on one side and co-cultured astrocytes, which positively influence the quality of the endothelium, on the other side. The TEER electrodes are thin-film AgCl electrodes that were deposited on glass slides. Reproduced from ref. 80 with permission. | ||
Several strategies to perform on-chip TEER measurements have been developed, including direct integration of electrodes in the chip50,51,65,80,86 (Fig. 3B), insertion of small-diameter electrodes through the apical and basolateral channels,62,63 or through access holes.79,84 Ag/AgCl-electrodes and platinum-electrodes are most commonly used,50,51,79,80,84 but also gold-plated electrodes covered with a conducting polymer (polypyrrole doped with polystyrene sulfonate) in order to reduce the influence of the double layer capacitance have been applied.65 In order to validate on-chip TEER-measurement techniques, tests in which the integrity of the epithelial monolayer is intentionally compromised should be performed. This can be done by Ca2+-removal, which reversibly dissolves cell–cell contacts, and is achieved by washing with Ca2+-free medium and Ca2+-chelation with compounds such as ethylene glycol tetraacetic acid (EGTA)65 or ethylenediaminetetraacetic acid (EDTA).50 Alternatively, irreversible disintegration of the monolayer by the treatment with a detergent such as Triton X-100 can be performed.65
On-chip TEER measurements are ideally suited for monitoring in real-time how the tightness of epithelial and endothelial barriers is affected by stimuli. In a microfluidic model of the BBB, Booth et al. demonstrated by TEER measurements that co-culture with astrocytes increases the barrier tightness.80 Furthermore, they showed that histamine, an inflammatory mediator that has been shown to lead to the formation of transient gaps between endothelial cells in vivo,87 also transiently lowered TEER in their microfluidic BBB model. In a different microfluidic model of the BBB, Griep et al.84 utilised TEER monitoring to directly show that fluid shear stress positively influences the BBB tightness, whereas tumour necrosis factor α (TNF-α), a pro-inflammatory cytokine,88 negatively affects the barrier tightness. In a microfluidic model of the intestinal epithelium based on Caco-2 cells, Kim et al. utilised TEER monitoring to show that luminal co-culture with a probiotic strain of bacteria, Lactobacillus rhamnosus GG, increases the intestinal barrier tightness.63
4.2 Tools to characterise trans-epithelial transport
Epithelial barriers are the major gatekeepers that control the exchange of substances between different compartments of multicellular eukaryotes. According to physiological requirements, epithelial barriers regulate the exchange of molecules and ions between the luminal space or blood and interstitial fluid. The lipid bilayer of cellular plasma membranes and the tight junctions prevent the passive transport of hydrophilic substances across epithelial cell layers. In order to transport specific molecules across epithelia in a regulated manner, three major trans-epithelial transport pathways exist (Fig. 4A). First, epithelial cells exhibit polarised expression of dedicated transporters, pumps, channels and carriers in their apical and basolateral plasma membranes, through which specific molecules are transported to the other side of the epithelium via the cytosol of the cells. This mode of transport is for example utilised to absorb nutrients in the small intestine or to reabsorb molecules in the kidney. Second, epithelial cells possess machineries to transport specific molecules via sequential endocytosis, vesicular transport, and exocytosis to the other side, which is termed transcytosis. As the first two pathways traverse individual cells, they are collectively termed transcellular pathways. Third, the tight junctions can be selectively permeable for certain substances, which allows regulated transport along the paracellular pathway. Furthermore, epithelial cell layers form the basic functional component of glands and can produce and secrete specific molecules in a vectorial manner.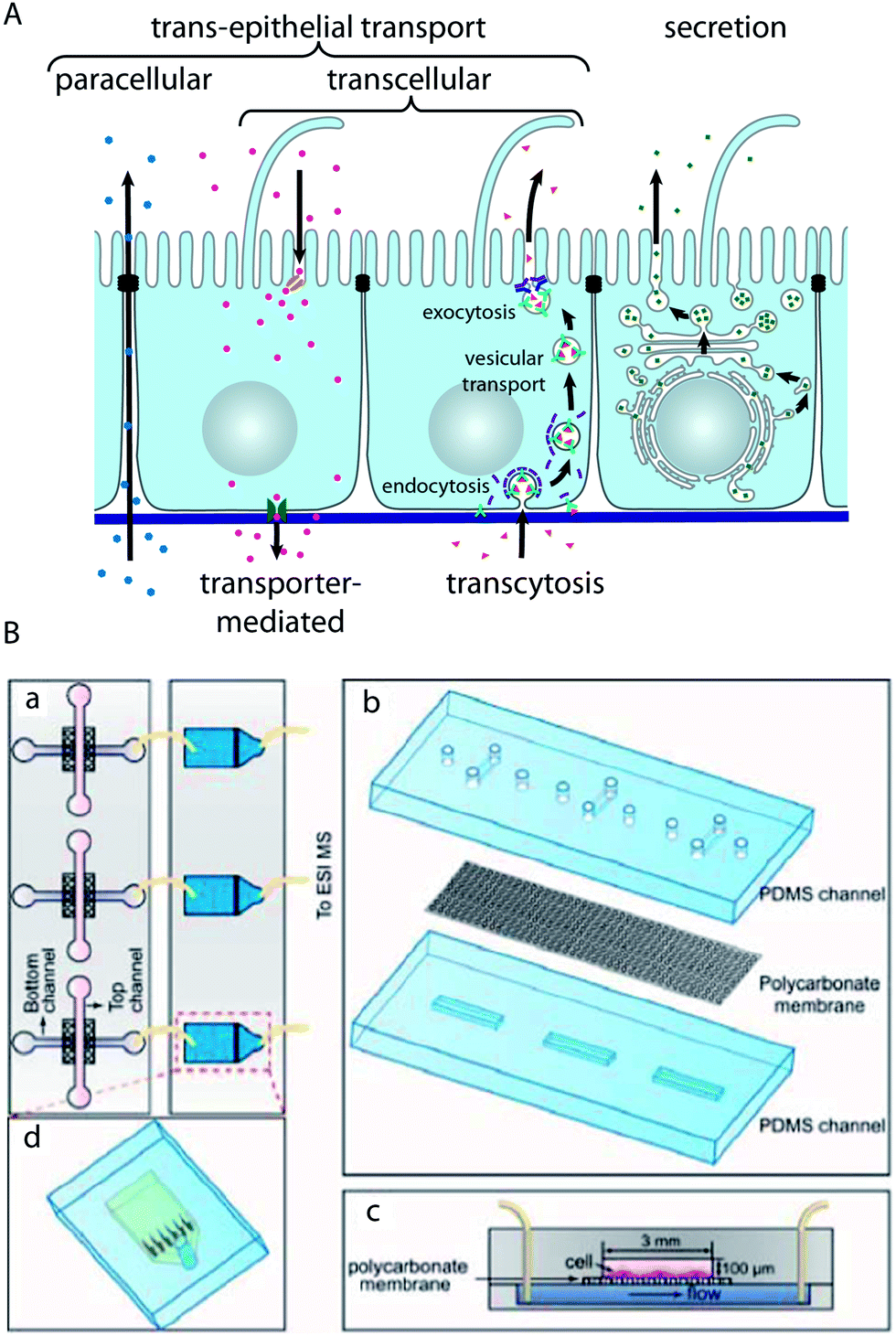 | ||
| Fig. 4 Characterisation of trans-epithelial transport in microfluidic biochips. (A) Schematic drawing showing trans-epithelial and secretory pathways. Trans-epithelial transport occurs via paracellular and transcellular routes. Transcellular transport is possible via transporter-mediated pathways (e.g. in the case of absorption or reabsorption of nutrients) or via transcytosis. In transcytosis, molecules bind to specific receptors, which are subsequently endocytosed. After vesicular transport through intracellular compartments, the molecules are released on the other side by exocytosis.91 (B) Microfluidic biochip design to carry out mass spectrometry-based analysis of substances that are transported across an epithelial barrier. (a) The chip consists of two parts: in the first part (left) epithelial cells are cultured on a porous membrane. The second part (right) contains microfluidic solid-phase extraction columns for sample pre-treatment that are connected to a mass spectrometer. (b) Components of the cell culture chip. (c) Schematic cross-section through the cell culture chip. (d) Schematic outline of the sample pre-treatment chip. Reproduced from ref. 73 with permission. | ||
Malfunctions of the highly specific transport mechanisms of epithelia are the cause for a plethora of diseases.1,23,89 However, the affected tissues are mostly inaccessible in living organisms, which makes in vivo studies of trans-epithelial transport intrinsically difficult. Microfluidic models of epithelial transport functions are therefore a valuable contribution to shed light on transport-related disease mechanisms.
In microfluidic biochips that incorporate porous membranes as growth substrates for the epithelial cell layer, the apical and basolateral sides are independently accessible via microfluidic channels. This allows direct monitoring of tracer molecules undergoing trans-epithelial transport. Furthermore, by applying a constant flow of solution, microfluidic approaches enable to keep the tracer concentration in the donor channel constant, which is not possible in traditional Transwell filter-based assays with their static reservoirs. It has become standard to use non-interacting tracer molecules (e.g. dextrans or inulin), which can passively diffuse through leaks, as means to assay the quality of chip-grown epithelial cell layers. This is done by applying a tracer solution to one side of the epithelial cell layer and collecting fluid aliquots from the exit port of the channel connected to the opposite side. The collected fluid aliquots are mostly assayed off-chip, which has the advantage that conventional detection methods can be applied. However, off-chip analysis typically requires large volumes. Thus, the advantages of microfluidic models, to resemble in vivo ratios of extracellular fluid volume to cell volume and to limit the amount of applied tracer molecules, are lost. This problem is resolved by on-chip detection systems, for which only a few examples in microfluidic biochips for epithelial cell layer characterisation have been demonstrated.39,41–43,47,73,90
One possibility is tracer detection via fluorescence, which has the disadvantage that the molecule of interest has to be fluorescent or fluorescently tagged, but has the advantage that very high detection sensitivities can be achieved. Young et al. demonstrated the on-chip detection of fluorescence signals with a light microscope in a microfluidic model of an epithelium,90 whereas Kimura et al. incorporated holes for inserting optical fibres to enable fluorescence detection.47
Gao et al. have developed a strategy to perform the on-line mass spectrometry detection of trans-epithelial transport in a microfluidic model of the intestinal epithelium based on Caco-2 cells73 (Fig. 4B). To this end, microfluidic solid-phase extraction columns were directly attached to perform sample purification for detection with a connected electrospray ionisation quadrupole time-of-flight mass spectrometer. The functionality was demonstrated by characterising the trans-epithelial transport of the model drug curcumin. Mass spectrometry has the capability of label-free detection of many molecules in parallel, which makes such a system very interesting for large-scale drug screening applications.
Another alternative detection approach is spectrophotometry.41–43 This method also allows the label-free and parallel detection of a few tracer molecules, given that their spectra are sufficiently different.
Ramadan et al. described a preliminary strategy to integrate an immunomagnetic assay, which is based on magnetic beads bearing antibodies in order to capture and detect molecules.39 This is a promising approach because large collections of antibodies against physiologically relevant molecules are available.
4.3 Stimulation of epithelial cell layers by fluid flow and mechanical stretching
Epithelial cell layers that line ducts, and especially endothelial cell layers that line the blood vessels, are subjected to a constantly changing fluid flow that causes shear strain in the cells (Fig. 5A). It is interesting to note that epithelial and endothelial cells actively sense a fluid flow. Their primary flow sensor is believed to be the primary cilium, because it has been shown that fluid flow-mediated bending of the primary cilium elicits intracellular Ca2+-signalling.92,93 By design, channels in microfluidic devices can readily be subjected to fluid flows of defined durations and velocities. Thus, microfluidic biochips are powerful tools to investigate the effects of fluid flow on cells and have helped to reveal that other cellular flow sensors might exist. Rahimzadeh et al. showed that the fluid flow exerts mechanical stress on the actin cytoskeleton of MDCK cells,94 which may constitute the basis for a flow-sensing mechanism. Furthermore, Tkachenko et al. demonstrated that under strong fluid shear, when endothelial cells disassemble primary cilia, mechanical strain due to the displacement of nuclei under hydrodynamic drag functions as a flow sensor.34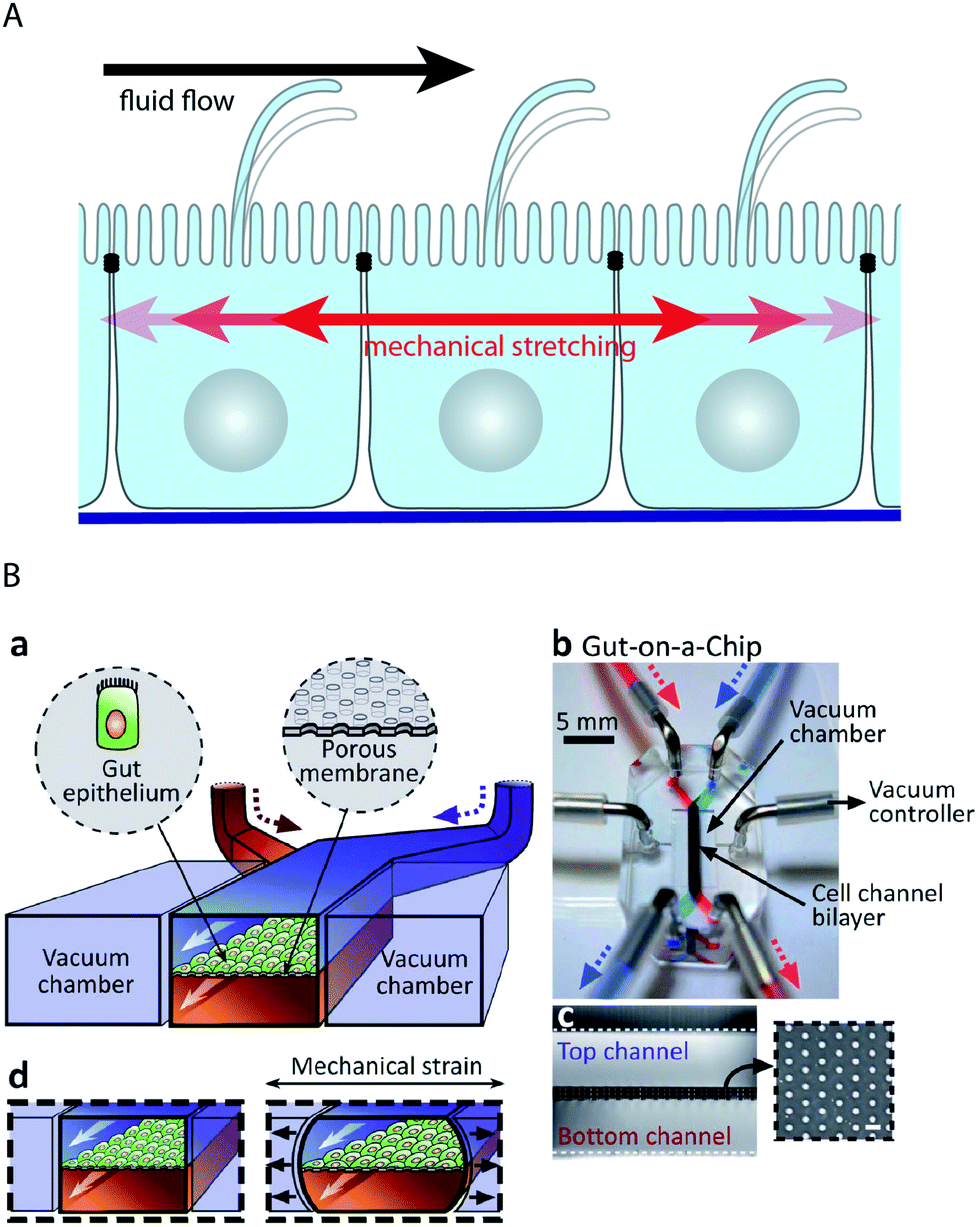 | ||
| Fig. 5 Microfluidic biochip approaches to stimulate epithelial cell layers with fluid flow and mechanical stretching. (A) Schematic drawing showing how fluid flow and mechanical stretching affect epithelial cell layers. (B) Microfluidic biochip to perform mechanical stretching of an epithelium grown on a flexible porous membrane. (a) Outline of the biochip. The chip contains a central chamber that is separated by a porous membrane. Next to the central chamber two vacuum chambers are located, which enable mechanical stretching of the porous membrane. (b) Photograph of the biochip. (c) Cross-sectional view of the chip that shows the porous membrane and the top and bottom channels. The inset shows a lateral view of the porous membrane. (d) Functional principle of mechanical stretching. When vacuum is applied to the two vacuum chambers, the flexible porous membrane carrying the epithelial cell layer is stretched. Reproduced from ref. 63 with permission. | ||
In addition, microfluidic biochips have allowed solidifying the hypothesis that fluid shear stress is required for complete differentiation of epithelia and endothelia, which are subjected to constant fluid flow in vivo. By using microfluidic models of the kidney it was demonstrated that kidney epithelial cells respond to fluid shear stress by increasing their cell height and actin cytoskeleton rearrangement, which results in the formation of tighter cell layers.44–46 In addition, other differentiation indicators, including albumin transport, glucose reabsorption, brush border alkaline phosphatase activity, cisplatin toxicity, and Pgp efflux transporter activity, showed values closer to in vivo values in fluid flow-treated cells when compared to cells cultured in traditional Transwell filters without fluid flow.45 Similar positive effects of fluid shear stress on barrier differentiation were also observed in microfluidic models of the BBB.80,84
Endothelia in arteries experience pulsatile and oscillatory shear stress due to temporal variation of the blood flow. Shao et al. developed a microfluidic biochip in which endothelial cells can be subjected to pulsatile and oscillatory fluid flow via an integrated pneumatic micropump.70 In this chip, endothelial cells are trapped in a microgap and then proliferate to form a functional endothelial barrier along the microgap. This elegant approach allowed monitoring of trans-endothelial permeability under in vivo-like pulsatile flow conditions.
Many epithelial cell layers, as e.g. in the lung or in the intestine, experience cyclic mechanical strain by stretching (Fig. 5A). Mechanical stretching of epithelial cell layers can be performed in microfluidic biochips by growing the cell layer on flexible porous PDMS membranes62–64,95 (Fig. 5B). Kim et al. reported that the combination of fluid flow and cyclic mechanical strain significantly improved the differentiation status in a microfluidic model of the intestinal epithelium based on Caco-2 cells.63,64 Upon combinatorial stimulation, the cells formed intestinal villi and also developed basal proliferative crypts. Furthermore, the cells differentiated into absorptive, mucus-secretory, enteroendocrine, and Paneth cells that were found at the same positions as in vivo.64
Moreover, microfluidic models of the lung that include means to mechanically stretch epithelial cell layers have been developed.62,95 The lung-related models are described in more detail in Section 4.5.
4.4 Novel light microscopy techniques for epithelial cell layers enabled by biochip technology
The polar organisation of epithelial cells is established and maintained by a remarkably complex vesicular trafficking machinery, which sorts and transports membrane proteins to apical and basolateral plasma membrane domains via exocytic and recycling routes.1 The concluding step in plasma membrane delivery is the fusion of cargo-bearing vesicles with the plasma membrane. Total internal reflection fluorescence (TIRF) microscopy, which utilizes a shallow evanescent wave to spatially limit the excitation volume, offers sufficient resolution in space and time to directly visualise vesicle fusion events.96However, because of the physical requirements to generate an evanescent wave,97 TIRF microscopy has been classically limited to image processes at or close to those parts of the plasma membrane that are attached to a support with high refractive index, such as a glass cover slip.98 This limitation can be overcome by a microfluidic biochip that allows TIRF imaging also at the non-support attached apical plasma membrane of epithelial cells by moving the apical plasma membrane into the region of the evanescent wave.72 The functional principle of the biochip is outlined in Fig. 6A. A polarised monolayer of epithelial cells is grown on a moveable platform within the biochip (Fig. 6A(a)). The platform is attached to the central area of a thin PDMS membrane that covers a microfluidic channel network. When pressure is applied to the channel network, the covering membrane will bulge and therefore allow positioning of the platform with sub-micrometer precision. For imaging, the whole biochip is inverted and the apical plasma membrane of the cells is precisely approached to a glass cover slip, thus allowing TIRF microscopy at the apical plasma membrane (Fig. 6A(b)). The biochip enabled the first direct visualisation of vesicle fusion events at the apical plasma membrane of polarised epithelia72 (see Fig. 6B for an example of an apical TIRF recording of a fusion event), as well as to show that vesicle fusion events occur at the base of apical microvilli.99
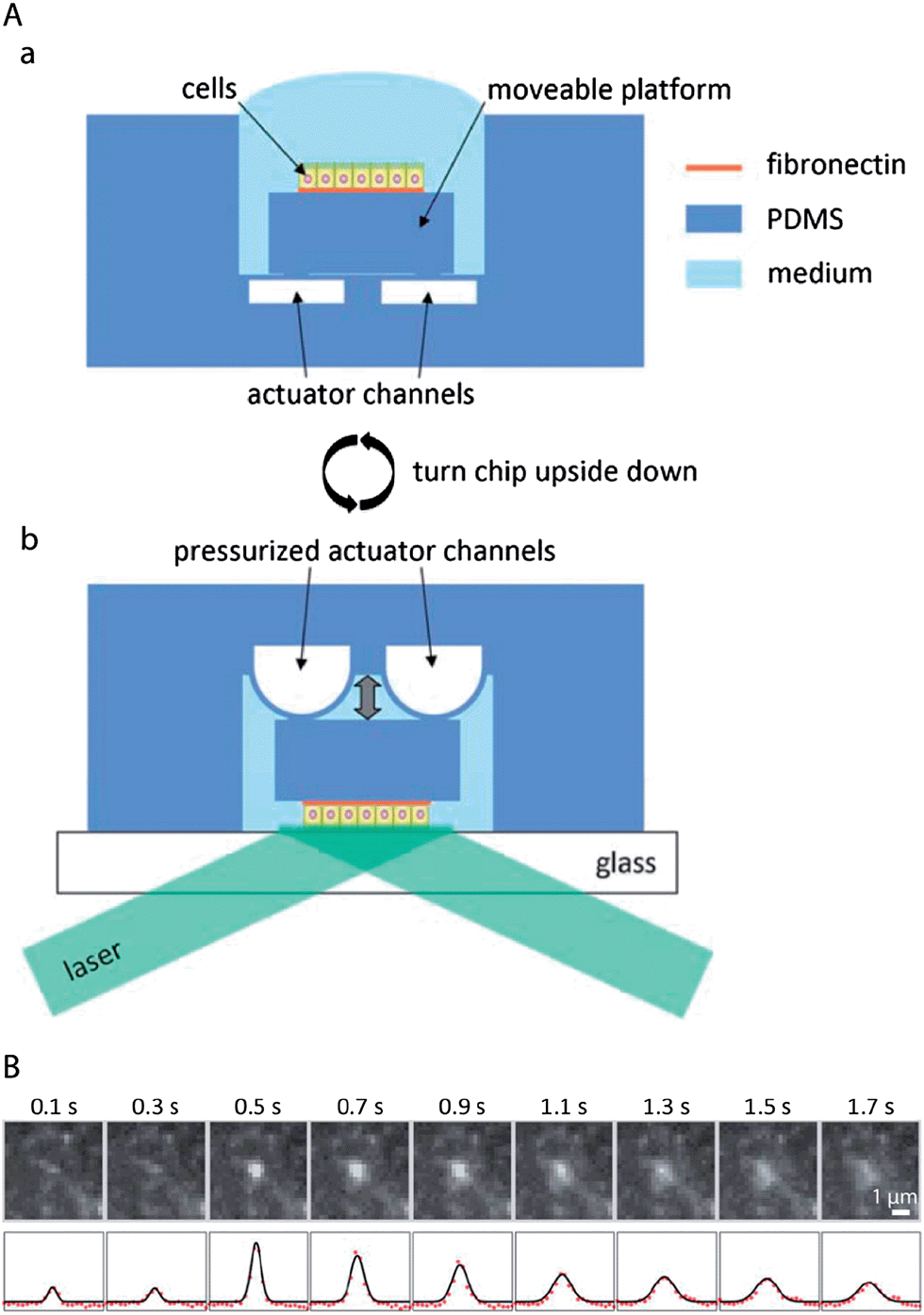 | ||
| Fig. 6 Microfluidic biochips enable novel microscopy techniques in order to image vesicular membrane trafficking in polarised epithelial cell layers. (A) Microfluidic biochip to perform TIRF microscopy at the apical plasma membrane of polarised epithelial cells. (a) The chip contains a moveable platform on which a polarised epithelial cell layer can be cultivated on a fibronectin-coated area. The moveable platform is attached to the central part of a thin flexible PDMS membrane covering the actuator channels. (b) For performing apical TIRF microscopy the chip is turned upside down and placed on a glass cover slip on a TIRF microscope. By applying pressure to the actuator channels, the platform carrying the cells can be precisely approached to the glass cover slip. In this way the apical plasma membrane of the cells is positioned within the evanescent wave of a totally internally reflected laser beam in order to carry out apical TIRF microscopy. (B) Fusion of a vesicle bearing the apical marker protein GPI-GFP with the apical plasma membrane. Top row: apical TIRF image sequence. Bottom row: intensity profiles along a horizontal cross-section through the centre of the intensity peaks (red dots) and through a two-dimensional Gaussian fitted to the peaks (black line). Adapted from ref. 72 with permission. | ||
4.5 Towards ‘organs-on-chips’: microfluidic models of the lung
Lung epithelial cell layers face unique environmental challenges. Their apical surface is exposed to air, only protected by a thin mucus layer (in the upper airway) or surfactant layer (in the lung alveoli). Due to breathing, lung epithelium is periodically stretched. Furthermore, pathogens as well as small pollutants have direct access to the lung epithelium. Several tools to reconstitute lung-typical environmental challenges have been integrated in microfluidic biochips.62,95,100–103 Microfluidic lung models have contributed significantly to the understanding of conditions related to these challenges.One example is airway reopening. This is an event that occurs in a variety of lung diseases that cause instabilities of the pulmonary surfactant, which then leads to the formation of small liquid plugs that block the small airways. The liquid plugs move with the air stream over the lung epithelium until the plugs eventually rupture, allowing the airways to be reopened.100 In order to reconstitute airway reopening, a microfluidic biochip has been developed in which a lung epithelial cell layer is grown on a porous polyester membrane and can be challenged by a liquid plug flow that is produced with an integrated plug flow generator.100,103,104 In the plug flow generator, a liquid stream is focused by an air stream to form a stratified air–liquid two-phase flow. Briefly switching off the air stream causes the generation of a liquid plug.100,104 This microfluidic model allowed to reveal that the forces exerted by moving air–liquid interphases of a propagating plug, as well as plug rupture, can have detrimental effects on lung epithelial cells.100,103 Interestingly, addition of surfactants significantly reduced cell injury due to propagating liquid plugs.103 In another microfluidic model of airway reopening, lung epithelial cells were grown on a thin flexible PDMS membrane, which allowed additional stretching of the cell layer.95 This approach revealed that the combination of fluid mechanical stress through propagating liquid plugs and solid mechanical stress through cyclic stretching further increased cell death, whereas surfactant-enriched growth medium had a protective effect.
Moreover, an endothelial–epithelial co-culture chip, based on a stretchable and porous PDMS membrane, has been developed.62 A layer of alveolar epithelial cells was cultivated on top of the porous membrane, whereas a layer of endothelial cells was cultivated on the other side. This approach allowed resembling several organ-level functions of the lung. Introducing an air–liquid interface at the apical membrane of the alveolar cells increased the TEER and reduced the albumin transport across the barrier, thus improving the differentiation status of the barrier. TNF-α stimulation activated the endothelium and induced adhesion of neutrophils to the activated endothelial cells. The neutrophils subsequently transmigrated through the capillary–alveolar barrier via the membrane pores. Application of the bacterium Escherichia coli to the alveolar cells could also induce neutrophil adhesion, transmigration, and clearance of the bacteria by neutrophils, thus mimicking the innate cellular response to bacterial infection. Interestingly, silica nanoparticles likewise activated the underlying endothelium, and breathing motions increased the inflammatory response as well as the absorption of nanoparticles.
5 Conclusions and future directions
Microfluidic devices have enabled reconstitution of important aspects of epithelial physiology by precisely resembling the in vivo microenvironment of epithelial cell layers. A particularly successful approach was to devise microfluidic designs that allow culturing of epithelial cell layers with supporting cell layers underneath. These multi-layer structures led to improved apico-basal polarisation and tightness of the epithelial barriers.40,62,80 However, epithelial cell layers exhibit a much more complex organisation in vivo. In particular, most epithelia consist of many specialised cell types that fulfil distinct functions. The different cell types show a well-controlled organisation within the plane of the cell layer. A remaining challenge is to reproduce such planarly structured epithelial cell layers on-chip.An intriguing observation is that on-chip stimulation of Caco-2 cells with fluid flow and cyclic stretching caused spontaneous self-organised differentiation into specialised cell types of the intestine, which also assumed proper planar organisation.64 This suggests that specific cues might be sufficient to trigger self-organised differentiation and planar structuring in other cell lines. Because of their ability to subject cell layers to multiple well-controlled cues, microfluidic biochips are ideally suited to identify more examples of self-organised differentiation and planar structuring in epithelial models.
Even in cases in which self-organised planar patterning might not occur spontaneously, microfluidic models could be combined with approaches to control the local cell environment in order to generate confined regions in which cells are stimulated to undergo further differentiation. In particular, local engineering of differentiation-relevant environmental parameters is a promising approach to produce epithelia that comprise a natural composition of cell types directly from stem cells. Multiple tools to locally adjust microenvironments have already been developed and await their integration into microfluidic models of epithelia. This includes tools that allow generating local variations of substrate properties, for example patterning of ECM-components or other immobilised molecules that influence cell differentiation,105–107 or local control of the substrate stiffness.105,108 Furthermore, several microfluidic methods exist to generate gradients of soluble molecules within the cell culture medium.109–111 This could be used to locally adjust the concentration of soluble factors influencing cell differentiation as well as to resemble morphogen gradients in order to resemble aspects of planar cell polarity.
However, most cell lines or primary cells have lost their capabilities to undergo further differentiation. In such cases, planar structuring of epithelia could be achieved by seeding different cell types in defined intercalating patterns within microfluidic biochips. To this end, a plethora of methods to pattern cells is available.111,112
Taken together, by controlling the planar organisation of epithelia, microfluidic models that much closer resemble all in vivo functions of epithelia could be engineered. This might, for example, enable creating secretory epithelia or whole glands that secrete fluids with a physiologically correct composition.
In addition to their transport functions, epithelial cell layers have several other essential functions. For example, epithelia are an important line of defence that prevents the intrusion of pathogens. However, several bacteria have evolved strategies to overcome epithelial barriers by e.g. hijacking trans-cellular transport processes or compromising tight junctions.113,114 Thus, microfluidic models of epithelia could provide powerful approaches to study bacterial intrusion.
The unique advantage of microfluidic approaches lies in the possibility to directly integrate analytical tools that enable measurement of biologically relevant parameters. Many analytical tools have been developed and integrated in microfluidic models for epithelia, as described in Section 4 of this review, and many more tools will be made available in this rapidly developing field. However, the analytical tools are usually integrated in a customised way. This can hamper a more widespread use of microfluidic models of epithelia, especially in biology labs, because such designs typically require homemade accessories and expert knowledge to operate the tools. This could be overcome by designing a modular microfluidic system of standardised analytical components, especially for tools that probe molecules in the cell supernatant (e.g. fluorescent readout, mass spectrometry, HPLC, immunoassays, etc.). An interesting option would be to design small microfluidic chips, each performing a distinct analytical readout, which can be connected to the main cell culture chip via short tubing. This will require the development of standardised connection interfaces.115 Such modular systems would enable to routinely monitor multiple different readouts in parallel during experiments with epithelial cell layers. This would allow more unbiased experimental approaches, because the chance to miss unexpected effects is drastically minimised. Another interesting perspective of standardised components is to connect different epithelial models, each resembling the function of a specific organ. This would enable to build models of whole organisms, which offers novel possibilities to study the effects of drugs or toxins with artificial systems.116–120
Acknowledgements
This work was supported by the Excellence Initiative of the German Research Foundation (EXC 294) and by a starting grant of the European Research Council (Programme “Ideas” – call identifier: ERC-2011-StG 282105) to WR. Research in ERB's lab was supported by NIH grants and funds from the Research to Prevent Blindness and Dyson foundations.References
- E. Rodriguez-Boulan, G. Kreitzer and A. Musch, Nat. Rev. Mol. Cell Biol., 2005, 6, 233–247 CrossRef CAS PubMed.
- B. Tanos and E. Rodriguez-Boulan, Oncogene, 2008, 27, 6939–6957 CrossRef CAS PubMed.
- T. Muller, M. W. Hess, N. Schiefermeier, K. Pfaller, H. L. Ebner, P. Heinz-Erian, H. Ponstingl, J. Partsch, B. Rollinghoff, H. Kohler, T. Berger, H. Lenhartz, B. Schlenck, R. J. Houwen, C. J. Taylor, H. Zoller, S. Lechner, O. Goulet, G. Utermann, F. M. Ruemmele, L. A. Huber and A. R. Janecke, Nat. Genet., 2008, 40, 1163–1165 CrossRef PubMed.
- T. Sato, S. Mushiake, Y. Kato, K. Sato, M. Sato, N. Takeda, K. Ozono, K. Miki, Y. Kubo, A. Tsuji, R. Harada and A. Harada, Nature, 2007, 448, 366–369 CrossRef CAS PubMed.
- M. Keiser, M. Alfalah, M. J. Propsting, D. Castelletti and H. Y. Naim, J. Biol. Chem., 2006, 281, 14393–14399 CrossRef CAS PubMed.
- C. A. Bertrand and R. A. Frizzell, Am. J. Physiol.: Cell Physiol., 2003, 285, C1–C18 CrossRef CAS PubMed.
- M. Fliegauf, T. Benzing and H. Omran, Nat. Rev. Mol. Cell Biol., 2007, 8, 880–893 CrossRef CAS PubMed.
- C. Boehlke, F. Kotsis, V. Patel, S. Braeg, H. Voelker, S. Bredt, T. Beyer, H. Janusch, C. Hamann, M. Godel, K. Muller, M. Herbst, M. Hornung, M. Doerken, M. Kottgen, R. Nitschke, P. Igarashi, G. Walz and E. W. Kuehn, Nat. Cell Biol., 2010, 12, 1115–U1126 CrossRef CAS PubMed.
- P. C. Harris and V. E. Torres, Annu. Rev. Med., 2009, 60, 321–337 CrossRef CAS PubMed.
- J. Z. Chuang and C. H. Sung, J. Cell Biol., 1998, 142, 1245–1256 CrossRef CAS PubMed.
- C. H. Sung and S. W. Tai, Int. Rev. Cytol., 2000, 195, 215–267 CAS.
- N. A. Zaghloul and N. Katsanis, J. Clin. Invest., 2009, 119, 428–437 CAS.
- A. L. Daugherty and R. J. Mrsny, Pharm. Sci. Technol. Today, 1999, 4, 144–151 CrossRef PubMed.
- P. H. Johnson, D. Frank and H. R. Costantino, Drug Discovery Today, 2008, 13, 261–267 CrossRef CAS PubMed.
- W. M. Pardridge, NeuroRx, 2005, 2, 3–14 CrossRef PubMed.
- D. M. Bryant and K. E. Mostov, Nat. Rev. Mol. Cell Biol., 2008, 9, 887–901 CrossRef CAS PubMed.
- L. E. O'Brien, M. M. P. Zegers and K. E. Mostov, Nat. Rev. Mol. Cell Biol., 2002, 3, 531–537 CrossRef PubMed.
- J. Debnath and J. S. Brugge, Nat. Rev. Cancer, 2005, 5, 675–688 CrossRef CAS PubMed.
- G. J. Pazour and G. B. Witman, Curr. Opin. Cell Biol., 2003, 15, 105–110 CrossRef CAS PubMed.
- V. Singla and J. F. Reiter, Science, 2006, 313, 629–633 CrossRef CAS PubMed.
- G. Bazzoni, Thromb. Haemostasis, 2006, 95, 36–42 CAS.
- H. Wolburg and A. Lippoldt, Vasc. Pharmacol., 2002, 38, 323–337 CrossRef CAS PubMed.
- I. Mellman and W. J. Nelson, Nat. Rev. Mol. Cell Biol., 2008, 9, 833–845 CrossRef CAS PubMed.
- F. Martin-Belmonte and M. Perez-Moreno, Nat. Rev. Cancer, 2012, 12, 23–38 CAS.
- M. Simons and M. Mlodzik, Annu. Rev. Genet., 2008, 42, 517–540 CrossRef CAS PubMed.
- E. K. Vladar, R. D. Bayly, A. M. Sangoram, M. P. Scott and J. D. Axelrod, Curr. Biol., 2012, 22, 2203–2212 CrossRef CAS PubMed.
- P. A. Lawrence, G. Struhl and J. Casal, Nat. Rev. Genet., 2007, 8, 555–563 CrossRef CAS PubMed.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed.
- J. Melin and S. R. Quake, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 213–231 CrossRef CAS PubMed.
- S. K. Sia and G. M. Whitesides, Electrophoresis, 2003, 24, 3563–3576 CrossRef CAS PubMed.
- E. Berthier, E. W. K. Young and D. Beebe, Lab Chip, 2012, 12, 1224–1237 RSC.
- F. Kotsis, R. Nitschke, M. Doerken, G. Walz and E. W. Kuehn, Pflügers Archiv – European Journal of Physiology, 2008, 456, 1025–1035 CrossRef CAS PubMed.
- S. Rydholm, T. Frisk, J. M. Kowalewski, H. A. Svahn, G. Stemme and H. Brismar, Biomed. Microdevices, 2008, 10, 555–560 CrossRef PubMed.
- E. Tkachenko, E. Gutierrez, S. K. Saikin, P. Fogelstrand, C. Kim, A. Groisman and M. H. Ginsberg, Biol. Open, 2013, 2, 1007–1012 CrossRef CAS PubMed.
- A. D. van der Meer, A. A. Poot, J. Feijen and I. Vermes, Biomicrofluidics, 2010, 4, 011103 CrossRef CAS PubMed.
- E. M. Frohlich, X. Zhang and J. L. Charest, Integr. Biol., 2012, 4, 75–83 RSC.
- J. S. Choi, Y. Piao and T. S. Seo, Bioprocess Biosyst. Eng., 2013, 36, 1871–1878 CrossRef CAS PubMed.
- L. K. Fiddes, N. Raz, S. Srigunapalan, E. Tumarkan, C. A. Simmons, A. R. Wheeler and E. Kumacheva, Biomaterials, 2010, 31, 3459–3464 CrossRef CAS PubMed.
- Q. Ramadan, H. Jafarpoorchekab, C. B. Huang, P. Silacci, S. Carrara, G. Koklu, J. Ghaye, J. Ramsden, C. Ruffert, G. Vergeres and M. A. M. Gijs, Lab Chip, 2013, 13, 196–203 RSC.
- C. M. Puleo, W. M. Ambrose, T. Takezawa, J. Elisseeff and T. H. Wang, Lab Chip, 2009, 9, 3221–3227 RSC.
- C. Desrousseaux, J. M. Prot, M. Dufresne, P. Paullier and E. Leclerc, Sens. Actuators, B, 2012, 174, 465–472 CrossRef CAS.
- A. Ould-Dris, P. Paullier, L. Griscom, C. Legallais and E. Leclerc, J. Membr. Sci., 2010, 352, 116–125 CrossRef CAS.
- C. Ramello, P. Paullier, A. Ould-Dris, M. Monge, C. Legallais and E. Leclerc, Toxicol. in Vitro, 2011, 25, 1123–1131 CrossRef CAS PubMed.
- K. J. Jang, H. S. Cho, D. H. Kang, W. G. Bae, T. H. Kwon and K. Y. Suh, Integr. Biol., 2011, 3, 134–141 RSC.
- K. J. Jang, A. P. Mehr, G. A. Hamilton, L. A. McPartlin, S. Y. Chung, K. Y. Suh and D. E. Ingber, Integr. Biol., 2013, 5, 1119–1129 RSC.
- K. J. Jang and K. Y. Suh, Lab Chip, 2010, 10, 36–42 RSC.
- H. Kimura, T. Yamamoto, H. Sakai, Y. Sakai and T. Fujii, Lab Chip, 2008, 8, 741–746 RSC.
- A. A. Epshteyn, S. Maher, A. J. Taylor, A. B. Holton, J. T. Borenstein and J. D. Cuiffi, Biomicrofluidics, 2011, 5, 046501 CrossRef PubMed.
- B. H. Chueh, D. Huh, C. R. Kyrtsos, T. Houssin, N. Futai and S. Takayama, Anal. Chem., 2007, 79, 3504–3508 CrossRef CAS PubMed.
- N. Ferrell, R. R. Desai, A. J. Fleischman, S. Roy, H. D. Humes and W. H. Fissell, Biotechnol. Bioeng., 2010, 107, 707–716 CrossRef CAS PubMed.
- S. Hediger, A. Sayah, J. D. Horisberger and M. A. M. Gijs, Biosens. Bioelectron., 2001, 16, 689–694 CrossRef CAS PubMed.
- K. Atsuta, H. Noji and S. Takeuchi, Lab Chip, 2004, 4, 333–336 RSC.
- D. Huh, H. J. Kim, J. P. Fraser, D. E. Shea, M. Khan, A. Bahinski, G. A. Hamilton and D. E. Ingber, Nat. Protoc., 2013, 8, 2135–2157 CrossRef CAS PubMed.
- R. J. Jackman, D. C. Duffy, O. Cherniavskaya and G. M. Whitesides, Langmuir, 1999, 15, 2973–2984 CrossRef CAS.
- X. Li, K. G. Klemic, M. A. Reed and F. J. Sigworth, Nano Lett., 2006, 6, 815–819 CrossRef CAS PubMed.
- Y. Luo and R. N. Zare, Lab Chip, 2008, 8, 1688–1694 RSC.
- H. Wei, B.-h. Chueh, H. Wu, E. W. Hall, C.-w. Li, R. Schirhagl, J.-M. Lin and R. N. Zare, Lab Chip, 2011, 11, 238–245 RSC.
- D. C. Duffy, J. C. McDonald, O. J. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- K. Haubert, T. Drier and D. Beebe, Lab Chip, 2006, 6, 1548–1549 RSC.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- G. M. Whitesides and S. K. Y. Tang, Proc. SPIE, 2006, 6329, U61–U73 CrossRef.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Lab Chip, 2012, 12, 2165–2174 RSC.
- H. J. Kim and D. E. Ingber, Integr. Biol., 2013, 5, 1130–1140 RSC.
- T. Sun, E. J. Swindle, J. E. Collins, J. A. Holloway, D. E. Davies and H. Morgan, Lab Chip, 2010, 10, 1611–1617 RSC.
- E. M. Frohlich, J. L. Alonso, J. T. Borenstein, X. Zhang, M. A. Arnaout and J. L. Charest, Lab Chip, 2013, 13, 2311–2319 RSC.
- M. B. Esch, J. H. Sung, J. Yang, C. H. Yu, J. J. Yu, J. C. March and M. L. Shuler, Biomed. Microdevices, 2012, 14, 895–906 CrossRef CAS PubMed.
- C. P. Ng, Y. Zhuang, A. W. H. Lin and J. C. M. Teo, International J. Tissue Eng., 2012, 2013, 319476 Search PubMed.
- B. Prabhakarpandian, M. C. Shen, J. B. Nichols, I. R. Mills, M. Sidoryk-Wegrzynowicz, M. Aschner and K. Pant, Lab Chip, 2013, 13, 1093–1101 RSC.
- J. B. Shao, L. Wu, J. Z. Wu, Y. H. Zheng, H. Zhao, Q. H. Jin and J. L. Zhao, Lab Chip, 2009, 9, 3118–3125 RSC.
- Y. Imura, Y. Asano, K. Sato and E. Yoshimura, Anal. Sci., 2009, 25, 1403–1407 CrossRef CAS PubMed.
- R. Thuenauer, K. Juhasz, R. Mayr, T. Fruhwirth, A. M. Lipp, Z. Balogi and A. Sonnleitner, Lab Chip, 2011, 11, 3064–3071 RSC.
- D. Gao, H. X. Liu, J. M. Lin, Y. N. Wang and Y. Y. Jiang, Lab Chip, 2013, 13, 978–985 RSC.
- S. Y. C. Chen, P. J. Hung and P. J. Lee, Biomed. Microdevices, 2011, 13, 753–758 CrossRef CAS PubMed.
- S. Bersini, J. Jeon, M. Moretti and R. Kamm, Drug Discovery Today DOI:10.1016/j.drudis.2013.12.006.
- M. Verhulsel, M. v. Vignes, S. p. Descroix, L. Malaquin, D. M. Vignjevic and J.-L. Viovy, Biomaterials, 2014, 35, 1816–1832 CrossRef CAS PubMed.
- K. H. K. Wong, J. M. Chan, R. D. Kamm and J. Tien, Annu. Rev. Biomed. Eng., 2012, 14, 205–230 CrossRef CAS PubMed.
- E. W. K. Young, J. Lab. Autom., 2013, 18, 427–436 CrossRef PubMed.
- N. J. Douville, Y. C. Tung, R. Li, J. D. Wang, M. E. H. El-Sayed and S. Takayama, Anal. Chem., 2010, 82, 2505–2511 CrossRef CAS PubMed.
- R. Booth and H. Kim, Lab Chip, 2012, 12, 1784–1792 RSC.
- S. Deborde, E. Perret, D. Gravotta, A. Deora, S. Salvarezza, R. Schreiner and E. Rodriguez-Boulan, Nature, 2008, 452, 719–U713 CrossRef CAS PubMed.
- L. W. Leung, R. G. Contreras, C. Flores-Maldonado, M. Cereijido and E. Rodriguez-Boulan, Am. J. Physiol.: Cell Physiol., 2003, 284, C1021–C1030 CrossRef CAS PubMed.
- K. Benson, S. Cramer and H.-J. Galla, Fluids Barriers CNS, 2013, 10, 5 CrossRef PubMed.
- L. M. Griep, F. Wolbers, B. de Wagenaar, P. M. ter Braak, B. B. Weksler, I. A. Romero, P. O. Couraud, I. Vermes, A. D. van der Meer and A. van den Berg, Biomed. Microdevices, 2013, 15, 145–150 CrossRef CAS PubMed.
- J. Wegener, D. Abrams, W. Willenbrink, H. J. Galla and A. Janshoff, Biotechniques, 2004, 37, 590–597 CAS.
- P. A. Vogel, S. T. Halpin, R. S. Martin and D. M. Spence, Anal. Chem., 2011, 83, 4296–4301 CrossRef CAS PubMed.
- N. Z. Wu and A. L. Baldwin, Am. J. Physiol., 1992, 262, H1238–H1247 CAS.
- N. J. Abbott, L. Ronnback and E. Hansson, Nat. Rev. Neurosci., 2006, 7, 41–53 CrossRef CAS PubMed.
- A. S. Verkman, Annu. Rev. Med., 2012, 63, 303–316 CrossRef CAS PubMed.
- E. W. K. Young, M. W. L. Watson, S. Srigunapalan, A. R. Wheeler and C. A. Simmons, Anal. Chem., 2010, 82, 808–816 CrossRef CAS PubMed.
- T. Su, D. M. Bryant, F. Luton, M. Verges, S. M. Ulrich, K. C. Hansen, A. Datta, D. J. Eastburn, A. L. Burlingame, K. M. Shokat and K. E. Mostov, Nat. Cell Biol., 2010, 12, 1143–U1160 CrossRef CAS PubMed.
- H. A. Praetorius and K. R. Spring, J. Membr. Biol., 2001, 184, 71–79 CrossRef CAS PubMed.
- H. A. Praetorius and K. R. Spring, J. Membr. Biol., 2003, 191, 69–76 CrossRef CAS PubMed.
- J. Rahimzadeh, F. J. Meng, F. Sachs, J. B. Wang, D. Verma and S. Z. Hua, Am. J. Physiol.: Cell Physiol., 2011, 301, C646–C652 CrossRef CAS PubMed.
- N. J. Douville, P. Zamankhan, Y. C. Tung, R. Li, B. L. Vaughan, C. F. Tai, J. White, P. J. Christensen, J. B. Grotberg and S. Takayama, Lab Chip, 2011, 11, 609–619 RSC.
- J. Schmoranzer, M. Goulian, D. Axelrod and S. M. Simon, J. Cell Biol., 2000, 149, 23–32 CrossRef CAS PubMed.
- D. Axelrod, Traffic, 2001, 2, 764–774 CrossRef CAS PubMed.
- J. K. Jaiswal and S. M. Simon, Nat. Chem. Biol., 2007, 3, 92–98 CrossRef CAS PubMed.
- R. Thuenauer, Y. C. Hsu, J. M. Carvajal-Gonzalez, S. Deborde, J. Z. Chuang, W. Römer, A. Sonnleitner, E. Rodriguez-Boulan and C. H. Sung, Proc. Natl. Acad. Sci. U.S.A, 2014, 111, 4127–4132 CrossRef CAS PubMed.
- D. Huh, H. Fujioka, Y. C. Tung, N. Futai, R. Paine, J. B. Grotberg and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18886–18891 CrossRef CAS PubMed.
- C. Long, C. Finch, M. Esch, W. Anderson, M. Shuler and J. Hickman, Ann. Biomed. Eng., 2012, 40, 1255–1267 CrossRef CAS PubMed.
- D. D. Nalayanda, C. Puleo, W. B. Fulton, L. M. Sharpe, T. H. Wang and F. Abdullah, Biomed. Microdevices, 2009, 11, 1081–1089 CrossRef CAS PubMed.
- H. Tavana, P. Zamankhan, P. J. Christensen, J. B. Grotberg and S. Takayama, Biomed. Microdevices, 2011, 13, 731–742 CrossRef PubMed.
- H. Tavana, C. H. Kuo, Q. Y. Lee, B. Mosadegh, D. Huh, P. J. Christensen, J. B. Grotberg and S. Takayama, Langmuir, 2010, 26, 3744–3752 CrossRef CAS PubMed.
- D. H. Kim, P. K. Wong, J. Park, A. Levchenko and Y. Sun, Annu. Rev. Biomed. Eng., 2009, 11, 203–233 CrossRef CAS PubMed.
- S. Kobel and M. P. Lutolf, Curr. Opin. Biotechnol., 2011, 22, 690–697 CrossRef CAS PubMed.
- F. L. Yap and Y. Zhang, Biosens. Bioelectron., 2007, 22, 775–788 CrossRef CAS PubMed.
- Y. Li, G. Huang, X. Zhang, L. Wang, Y. Du, T. J. Lu and F. Xu, Biotechnol. Adv., 2014, 32, 347–365 CrossRef CAS PubMed.
- T. M. Keenan and A. Folch, Lab Chip, 2008, 8, 34–57 RSC.
- S. Kim, H. J. Kim and N. L. Jeon, Integr. Biol., 2010, 2, 584–603 RSC.
- M. Tehranirokh, A. Z. Kouzani, P. S. Francis and J. R. Kanwar, Biomicrofluidics, 2013, 7, 51502 CrossRef PubMed.
- R. S. Kane, S. Takayama, E. Ostuni, D. E. Ingber and G. M. Whitesides, Biomaterials, 1999, 20, 2363–2376 CrossRef CAS PubMed.
- J. Engel and Y. Eran, Frontiers in microbiology, 2011, 2, 114 CrossRef PubMed.
- B. I. Kazmierczak, K. Mostov and J. N. Engel, Annu. Rev. Microbiol., 2001, 55, 407–435 CrossRef CAS PubMed.
- H. van Heeren, Lab Chip, 2012, 12, 1022–1025 RSC.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–754 CrossRef CAS PubMed.
- D. Huh, Y. S. Torisawa, G. A. Hamilton, H. J. Kim and D. E. Ingber, Lab Chip, 2012, 12, 2156–2164 RSC.
- C. Moraes, G. Mehta, S. C. Lesher-Perez and S. Takayama, Ann. Biomed. Eng., 2012, 40, 1211–1227 CrossRef PubMed.
- M. L. Shuler and M. B. Esch, Pure Appl. Chem., 2010, 82, 1635–1645 CrossRef CAS.
- J. H. Sung, M. B. Esch, J. M. Prot, C. J. Long, A. Smith, J. J. Hickman and M. L. Shuler, Lab Chip, 2013, 13, 1201–1212 RSC.
| This journal is © The Royal Society of Chemistry 2014 |