 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of dsDNA fragment length on particle binding in an evanescent field biosensing system
Marjo
Koets
,
Kim
van Ommering
,
Liqin
Wang
,
Emilie
Testori
,
Toon H.
Evers
and
Menno W. J.
Prins
*
Philips Research Europe, High Tech Campus 11, 5656 AE Eindhoven, The Netherlands. E-mail: menno.prins@philips.com
First published on 14th January 2014
Abstract
Particle labels are widely used in affinity-based biosensing due to the high detection signal per label, the high stability, and the convenient biofunctionalization of particles. In this paper we address the question how the time-course of particle binding and the resulting signals depend on the length of captured target molecules. As a model system we used fragments of dsDNA with lengths of 105 bp (36 nm), 290 bp (99 nm) and 590 bp (201 nm), detected in an evanescent-field optomagnetic biosensing system. On both ends the fragments were provided with small-molecule tags to allow binding of the fragments to protein-coated particles and to the capture molecules at the sensor surface. For isolated single particles bound to the surface, we observe that the optical scattering signal per particle depends only weakly on the fragment length, which we attribute to the pivoting motion that allows the particles to get closer to the surface. Our data show a strong influence of the fragment length on the particle binding: the binding rate of particles to the sensor surface is an order of magnitude higher for the longest dsDNA fragments compared to the smallest fragment studied in this paper. We attribute the enhanced binding rate to the length and motional freedom of the fragments. These results generate a new dimension for the design of assays and systems in particle-based biosensing.
Introduction
Micro- and nanoparticles are frequently applied as labels in affinity biosensing for the high detection signals per label, the high stability, and the convenient biofunctionalization of particles.1–5 A disadvantage of using particles as labels can be that they diffuse more slowly compared to molecular labels. One way to resolve the kinetics limitations is by using superparamagnetic particles which can be actuated by magnetic fields. The actuation by magnetic fields has the advantage that transport and binding are accelerated, and that well-controlled stringency forces can be applied to the particles, resulting in rapid and compact biosensing systems.6–8 An interesting question in the field of particle-based biosensors is how the particle-to-surface binding and the limit of detection depend on the size of the captured molecules. The influence of the molecular length on particle-to-surface binding has been studied before using polyethylene glycol spacers of different molecular weights9 and by comparing binding pairs of different sizes.10 These studies were performed in the perspective of in vivo particle targeting applications, where particles are coated with high densities of molecular targeting moieties and therefore multivalency effects play a very important role. The reported size effects cannot be generalized to the case of diagnostic biosensing assays, because such assays generally deal with very low target concentrations and therefore very low densities of binding moieties on the particles and/or the surface.In this paper we study the influence of the molecular size on the particle binding rate, dose–response curve, and signal per particle in an optomagnetic biosensing assay. The biosensor is based on superparamagnetic particles that are magnetically actuated and optically detected.6 We use dsDNA as a molecular model system, with fragment lengths of 105 bp (36 nm), 290 bp (99 nm) or 590 bp (201 nm). The fragments are tagged on both ends, so that they can be captured by magnetic particles and thereafter bind to a functionalized biosensor surface, as sketched in Fig. 1. The bound particles are detected at the surface by light scattering in an optical evanescent field. In the subsequent sections we will describe the experimental system in detail and we will report the influence of the fragment length on the particle-based biosensing assay.
 | ||
| Fig. 1 A schematic view of the assay configuration and the optomagnetic detection system. Target molecules (dsDNA fragments, with a biotin tag and a Texas Red tag) are captured by streptavidin-coated magnetic particles. Subsequently the particles bind to the anti-Texas Red coated sensor surface via the DNA fragments, and the bound particles are detected by light scattering in an evanescent optical field.6 The dsDNA fragments have a length of 105 bp (36 nm), 290 bp (99 nm) or 590 bp (201 nm). Part of the image is reproduced with permission from the Royal Society of Chemistry. | ||
Experimental
Preparation of tagged-dsDNA fragments
We used a DNA-immunoassay format based on the capturing of double-tagged dsDNA fragments.7,11–13 Double tagged dsDNA fragments with different lengths were prepared by amplification of bacterial genus- and species-specific fragments. The forward- and reverse primers were end-tagged with Texas Red and biotin, respectively (Biolegio, Nijmegen, The Netherlands). The following primer sets were used: Staphylococcus aureus ATCC 29213: (5′-biotin-CGGCTTTTACATACAGAACACAATGT-3′ and 5′-Texas Red-CGCTCATTCGCGATTTTATAAAT-3′), Salmonella enterica S398 (ref. 14): (5′-Texas Red-GCAGCGCAATGACATTCTTG-3′ and 5′-biotin-CATCCTTCGGCGCGATTTTG-3′) and Escherichia coli ATCC 35218: (5′-Texas Red-GAGGGTTGCTGGAGGTATCA-3′ and 5′-biotin-CCACTTTCGTGTTTGCACAG-3′). Amplification using these primer sets resulted in fragments of 105 bp for S. aureus, 290 bp for Salmonella and 590 bp for E. coli. The PCR reactions were carried out as described by Koets et al.7 Excess tagged primers were removed from the PCR solutions using the ChargeSwitch PCR Clean-up Kit (Invitrogen, Breda, The Netherlands). The PCR products were analysed with an Agilent 2100 Bioanalyzer using the DNA 1000 kit (Agilent Technologies, Amstelveen, The Netherlands).Preparation of the biosensor surface and cartridge
Experiments were performed on polystyrene surfaces onto which four antibody containing spots were deposited with a diameter of 200 μm. Anti-Texas Red polyclonal antibodies (Molecular Probes, The Netherlands) were diluted to 0.15 mg protein mL−1 in Phosphate Buffered Saline (PBS), followed by printing 2 nL of antibody per spot on the sensor surface as described previously.7 The printed surfaces were dried for three minutes at 37 °C and then washed three times with 500 μL PBS containing 0.05% (v/v) Tween-20. Closed microfluidic cartridges were made using 180 μm double-side adhesive tape, resulting in a reaction channel with a total volume of 10 μL and a 1 μL assay chamber. The cartridges were blocked in PBS containing 1% (w/v) BSA and 10% (w/v) sucrose; and were stored at 4 °C until used.Detection of bound particles by evanescent optical fields
Two experimental setups were used for detection of bound particles in an optical evanescent field: (i) detection by frustrated total internal reflection (f-TIR6), which is an optical bright-field detection method, with results in Fig. 1, 2, and 3b and (ii) detection by microscopic particle imaging,15 which is an optical dark-field detection method, with results in Fig. 3. In both cases the light penetrates into the fluid by a sub-wavelength distance and is scattered by the presence of the magnetic particles at the sensor surface. The intensity of the evanescent field and therefore also the scattering signal decay as a function of distance from the sensor surface.16 f-TIR detection was applied to record biosensor dose–response curves. Microscopic imaging was applied to record the scattering behaviour of individual particles at the surface. | ||
| Fig. 2 Assay time traces and dose–response curves for different fragment lengths. (a) Time curves of the target-induced binding of magnetic particles to the sensor surface. The signals were recorded by frustrated total internal reflection, as sketched in the inset. Targets were dsDNA fragments with lengths of 105, 290 or 590 base pairs. Curves have been recorded at target concentrations of 12 pM and 150 pM. The fluctuations on the curves are caused by the pulsed magnetic fields that were applied during the binding process. At the end a magnetic wash was applied, which stabilizes the signal. (b) Dose–response curves of dsDNA fragments in a one-step assay, recorded for different fragment lengths. Signals were obtained by measuring two cartridges with four spots each. The inset shows the dose–response curves on a log–log scale, in order to highlight the signals at low fragment concentrations. The dotted horizontal line in the inset shows the signal of the blank plus three times the standard deviation of the blank, resulting in a limit of detection of 0.62% signal change; this level corresponds to fragment concentrations of 2 pM for 290 bp and 590 bp, and 25 pM for 105 bp. In both figure panels, the assumed particle arrangements are sketched next to the curves. (c) Enlarged sketches of the assumed particle arrangements. At low surface densities, bound particles have the freedom to pivot. At high surface densities, the motion of particles is restricted and the fragments are aligned. | ||
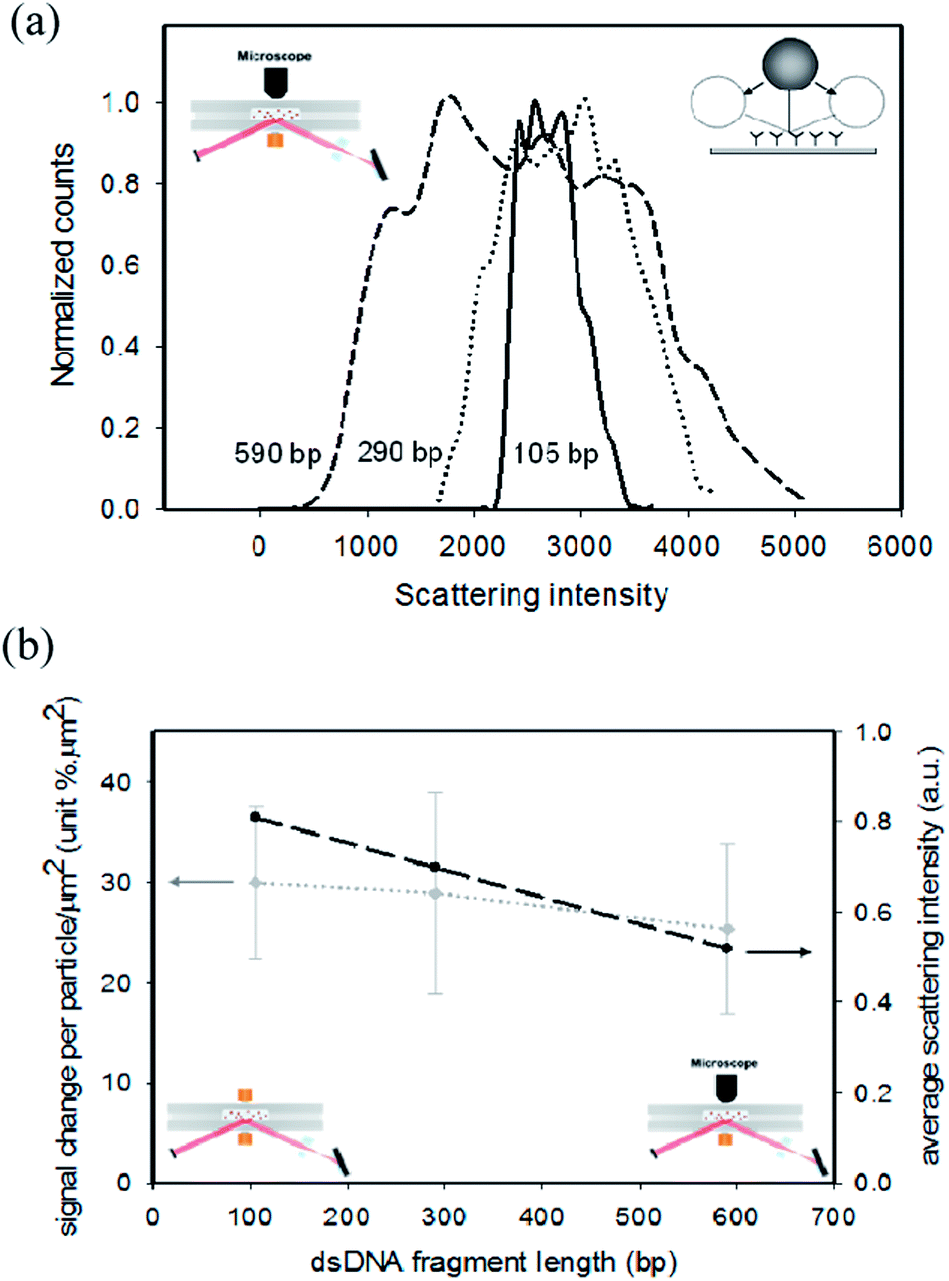 | ||
| Fig. 3 Optical signals as a function of fragment length. (a) Histograms of the scattering intensity of single polystyrene particles (760 nm diameter). The intensity of the scattered light was recorded by using a microscope (see the sketch on the top left). Particles were attached to the sensor surface by a dsDNA fragment with a length of 105 bp (36 nm), 290 bp (99 nm) or 590 bp (201 nm). The width of the histogram reflects the intensity fluctuations caused by the thermal motional freedom of the fragment-bound particles (see the sketch on the top right). The histograms have been scaled to a maximum value of unity. The lines are guides to the eye; the irregularities in the lines are caused by limited statistics in the data analysis. (b) Optical signal as a function of dsDNA fragment length. Left axis: signal measured using magnetic particles and f-TIR detection, expressed as signal per particle surface density (% μm2). The number of bound particles was determined by microscopic imaging. Right axis: average scattering intensity of polystyrene particles, calculated from the histograms in panel (a). The dashed lines are guides to the eye. | ||
dsDNA sandwich assay with magnetic particles as labels
A dsDNA sandwich assay was performed as sketched in Fig. 1. Streptavidin coated magnetic particles (500 nm; Ademtech, Pessac, France) were washed with assay buffer (100 mM borate buffer (pH 8.5) containing 1% (w/v) BSA and 0.05% (v/v) Tween-20). The dsDNA fragments were diluted in assay buffer containing 0.1% (w/v) end-concentration of streptavidin magnetic particles. The magnetic particles were pre-incubated with the dsDNA fragments for 1 min at room temperature and thereafter applied to the cartridge for magnetic actuation and optical detection. Experiments have shown that within 1 min equilibrium was reached between the magnetic particles and the dsDNA fragments, because longer pre-incubation did not increase the signal change obtained. Just before use the cartridge was washed with 100 mM borate buffer (pH 8.5) containing 0.05% (v/v) Tween-20 and blown dry. Thereafter the dsDNA–magnetic particle solution was inserted into the cartridge. All measurements were performed in duplicate, recording four spots per cartridge. Fragment concentrations ranged between 1 and 200 pM dsDNA. The molar ratio of particles-to-fragments is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1 at 1 pM and 1:20 at 200 pM fragment concentrations. In the f-TIR experiments, the magnetic field was alternatively switched between the lower magnet (28 kA m−1) and the upper magnet (8 kA m−1) for 250 s. In a final step, the upper magnet was powered (10 kA m−1) to remove unbound particles from the sensor surface for 20 s. The total assay time was approximately 5 min. The signal was determined by relating the reflected light intensity to the measured light intensity prior to the binding of particles, using the formula: signal (t) = [R(0) − R(t)]/R(0), where R(0) is the reflected light intensity in the absence of magnetic particles at the sensor surface and R(t) is the reflected light intensity during the assay. The signal due to the binding of particles to the sensor surface was calculated for each spot, averaging over an area of 40 pixels × 40 pixels.
:0.1 at 1 pM and 1:20 at 200 pM fragment concentrations. In the f-TIR experiments, the magnetic field was alternatively switched between the lower magnet (28 kA m−1) and the upper magnet (8 kA m−1) for 250 s. In a final step, the upper magnet was powered (10 kA m−1) to remove unbound particles from the sensor surface for 20 s. The total assay time was approximately 5 min. The signal was determined by relating the reflected light intensity to the measured light intensity prior to the binding of particles, using the formula: signal (t) = [R(0) − R(t)]/R(0), where R(0) is the reflected light intensity in the absence of magnetic particles at the sensor surface and R(t) is the reflected light intensity during the assay. The signal due to the binding of particles to the sensor surface was calculated for each spot, averaging over an area of 40 pixels × 40 pixels.
Single-particle scattering experiments
Microscopic imaging was applied to record the behaviour of individual particles at the surface with nanometer precision.15 The goal was to determine the influence of dsDNA fragment length on the scattering histograms, in order to be able to reveal how the scattering signal per particle depends on the dsDNA fragment length. In these experiments, the material of which the particles are made is not important, because the dynamics of sub-micrometer particles is determined by Brownian motion, which is independent of the mass density of the particles. Polystyrene particles were selected because these particles are spherical and highly monodisperse; therefore the scattering does not depend on the particle orientation and the particle-to-particle variation is very low.15 Streptavidin-coated polystyrene particles were used (760 nm, Spherotech, Lake Forest, USA) (0.25% w/v). 5 pM dsDNA fragments were applied to the cartridge and were incubated for 3–8 minutes in order to obtain a low density of bound particles at the sensor surface (∼0.01 particles per μm2). Unbound particles were removed by washing with 100 mM borate buffer (pH 8.5) containing 0.05% (v/v) Tween-20. The scattering signal per particle was calculated by integrating pixel signals belonging to a single particle. Signals were recorded as a function of time, so that intensity histograms could be established (Fig. 3a) as well as a time-averaged scattering intensity per particle (Fig. 3b).Measurement of the signal per particle in f-TIR
Particle-counting microscopy experiments were combined with f-TIR measurements in order to quantify the f-TIR signal change per particle surface density. 0.1% (w/v) particles were incubated with 10 pM dsDNA fragments with different lengths, resulting in approximately 1 fragment per particle. In the f-TIR biosensing system, magnetic attraction was applied and followed by a magnetic wash step, in order to generate a signal between 1 and 5% signal change. Reliable counting of bound particles by microscopy requires that unbound particles are removed. Removal of unbound particles was performed by repetitively flushing PBS buffer through the cartridge. Thereafter the f-TIR signal change was recorded and the cartridge was moved from the f-TIR setup to the microscope setup. In the microscope a movie was recorded at 20× magnification. Movie frames were averaged so that remaining unbound particles were smeared out by their motion and became invisible for the particle counting software. An in-house written MatLab algorithm was used to recognize and count the bound particles, giving the density of bound particles on the sensor surface.Results and discussion
Fragment length-dependence of binding kinetics
Fig. 2a shows the time-dependent signals measured for double-stranded DNA fragments with different lengths, recorded at fragment concentrations of 12 pM and 150 pM. During the first 250 s of the assay a pulsating magnetic field is applied in order to attract and bind the magnetic particle–DNA complexes to the sensor surface.6 For clarity, the high-amplitude pulsating signals due to the magnetic field pulses are not shown; only the envelopes of the curves are shown, corresponding to the measurement of surface-bound particles as a function of time. At 250 s a reversed magnetic force is applied (so-called magnetic wash) in order to remove non-bound and weakly bound magnetic particles from the sensor surface. We first discuss the curves for the low fragment concentration (12 pM). The signals rise as a function of time and the signals are larger for long fragments than for short fragments. The optical signals that we observe are a product of the particle density on the sensor surface and of the signal generated per bound particle. The signal per particle is expected to decrease for longer fragments due to the distance dependence of the evanescent field (in the next section we will quantify the influence of the fragment length on the signal per particle). Therefore the observed increase of the signal for longer fragments must be due to a higher density of particles bound to the surface. This means that particles with longer fragments have a higher binding rate to the sensor surface (kon). We attribute the higher binding rate of particles with longer fragments to the improved steric availability of the Texas Red tags for binding to the sensor surface, caused by the outreach on longer dsDNA fragments.The curves at high concentration (150 pM) are remarkably different. At short times (<50 s) the long fragments give a larger signal than the short fragments, again caused by the higher binding rate of longer DNA fragments. However, beyond 50 s, the 290 bp and 590 bp curves bend downward and show a decrease of signal as a function of time. We attribute this behaviour to the density-dependence of the particle arrangement on the sensor surface. From prior single-particle studies15 we know that individual particles attached to a surface via dsDNA with antibody–ligand binding (as in Fig. 1) are able to perform a pivoting motion, as sketched in Fig. 2a in the low-signal regime of the curves, and as sketched in Fig. 2c, left-hand side. However, at a high particle density the pivoting motion is sterically hindered, as is sketched in Fig. 2a in the high-signal regimes, and as sketched in Fig. 2c, right-hand side. The steric hindrance reduces the motional freedom of bound particles and as a result the net particle-to-surface distance increases. This hypothesis can explain why the optical scattering signal per bound particle goes down as a function of time and why the end signals are lowest for long dsDNA fragments. The maximum alignment-induced increase of the particle-to-surface distance depends on the fragment length, being respectively ∼36 nm (105 bp), ∼99 nm (290 bp) and ∼201 nm (590 bp). The change of optical scattering signal depends on the ratio between the distance change and the depth of the evanescent field.6,15 In the optomagnetic biosensor, the evanescent field depth is about 90 nm. Therefore, the alignment is expected to give appreciable reductions of signal change for 290 bp and 590 bp, but not for 105 bp fragments, in agreement with the data in Fig. 2a.
Interestingly, the curves measured at 150 pM are smoother than the curves recorded at 12 pM, which have short-timescale fluctuations. The fluctuations on the curves are caused by the pulsed magnetic fields that were applied during the binding process.6 We attribute the smoothness of the 150 pM curves to the molar ratio of particles-to-fragments; the molar ratio is 1:15 at 150 pM and 1:1.2 at a 12 pM fragment concentration. At 150 pM, the particles have captured many dsDNA fragments and are expected to bind to the sensor surface within a short time of encounter (hit-and-stick behavior). At 12 pM, an order of magnitude fewer fragments have been bound to the particles, so the particles are expected to have a much lower sticking probability and need repeated encounters with the sensor surface before binding takes place. Thus, we attribute the difference in short-timescale fluctuations on the curves to the different binding properties, caused by the different molar ratios. Finally, the 150 pM curves all show a small increase of the signal after the magnetic wash. This may be caused by mutually repulsive dipolar interactions between bound particles.
Curves shown in Fig. 2a have been recorded for a range of concentrations and the final signal values are plotted in Fig. 2b as a dose–response curve. The inset highlights the behaviour at low concentrations, showing that the limit of detection is reduced by about one order of magnitude. The 290 bp and 590 bp curves rise at lower concentrations than the 105 bp curve, caused by the higher binding rate of particles with the longer fragment lengths. At concentrations above 50 pM the curves for the longer fragments bend downward, which we attribute to the steric alignment of particles at the sensor surface due the high density of bound particles, as discussed above. At low concentrations the individual particles are spaced apart and have the freedom to pivot around the binding point. At high particle surface densities, steric hindrance occurs, which reduces the motional freedom and thereby the average signal per particle.
The data in Fig. 2b show that the longer dsDNA fragments give a left-shifted and thereby more sensitive dose–response curve. The limit of detection of the assay differs by an order of magnitude between the long and short DNA fragments: 2 pM for 290 bp and for 590 bp, and 25 pM for 105 bp (we note that the assay has not been optimized for the lowest possible limit of detection, and the significance lies in the fragment-length dependence of the results). In order to be able to interpret the difference in terms of the underlying processes, we will determine in the next section how the fragment length influences the optical signal per particle. Thereafter we will be able to draw a quantitative conclusion on the influence of the fragment length on the binding kinetics of the particles.
Molecular arrangement and signals as a function of fragment length
We have conducted experiments to determine the scattering signals of particles in the limit of low surface densities as a function of the fragment length. Fig. 3a shows data of experiments in which particles were detected with single-particle resolution. The particles were illuminated by an evanescent optical field and the optical scattering was detected with a microscope objective (see the left inset). The figure shows histograms of the scattered light intensity for the three fragment lengths. The histograms clearly broaden for increasing fragment length, which means that particles tethered by longer fragments assume a wider distribution of distances from the surface. We attribute these results to the mobility of the particles on the surface (see the right inset). Interestingly, the maximum scattering intensity increases for longer fragments. This means that particles bound by long fragments are able to get closer to the surface than particles bound by short fragments, because the flexibility increases with longer fragment length.15The influence of the fragment length on the time-averaged scattering signal per particle was quantified in the two detection systems, namely f-TIR detection and microscopic single-particle detection, in the limit of low fragment concentrations and low particle surface densities, see Fig. 3b. The grey curve shows the signal per particle surface density measured by bright-field scattering in an f-TIR experiment with magnetic particles (see left inset and left axis). The dark curve shows the average scattering intensity measured by dark-field scattering with polystyrene particles (see right inset and right axis). These experiments were performed with the same cartridges, the same optical evanescent fields, and the same molecular fragments; but the particles and the detection methods were different. The data in Fig. 3b show that the dependencies on the fragment length of the average optical scattering of the polystyrene particles (right axis) and the magnetic particles (left axis) are consistent. Both experiments show that the signals do not depend very strongly on the fragment length, but the measurement uncertainties are quite large. There may be a weakly negative dependency of scattering signal on the fragment length, at most about 30% decrease of signal for an increase of fragment length from 100 bp to 600 bp. We attribute a slight decrease of signal with increasing fragment length to an increase of the time-averaged distance between the particle and the surface, which gives lower signals due to the decaying evanescent field. In contrast, the fragment-length dependence of the assay signals at low target concentrations is very different (see Fig. 2a, bottom curves): the fragment-length dependence is strong and is of opposite sign (longer fragments give higher signals). This proves that the fragment-length dependence of the rise of the curves in Fig. 2 (as a function of time in Fig. 2a, and as a function of concentration in Fig. 2b) is dominantly caused by an increase of the binding rate of the particles. Quantitatively, the increase of binding rate is about one order of magnitude for a fragment of 590 bp compared to a fragment of 105 bp (Fig. 2b). At high fragment concentrations the scattering signal per particle could not be determined, because at high densities the particle images overlap and therefore particles cannot be individually recognized and counted.
Conclusions
We have studied the influence of dsDNA fragment length on the binding rate, dose–response curve, and optical signals in a biosensor based on magnetic particle labels that are detected in an optical evanescent field. At low bound particle densities, we have been able to quantify the fragment-length dependence of the optical signal per particle (see Fig. 3). The fragment-length dependence of the optical signal is very small, which we attribute to the pivoting motional freedom of the fragment attachment points, as has been observed before.15 Knowing that the optical signal per particle depends very weakly on the fragment length, we have been able to extract the fragment dependence of the binding rate from the time-dependent curves. We have studied the assay signals as a function of time for a range of DNA concentrations, entering different regimes in terms of the number of DNA fragments per particle and density of particles bound to the sensor surface. At high bound particle densities reduced signals are observed, which we attribute to steric particle–particle interactions and alignment of the fragments, as sketched in Fig. 2. The fragment-length dependence of the optical signal is very small and of opposite sign compared to the fragment-length dependence of the kinetic signals. This gives hard proof that longer DNA fragments cause a higher binding rate of the particles to the biosensor surface; the binding rate increases by an order of magnitude for the longest dsDNA fragments (290 bp, 590 bp) compared to the smallest fragment (105 bp) studied in this paper.It is interesting to compare our work with the literature on linker molecules. In our experiments, we can view the dsDNA fragments as molecules that form a link between the magnetic particle and the Texas Red affinity tag. Viewed along this perspective, our experiments reveal the influence of the linker length on the kinetics of the binding between the Texas Red coupled to the particle and the anti-Texas Red coupled to the sensor surface. The positive influence of linkers on molecular binding is well known and many linker molecules have been studied before, e.g. dendrimers,17 thymidine units,18 different charged linkers,19 poly(ethylene glycol) spacers,9,20 peptide linkers,21 and nucleic acid linkers.22–24 In this paper we have studied the influence of long and rigid double-stranded DNA linkers in a particle-based biosensing assay. dsDNA has a mechanical persistence length of about 50 nm, corresponding to 150 bp.25 This means that dsDNA behaves like a rod over lengths shorter than 150 bp and shows bends over longer distances. The longest fragments that we have studied are 590 bp, which means that these linkers will show thermal bending fluctuations. Averaged over time, the end of the surface-bound dsDNA fragment (i.e. the Texas Red tag) will reach out from the particle surface by a distance of several times the persistence length. In addition, we know from earlier experiments15 that the dsDNA linkers have a pivoting motional freedom. Therefore, we expect that the dsDNA-linked Texas Red tags are able to statistically cover a hemisphere with a radius that relates to the length of the DNA linker, thereby increasing the particle-to-surface binding kinetics.
The observed ten-fold increase of binding kinetics is very relevant for point-of-care diagnostic applications. In a molecular diagnostic assay, the specificity of the assay is determined by the selected target sequences. Here we have shown that the fragment length has a strong influence on the binding kinetics and the limit of detection in the optomagnetic assay. This gives the opportunity to optimize the species-specific fragment length in order to achieve the best overall assay performance. In future research we will exploit this degree of freedom in a variety of biosensor assay designs.
Acknowledgements
This project was partly supported by the Casimir program of The Netherlands Ministry of Economic Affairs, the Ministry of Education, Culture and Science and by the Delta Plan for Science and Technology.Notes and references
- P. B. Monaghan, K. M. McCarney, A. Ricketts, R. E. Littleford, F. Docherty, W. E. Smith, D. Graham and J. M. Cooper, Anal. Chem., 2007, 79, 2844 CrossRef CAS PubMed.
- F. Qiu, D. Jiang, Y. Ding, J. Zhu and L. L. Huang, Angew. Chem., 2008, 120, 5087 CrossRef.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763 CrossRef CAS PubMed.
- M. Y. Sha, H. Xu, S. G. Penn and R. Cromer, Nanomedicine, 2007, 2, 725 CrossRef CAS PubMed.
- Y. Zhang and D. Zhou, Expert Rev. Mol. Diagn., 2012, 12, 565 CrossRef CAS PubMed.
- D. M. Bruls, T. H. Evers, J. A. H. Kahlman, P. J. W. van Lankvelt, M. Ovsyanko, E. G. M. Pelssers, J. J. H. B. Schleipen, F. K. ten Theije, C. A. Verschuren, T. van der Wijk, J. B. A. van Zon, W. U. Dittmer, A. H. J. Immink, J. H. Nieuwenhuis and M. W. J. Prins, Lab Chip, 2009, 9, 3504 RSC.
- M. Koets, T. van der Wijk, J. T. W. M. van Eemeren, A. van Amerongen and M. W. J. Prins, Biosens. Bioelectron., 2009, 24, 1893 CrossRef CAS PubMed.
- W. U. Dittmer, T. H. Evers, W. M. Hardeman, W. Huijnen, R. Kamps, P. de Kievit, J. H. M. Neijzen, M. J. J. Sijbers, J. H. Nieuwenhuis, D. W. C. Dekkers, M. H. Hefti and M. F. W. C. Martens, Clin. Chim. Acta, 2010, 411, 868 CrossRef CAS PubMed.
- A. S. W. Ham, A. L. Klibanov and M. B. Lawrence, Langmuir, 2009, 25, 10038 CrossRef CAS PubMed.
- J. B. Haun, L. R. Pepper, E. T. Boder and D. A. Hammer, Langmuir, 2011, 27, 13701 CrossRef CAS PubMed.
- P. F. Mens, H. M. de Bes, P. Sondo, N. Laochan, L. Keereecharoen, A. van Amerongen, J. Flint, J. R. S. Sak, S. Proux, H. Tinto and H. D. F. H. Schallig, J. Clin. Microbiol., 2012, 50, 3520 CrossRef CAS PubMed.
- S. Liébana, A. Lermo, S. Campoy, J. Barbé, S. Alegret and M. I. Pividori, Anal. Chem., 2009, 81, 5812 CrossRef PubMed.
- D. Chen, M. Mauk, X. Qiu, C. Liu, J. Kim, S. Ramprasad, S. Ongagna, W. R. Abrams, D. Malamud, P. L. A. M. Corstjens and H. H. Bau, Biomed. Microdevices, 2010, 12, 705 CrossRef CAS PubMed.
- A. H. A. M. van Hoek, I. M. J. Scholtens, A. Cloeckaert and H. J. M. Aarts, J. Microbiol. Methods, 2005, 62, 13 CrossRef CAS PubMed.
- K. van Ommering, P. A. Somers, M. Koets, J. J. H. B. Schleipen, L. J. van IJzendoorn and M. W. J. Prins, J. Phys. D: Appl. Phys., 2010, 43, 155501 CrossRef; K. van Ommering, M. Koets, R. Paesen, L. J. van IJzendoorn and M. W. J. Prins, J. Phys. D: Appl. Phys., 2010, 43, 385501 CrossRef.
- D. C. Prieve, Adv. Colloid Interface Sci., 1999, 82, 93 CrossRef CAS.
- V. Gubala, C. Crean, R. Nooney, S. Hearty, B. McDonnel, K. Heydon, R. O'Kennedy, B. D. MacCraith and D. E. Williams, Analyst, 2011, 136, 2533 RSC.
- S. Balamurugan, A. Obubuafo, R. L. McCarley, S. A. Soper and D. A. Spivak, Anal. Chem., 2008, 80, 9630 CrossRef CAS PubMed.
- M. Imanishi and Y. Sugiura, Biochemistry, 2002, 41, 1328 CrossRef CAS PubMed.
- L. Cohen-Tannoudji, E. Bertrand, J. Baudry, C. Robic, C. Goubault, M. Pellissier, A. Johner, F. Thalmann, N. K. Lee, C. M. Marques and J. Bibette, Phys. Rev. Lett., 2008, 100, 108301 CrossRef CAS.
- A. Gaebler, T. Schaefer, K. Fisher, D. Scharnweber and B. Mauth, Acta Biomater., 2013, 9, 4899 CrossRef CAS PubMed.
- A. E. Prigodich, O.-S. Lee, W. L. Daniel, D. W. Seferos, G. C. Schatz and C. A. Mirkin, J. Am. Chem. Soc., 2010, 132, 10638 CrossRef CAS PubMed.
- M. E. Leunissen, R. Dreyfus, R. Sha, N. C. Seeman and P. M. Chaikin, J. Am. Chem. Soc., 2010, 132, 1903 CrossRef CAS PubMed.
- Y.-H. M. Chan, B. van Lengerich and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 979 CrossRef CAS PubMed.
- A. J. Mastroianni, D. A. Sivak, P. L. Geissler and A. P. Alivisatos, Biophys. J., 2009, 97, 1408 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |