 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An antibody-free microfluidic paper-based analytical device for the determination of tear fluid lactoferrin by fluorescence sensitization of Tb3+†
Kentaro
Yamada
,
Shunsuke
Takaki
,
Nobutoshi
Komuro
,
Koji
Suzuki
and
Daniel
Citterio
*
Department of Applied Chemistry, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: citterio@applc.keio.ac.jp; Fax: +81 45 566 1568; Tel: +81 45 566 1568
First published on 3rd January 2014
Abstract
An inkjet-printed microfluidic paper-based analytical device (μPAD) for the detection of lactoferrin has been developed. The analyte concentration dependent fluorescence emission, caused by the sensitization of pre-deposited terbium (Tb3+) upon complexation with lactoferrin on the paper device, is captured using a digital camera. The dynamic response range (0.5–3 mg mL−1) and the limit of detection (0.30 mg mL−1) of the μPAD are suitable for the analysis of normal human tears and the detection of eye disorders. Finally, lactoferrin concentrations in human tear samples were analyzed by the μPADs and the assay results corresponded within 6% error to those obtained by an immunoassay (ELISA). The μPADs provide a simple, rapid and accurate method for lactoferrin detection in tear fluid. Results are obtained within 15 min of a single application of 2.5 μL of sample. To the best of our knowledge, this is the first report of a device for lactoferrin quantification relying neither on an immunoassay nor on high cost analytical instrumentation.
Introduction
Since their introduction by the Whitesides group in 2007, microfluidic paper-based analytical devices (μPADs) have gained significant attention as an analytical platform.1 Owing to the intrinsic properties of paper, μPADs feature several advantageous characteristics relevant to simple and low-cost analytical devices: (1) they are fabricated from low-cost materials. (2) They are lightweight, making them easy to transport and to distribute. (3) They are safely disposable by incineration. Compared to plastic microplates contaminated with biological substances in laboratory tests, incineration of used μPADs enables sanitary disposal, which eliminates hazardous biological substances. (4) Assays performed on μPADs require only low sample volumes, which is important for samples with limited availability such as tears, saliva, urine from newborn infants, and blood from finger pricks.2 (5) μPADs do generally not depend on external power sources. The need for pumps is eliminated, since capillary forces in the microporous cellulose fiber network of paper drive sample transport. In sum, μPADs are easy-to-handle and user-friendly analytical tools suitable for volume-limited samples, as has already been shown in various applications (e.g. blood test, food safety, and metal analysis).3–7In 2008, our group had demonstrated μPADs for (bio)chemical sensing fabricated by inkjet printing technology for the first time.8 Among the various reported printing technologies for the microfluidic patterning of paper substrates (plotting,9 wax printing,10,11 flexographic printing,12 wax screen printing13), inkjet printing is so far the only industrially applied technology that allows performing all processing steps required for the fabrication of complete μPADs.14,15
Human tear fluid is a mixture of various components such as water, proteins, enzymes, electrolytes and lipids. The proteomics of tear fluid has recently become an active area of research.16,17 Proteins in tears play a key role in the preservation of the ocular surface and in the adjustment of tear components. Therefore, disorders in tear protein secretion can be a cause of several diseases. It has been reported that analysis of the tear protein concentration enables their diagnosis.18 In particular, the down-regulation of lactoferrin, a glycoprotein existing in human tear fluid at relatively high concentrations, is strongly linked to disorders of the corneal epithelium,19 and the determination of its concentration is expected to facilitate the diagnosis of systemic autoimmune diseases. So far, most methods for lactoferrin quantification reported in the literature are based on immunoassays. Examples include a conventional enzyme linked immunosorbent assay (ELISA),20 a radial immunodiffusion assay (Lactoplate),21 and a colorimetric solid phase immunoassay (Lactocard).22 Although they are highly selective and sensitive, these assays have the drawbacks of being time-consuming and requiring multiple operational steps. To overcome these disadvantages, Karns and Herr have recently developed a homogeneous electrophoretic immunoassay on a microfluidic glass chip, which enables the quantification of lactoferrin in <1 μL of human tear fluid within 5 s.17 However, costs associated with the use of monoclonal anti-lactoferrin antibodies for lactoferrin capture remain an issue. In addition, the requirement for sophisticated high-tech instruments for signal detection (e.g. fluorescence microscope and cooled CCD camera) makes it difficult to realize a more simple and low-cost assay. Although iTRAQ (isobaric tag for relative and absolute quantitation) technology combined with 2D-nanoLC-nanoESI-MS/MS23 and SELDI-TOF-MS24 have been reported as analytical methods for lactoferrin quantification without employing antibodies, they require relatively large sample volumes and rely on high cost instrumentation not commonly found in small or medium sized clinical laboratories.
This work demonstrates a simple, low-cost, and rapid lactoferrin determination in human tear fluid using inkjet-printed μPADs based on fluorescence detection. Inkjet printing is used for both the patterning of microchannels and the deposition of the reagents required for sensing. The assay relies on the fluorescence emission from complexes formed between human lactoferrin in the sample solution and Tb3+ cations printed on the sensing area of μPADs. The concentration of lactoferrin in the sample is quantified by observing the color intensity of the fluorescence emitted from the complexes formed on the μPAD. This method allows for rapid analysis (15 min) of lactoferrin, based on a simple fluorometric assay without using costly antibodies. The achieved detection limit is sufficiently low for the detection of deviating lactoferrin levels in human tear fluid. To the best of our knowledge, this is the first report of a lactoferrin determination method relying neither on an immunoassay nor on high cost analytical instrumentation.
Experimental section
Reagents and instruments
All reagents were used as received. Terbium chloride hexahydrate (TbCl3·6H2O) and human lactoferrin were purchased from Sigma-Aldrich (St. Louis, MO). 1,10-Decanediol diacrylate was purchased from TCI (Tokyo, Japan). N-(2-Hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) was purchased from Dojindo Laboratories (Kumamoto, Japan). Sodium hydroxide and poly(vinyl alcohol) were purchased from Kanto Chemical (Tokyo, Japan). The human lactoferrin ELISA kit was purchased from EMD Chemicals, Inc. (San Diego, USA). All other reagents were purchased from Wako Pure Chemical Industries Ltd (Osaka, Japan). All solutions were prepared using 18 MΩ Milli-Q water. Circular filter paper sheets of 185 mm diameter (Advantec No. 5C) were obtained from Toyo Roshi Co., Ltd (Tokyo, Japan).Fluorescence spectra in solution were recorded on a SPEX Fluorolog-NIR spectrophotometer (HORIBA, Kyoto, Japan). Patterning of the microfluidic structures was performed on an unmodified piezoelectric EPSON PX-105 inkjet printer (Epson, Suwa, Japan), whereas the reagents for lactoferrin detection were deposited with a piezoelectric Dimatix DMP 2831 (Dimatix-Fujifilm Inc., Santa Clara, USA) material printer with 10 pL nominal droplet volume cartridges (DMC-11610). Photopolymerization of printed structures was performed under irradiation from a Hg–Xe lamp (Lightingcure LC-6, Hamamatsu Photonics, Hamamatsu, Japan) at a power of 4 mW cm−2 (measured at 365 nm). For fluorescence emission signal detection from μPADs, UV hand lamps (Funakoshi, Tokyo, Japan) were used as excitation light sources inside a Mini UV viewing cabinet (UVP, Upland, CA, USA), and the emission was captured with a DMC-FZ50 digital camera (Panasonic, Osaka, Japan) through a 520 nm longpass filter (Sigma Koki Co., Ltd, Tokyo, Japan).
Fluorescence emission measurements in solution
Fluorescence emission spectra were collected from HEPES buffered solutions (pH 7.4, 50 mM) containing 0 to 1 mg mL−1 lactoferrin in the presence of 100 μM TbCl3 and 3.75 mM NaHCO3. The excitation wavelength was set to 290 nm and the emission spectra were recorded between 480 nm and 640 nm through a 440 nm longpass filter (Sigma Koki Co., Ltd, Tokyo, Japan).Device fabrication
The microfluidic patterns on μPADs were fabricated with the inkjet printer and a UV-curable ink by a method similar to the one previously reported by our group.15 Briefly, filter paper taped onto a sheet of A4 copy paper with a circular cut-out area in the center was fed into the EPSON inkjet printer. The attachment of the filter paper to a sheet of copy paper is necessary, because the paper feeder of the inkjet printer used in this work is unable to handle round shapes. With the circular cut-out in the copy paper, both surfaces of the attached filter paper are accessible for printing. The ink cartridges were loaded with the UV-curable ink based on octadecyl acrylate and 1,10-decanediol diacrylate. On the topside of the paper, microfluidic patterns designed with PowerPoint (Microsoft) were printed. After the paper was ejected from the printer, it was placed on a cooling plate at 10 °C, while being exposed to the UV light source for 15 min. On the backside of the paper, the UV-curable ink was deposited covering the entire area patterned on the topside, followed by cooling and UV light exposure for 10 min. With this method, 72 microfluidic patterns were printed onto a 9 × 9 cm2 area of the filter paper in a single batch. The design of a single pattern consists of two square areas for sampling and sensing connected by a straight channel, as shown in Fig. 1.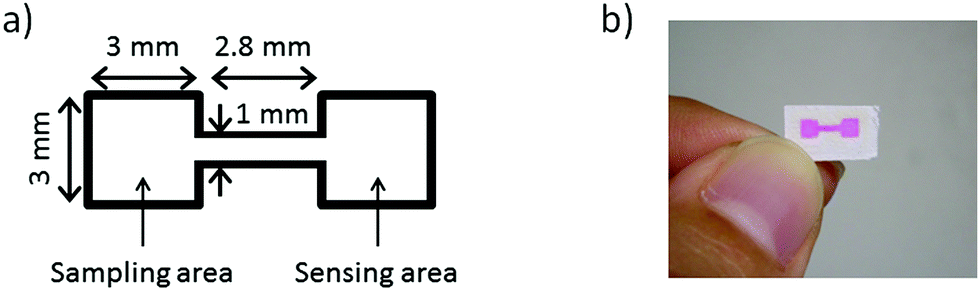 | ||
| Fig. 1 (a) Outline and dimensions of a single microfluidic pattern. The black line indicates the hydrophobic barrier composed of photopolymerized UV curable ink patterned by inkjet printing. (b) Photograph of a pattern (a red food colorant has been applied to visualize the patterned structure). | ||
Before cutting into single devices, all reagents required for lactoferrin detection were deposited onto the patterned paper substrates. First, 8 printing layers of 1 mM TbCl3 solution with 15 vol% ethylene glycol were deposited onto the sensing areas. To prevent the adsorption of lactoferrin to the paper surface, the entire paper was then soaked in 50 mL of 0.5 wt% poly(vinyl alcohol) for 5 min, followed by drying for 20 min at 37 °C. The soaking solution was replaced after every use. Next, 12 printing layers of 25 mM NaHCO3 solution were deposited onto the sampling areas. In all cases HEPES buffered solutions (pH 7.4, 50 mM) were used. In the last step, the completely processed substrate was cut into single μPADs. A schematic illustration of the reagent deposition procedure for fabricating the final μPADs is shown in Fig. 2.
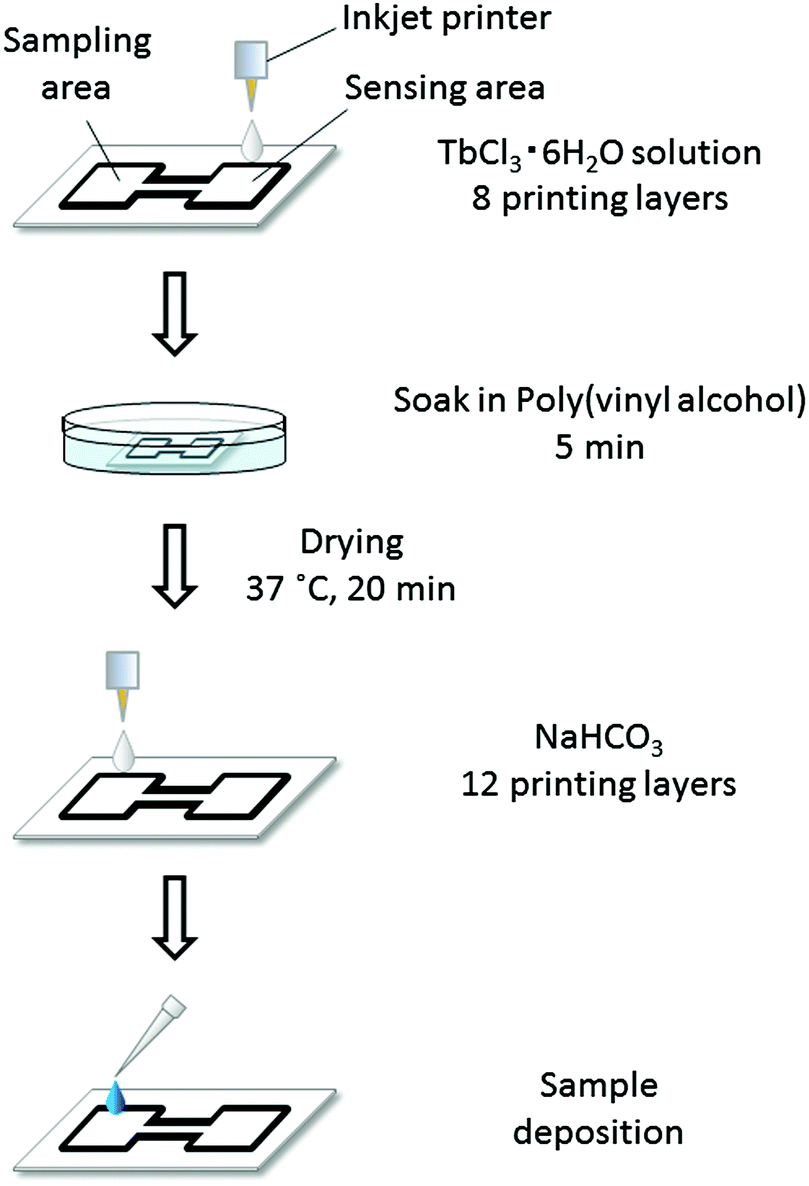 | ||
| Fig. 2 Schematic illustration of the reagent deposition process during fabrication of final μPADs: TbCl3 and NaHCO3 solutions were printed by using an inkjet printer, while surface treatment of the paper substrate was performed by soaking in poly(vinyl alcohol) solution. All reagents were dissolved in HEPES buffered solution (pH 7.4, 50 mM). | ||
Device calibration and quantitative data processing
For calibration and quantification of human lactoferrin levels in real samples, 2.5 μL of calibration solution (human lactoferrin in 50 mM HEPES pH 7.4 buffer) or tear sample (see below) was pipetted onto the sampling area of a μPAD. After complete drying at room temperature (10–12 min after sample application), the μPAD was placed between two UV hand lamps (λex = 254 nm) in a darkened UV viewing cabinet, and the emitted green fluorescence was imaged using the digital camera through the longpass filter to eliminate the influence of excitation light reflected from the paper substrate. The captured images were stored in JPEG format at 240 dpi and the green (G) intensity (RGB scale) in the sensing area was measured using the image processing software ImageJ (National Institutes of Health). All signals are reported as ΔG values (Gsample − Gblank). The setup for fluorescence signal capture is shown in Fig. S1 of the ESI.†Human tear fluid analysis
Human tear samples were collected from five volunteers with disposable polyethylene pipettes (AS ONE, Osaka, Japan) and stored in autoclaved Protein LoBind tubes (Eppendorf) at 4 °C until use and no longer than 3 days. For μPAD analysis of lactoferrin concentrations, undiluted human tear fluid was deposited into the sampling area. For ELISA analysis, human tear samples were 105-fold diluted in autoclaved Protein LoBind tubes with the sample diluting buffer provided in the kit. This step was necessary to adjust the lactoferrin concentrations to the dynamic response range of the kit (5–50 ng mL−1).Results and discussion
Assay principle
The glycoprotein lactoferrin is known for its capacity to reversibly bind two iron ions in their trivalent Fe3+ state. In this process, a bicarbonate ion acts as a synergistic anion by neutralizing a positive charge in the binding site of the protein.25 It has also been reported that various other metal ions can be substituted for iron, including lanthanides.26 On the other hand, the fluorescence emission of terbium ions is efficiently sensitized upon binding to certain peptides or proteins.27,28 More recently, it has been shown that Tb3+ ions bound to the metal ion binding site of lactoferrin emit pH-dependent fluorescence (λmax = 548 nm).29 The fluorescence intensity shows a sharp increase between pH 6 and 7 and flattens out at around pH 7.2. Therefore, observing the intensity of the green fluorescence from lactoferrin–terbium complexes at a constant pH value was expected to be applicable to the quantification of lactoferrin. While the pH-dependence of the fluorescence emission from lactoferrin–Tb3+ complexes has been reported, the dependence on the lactoferrin concentration has not been investigated so far. In a proof-of-concept experiment, the fluorescence emission spectra of aqueous TbCl3 solutions (100 μM) in the presence of increasing concentrations of human lactoferrin (0–1 mg mL−1) were recorded at a fixed pH of 7.4 (50 mM HEPES buffer) and a background of 3.75 mM NaHCO3. In analogy to the binding of Fe3+ to lactoferrin,25 it was assumed that the presence of the bicarbonate anion would also strengthen the binding of Tb3+ to the protein. In that way, NaHCO3 acts as an indirect fluorescence signal enhancer. The spectra shown in Fig. 3 clearly demonstrate that the intensity of the main Tb3+ emission peak at 548 nm is strongly enhanced by the presence of lactoferrin. While a protein-free solution of Tb3+ is non-fluorescent, fluorescence turns on upon protein binding due to an energy transfer process. This results in the characteristic emission peaks of Tb3+, which are observed as a green colored emission by the naked eye. The lactoferrin concentration dependent emission at 548 nm is shown in Fig. S2 (ESI†).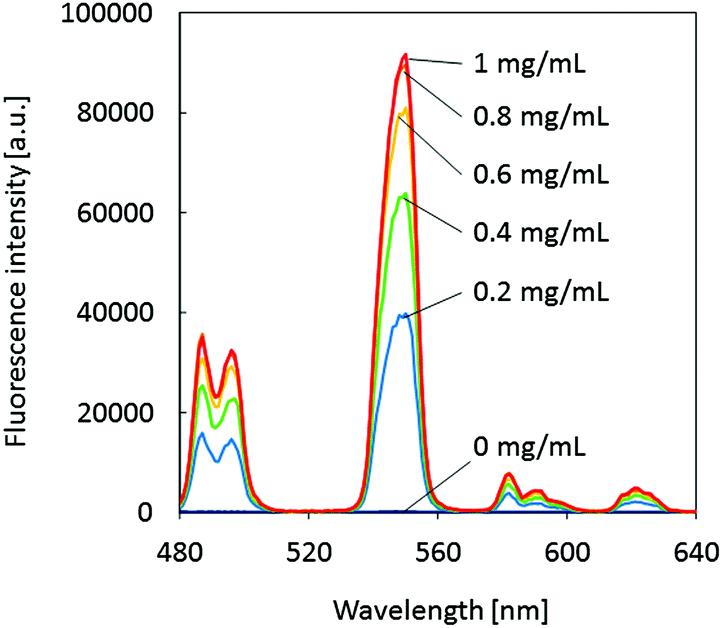 | ||
| Fig. 3 Fluorescence emission spectra of 100 μM TbCl3 solutions (50 mM HEPES, 3.75 mM NaHCO3, pH 7.4) in the absence and presence of human lactoferrin at various concentrations; λex = 290 nm. | ||
This newly developed fluorescence-based assay allows the quantification of lactoferrin without depending on antibodies or on labor-intensive and time-consuming analytical methods. However, the assay performed in solution, for example in a microtiter plate, requires sample volumes that are not readily available in the case of tear fluid. In addition, a costly fluorescence microplate reader is not a standard instrument for an ophthalmologist's clinic. The elaboration of an alternative format of the Tb3+-based assay was regarded as an essential step towards simpler and more convenient lactoferrin determination. Therefore, a μPAD for the quantification of lactoferrin concentrations, based on the green fluorescence emission (λmax = 548 nm) from lactoferrin–terbium complexes formed on the paper device, was developed.
μPAD design
The simple μPAD used throughout this work consists of two identical square areas connected by a single straight channel (Fig. 1a). One of the square areas serves as the sample deposition area (sampling area), while the second one acts as the fluorescence response area (sensing area). Inkjet patterning of the paper substrate with hydrophobic barriers using a UV curable ink composition was performed by a slightly modified version of our previously reported method.15 In order to enable the fabrication of a larger number of μPADs in a single photocuring cycle, the UV irradiated area has been enlarged by increasing the distance between the light source and the paper substrate. To compensate for the weaker irradiation power per unit area caused by the larger distance from the light source, the UV irradiation time was extended from the previously reported 1 min to 15 min. This irradiation time was experimentally confirmed to be sufficient for the formation of hydrophobic barriers. To prevent the spreading of the liquid ink, which would lead to loss of structural resolution during the prolonged photocuring process, the filter paper was placed on a cooling plate at 10 °C, while being exposed to the UV light source. By this modified method, 72 μPADs were produced simultaneously in every batch. A single inkjet printed microfluidic pattern visualized by a red food colorant is shown in Fig. 1b.To implement the fluorescence-based assay on the μPAD, three essential components were pre-deposited on the patterned paper substrate: (1) a Tb3+-salt, (2) a bicarbonate salt, and (3) a pH-buffer system. The storage of all required reagents in dry form on the paper device enabled lactoferrin analysis by simply applying the sample without any pretreatment. A schematic illustration of the reagent deposition process is shown in Fig. 2. Since all reagents for printing and surface treatment were dissolved in HEPES buffered solution (pH 7.4, 50 mM), the components remaining on the μPAD in dry form guarantee a constant pH value for the entire device, eliminating the requirement to adjust the pH of biological samples.
In the first printing step, 8 layers of TbCl3 solution were deposited into the sensing area. To reduce adsorption of lactoferrin to the paper surface during migration from the sampling area to the sensing area, μPADs were soaked in a solution of poly(vinyl alcohol) (PVA) after terbium deposition. Among five tested reagents (bovine serum albumin, poly(vinyl pyrrolidone), PVA, casein, and glycerol), PVA treated surfaces showed the best mobility of lactoferrin and little reduction of sample flow speed. The optimal PVA concentration in the solution was experimentally determined to be 0.5 wt%. Further details regarding the optimization of the surface treatment reagent are given in Fig. S3 (ESI†). It was found to be essential to deposit the Tb3+ reagent before the surface treatment step. In the case of reversing the order, only weak signals were observed (data not shown). It is assumed that Tb3+ is immobilized to the paper by strong electrostatic interactions with negative surface charges of untreated cellulose fibers.30 By modifying the paper surface with PVA before Tb3+ printing, the retention of the reagent is assumed to decrease.
In the final printing step, 12 layers of NaHCO3 solution were deposited into the sampling area. The concentrations of TbCl3 and NaHCO3 in the printing inks are kept relatively low to guarantee a stable jetting of liquids. Therefore, multiple printing cycles are required for reagent deposition. The optimal number of printing repetitions for TbCl3 and NaHCO3 was empirically investigated and the above-mentioned numbers were found to be the most suitable (tested number of printing layers: 4–12 layers for terbium, 11–15 layers for NaHCO3, data not shown).
Due to the insolubility of terbium carbonate in water, TbCl3 and NaHCO3 cannot be inkjet deposited in the form of a mixed solution. Furthermore, considering the synergistic role of the bicarbonate anion during metal cation binding to lactoferrin, it was assumed to be advantageous to have the bicarbonate anion and the terbium cation deposited into different areas of the μPAD. By doing so, the positive charge in the metal binding site of lactoferrin would be neutralized by bicarbonate before the protein interacts with Tb3+ cations. This resulted in the present design of a μPAD with separate sampling and sensing areas.
Fluorescence-based lactoferrin assay on μPADs
To confirm the fluorescence response of the developed μPADs, lactoferrin samples of various concentrations have been applied onto the sampling area of the devices. Because the normal tear fluid lactoferrin concentrations of humans are between 0.63 and 2.9 mg mL−1,31 calibration solutions were prepared in the 0.1–4 mg mL−1 range. After application by a micropipette, the sample solution reached the sensing area within less than 1 min by capillary force driven flow through the connecting channel. μPADs were allowed to completely dry for 10–12 min at room temperature, before images of the fluorescence signal emitted from the sensing area were captured under UV illumination. The calibration curve (Fig. 4a) shows a good correlation between the lactoferrin concentration and the green color intensity (on the RGB scale) in the sensing area recorded by the digital camera. The lactoferrin concentration dependent increase of the fluorescence intensity was also readily observable by the naked eye (Fig. 4b). It should be noted that the μPADs are single-use devices. For this reason, every data point in the calibration curve (Fig. 4a) has been measured with a separate μPAD. The assay (from the application of the sample to the fluorescence signal capture) takes no longer than 15 min, which is significantly shorter than the ELISA method requiring several hours. The limit of detection (LOD), calculated to be 0.30 mg mL−1 based on a sigmoidal curve fit and three times the standard deviation (3σ) of the intensity of a blank sample, is below the lower limit of lactoferrin concentrations found in tear fluid of healthy humans (0.63 mg mL−1). Additionally, the dynamic response range fully covers the normal lactoferrin concentration range of human tear fluid. A further strength of the μPAD is the possibility of performing quantitative lactoferrin analysis at very low cost. A simple material cost estimation is given in Table S1 (ESI†).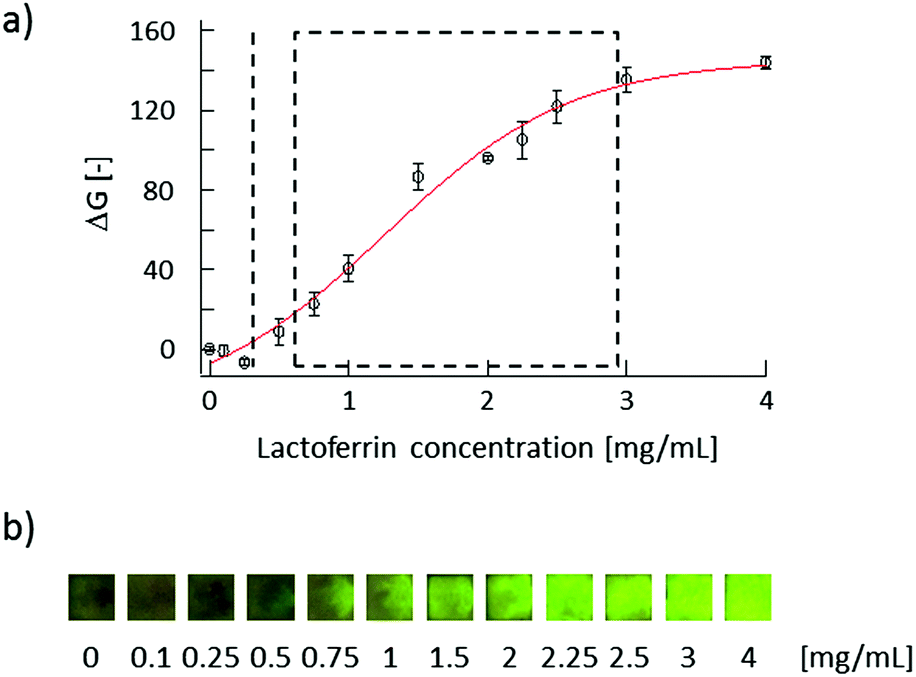 | ||
| Fig. 4 (a) μPAD calibration curve for human lactoferrin: the broken line indicates the limit of detection (LOD) of the μPAD, whereas the rectangular area represents the normal range of tear fluid lactoferrin (0.63–2.9 mg mL−1). The markers and error bars reflect the average and standard deviations of three measurements. (b) Images of sensing areas after application of lactoferrin samples captured under UV light (λ = 254 nm). | ||
Arrangement of pre-deposited reagents on μPADs
In a further series of experiments, the influence of the presence of NaHCO3 as a signal-enhancing additive and the advantage of separate sampling and sensing areas connected by a flow channel compared to a simple spot test have been evaluated. By depositing NaHCO3 solution onto the sampling area of the μPADs, lactoferrin interacts with the Tb3+ cations in the sensing area after being in contact with bicarbonate. It was experimentally confirmed that the presence of HCO3− in a distinct area from the sensing area improved the performance of the μPADs. For comparison purposes, simple spot tests with NaHCO3 and TbCl3 pre-deposited by inkjet-printing of separate solutions into a 3 × 3 mm2 area surrounded by inkjet printed hydrophobic barriers were fabricated (Fig. S4a†). Similarly to the μPAD arrangement, these spot tests showed increasing Tb3+ fluorescence emission depending on the lactoferrin concentrations. However, as shown in Fig. S4b (ESI†), the observed sensitivity, expressed as the total green intensity signal change between a blank sample and a sample containing 4 mg mL−1 of lactoferrin, was lower (ΔGmax = 135) than in the case of μPADs with separate sampling and sensing areas connected by a flow channel (ΔGmax = 144) (Fig. 4a). For a bicarbonate-free spot test arrangement with only the Tb3+ sensing reagent (Fig. S4c†), the sensitivity in terms of total signal change was further reduced (ΔGmax = 119). Fig. S5 (ESI†) compares a set of calibration curves for lactoferrin obtained with μPADs with separate sampling and sensing areas in the presence (red line) and absence (blue line) of NaHCO3 printed onto the sampling area. As in the case of the spot test, the measured ΔG signals are larger in the presence of the additive. Although the observed differences are not very large, the same trend was noted throughout all experiments performed with paper devices in this study. In all cases, the use of μPADs with separate sampling and sensing areas connected by a microfluidic channel, where NaHCO3 had been pre-deposited onto the sampling area, showed the highest sensitivities.Shelf life of μPADs
The shelf life of the developed μPADs was investigated. For this purpose, devices were stored at room temperature (25 °C) wrapped in aluminum foil to protect against ambient light for up to 100 days after fabrication. Alternatively, they were kept in a dark climate control chamber at 35 °C and 50% relative humidity for 10 days. Calibration curves obtained by applying lactoferrin samples to the μPADs stored under various conditions are shown in Fig. S6 (ESI†). Upon storing at room temperature, a reduction in sensitivity is observed after a period of 30 days (Fig. S6a†). In the case of storage at increased temperature (35 °C), the onset of decreasing sensitivity is observed after a 10-day period (Fig. S6b†). However, according to the calculated limits of detection (LOD) and limits of quantification (LOQ) shown in Table S2,† the μPADs remain functional for at least 45 days when stored at room temperature, as long as proper calibration is performed at the time of use. After a storage period of 100 days, a significant change in the calibration curve accompanied by a general deterioration of LOD and LOQ was observed. As for the reasons of degradation, a reduction in sample flow speed after extended storage has been experimentally observed. This assumedly results in a lower amount of lactoferrin reaching the sensing area, leading to a sensitivity decrease. The causes for the reduction in sample flow speed are so far not known. However, the fact that μPADs can be stored at room temperature is a clear advantage over the currently commercially available ELISA kits for lactoferrin detection, which require constant refrigeration to preserve the functionality of the used antibodies.Selectivity evaluation
Human tear fluid consists of various substances including proteins and electrolytes. Primary constituents and their concentrations are shown in Table 1. Before applying the μPADs to the analysis of human tear fluid samples, possible interference of these components was investigated. Fig. 5 summarizes the results of the interference study. None of the primary tear constituents except lactoferrin resulted in a significant fluorescence signal (indicated as ΔG) when applied as single components to the μPADs (Fig. 5, blue bars) at concentrations indicated in Table 1. This demonstrates the high selectivity of the sensitizing interaction between Tb3+ and lactoferrin. In addition, the major tear fluid constituents did not interfere with the terbium sensitization by lactoferrin. This was confirmed by the identical fluorescence signals observed in mixed solutions of Tb3+ and other tear fluid constituents (Fig. 5, orange bars). Thus, it has been clearly demonstrated that the μPADs respond to none of the major tear fluid constituents except lactoferrin, and that the presence of other constituents does not block the binding of lactoferrin to Tb3+.| Constituent | Concentrationa |
|---|---|
| a The bold print values (the upper limit of the range) were used for selectivity evaluation. b A mean concentration of 1.84 mg mL−1 lactoferrin was used for selectivity evaluation. | |
| Lactoferrin | 0.63–2.9b mg mL−1 (ref. 31) |
| Na+ | 80–170 mM (ref. 32) |
| K+ | 6–42 mM (ref. 32) |
| Ca2+ | 0.3–2.0 mM (ref. 32) |
| Mg2+ | 0.3–1.1 mM (ref. 32) |
| Lysozyme | 2.5–3.4 mg mL−1 (ref. 33) |
| Albumin | 6.0–15.2 μg mL−1 (normal)34 |
| 67–150 μg mL−1 (ocular diseases)34 | |
| Glucose | 1.0–6.2 mg per 100 mL (normal)35 |
| 7.2–26 mg per 100 mL (diabetes)35 | |
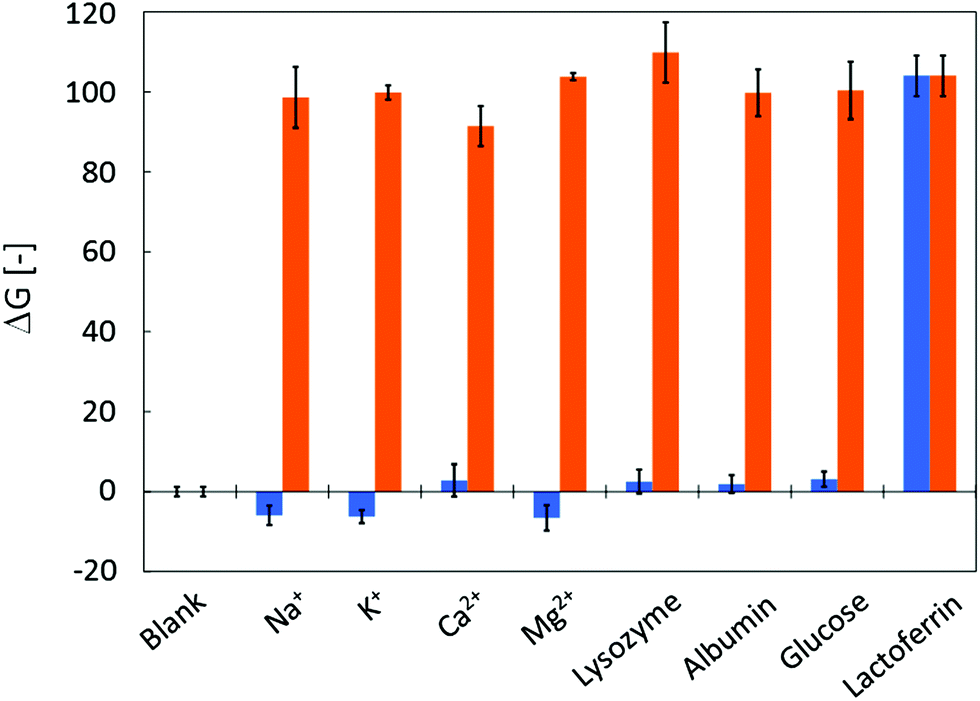 | ||
| Fig. 5 Selectivity of μPADs for lactoferrin: the graph shows the fluorescence response of μPADs to the application of single human tear fluid constituents (blue bars) or to mixtures of lactoferrin with the corresponding tear fluid constituent (orange bars). All concentrations are as shown in Table 1. The data reflect the average and standard deviations of three measurements. | ||
Quantitative measurements of lactoferrin concentrations in human tear fluid
Quantitative lactoferrin analysis in human tear fluid from several volunteers was performed. The concentrations of lactoferrin in tear fluids were determined by using the developed μPADs. For validation purposes, the assay results were compared to those obtained by the ELISA method. Human tear samples were collected from the inferior cul-de-sac of five volunteers. The results of human tear analysis shown in Table 2 indicate that all samples were correctly analyzed with the developed μPADs within 6% error of the ELISA method. The direct comparison of the two different methods (Fig. S7†) shows a linear correlation coefficient R2 of 0.9907, with a slope close to unity (0.9762) and a y-axis intercept close to zero (0.0725). A major conceptual difference between the μPAD assay and the ELISA method is the fact that the immunoassay detects the total concentration of lactoferrin, independent of its iron saturation state, while complex formation between lactoferrin and terbium on the μPAD occurs only with the iron free apo-lactoferrin. However, in the case of tear fluid, it is known that lactoferrin is essentially present only in its apo-form.36 This is confirmed by the identical results within 6% error of lactoferrin concentrations measured by μPADs and by the ELISA method.| Sample no. | Method; concentration [mean ± 1σ, mg mL−1] | ||
|---|---|---|---|
| μPADa | ELISAb | Errorc (%) | |
| a Measured values were calculated from a calibration curve and the green intensity in the sensing area obtained by applying 2.5 μL of tear samples. The data reflect the average values and standard deviations of three measurements. b Tear samples were diluted 105-fold before use with the sample diluting buffer provided in the kit. The data reflect the average values and standard deviations of four measurements. c Error (%) calculated as 100 × (μPAD − ELISA)/ELISA. | |||
| 1 | 1.82 ± 0.05 | 1.91 ± 0.28 | −4.8 |
| 2 | 1.78 ± 0.01 | 1.87 ± 0.05 | −4.8 |
| 3 | 2.13 ± 0.06 | 2.15 ± 0.18 | −0.9 |
| 4 | 1.79 ± 0.06 | 1.70 ± 0.09 | +5.2 |
| 5 | 3.58 ± 0.21 | 3.57 ± 0.10 | +0.4 |
In the case of lactoferrin analysis with μPADs, small standard deviations were observed, with the exception of sample 5, which showed a relatively large value owing to its concentration being at the upper limit of the dynamic response range of the μPAD. Upon two-fold dilution of this sample with HEPES buffered solution, the analysis resulted in a value of 1.74 ± 0.11 mg mL−1 with a significantly lower standard deviation.
Conclusions
A rapid, user-friendly and low-cost sensing device for analysis of lactoferrin in human tear fluid was successfully developed. Lactoferrin detection was achieved by measuring the fluorescence emitted from lactoferrin–terbium complexes formed on the paper devices. This is to the best of our knowledge the first report of a quantitative lactoferrin assay without the requirement of using antibodies or high cost analytical instrumentation. It has been confirmed that the fluorescence emission intensity increases in proportion to the lactoferrin levels in the sample, which even allows detection by the naked eye. By applying filter paper as the sensing platform, a low-cost, light-weight, and easily and safely disposable device has been realized.Although the achieved limit of detection was much higher compared to the one reported for the ELISA kit (1 ng mL−1), the developed μPAD is a prospective alternative method for simple lactoferrin determination at concentrations encountered in human tear fluid usable by non-trained personnel. Assay results can be obtained within 15 min by simply pipetting a freshly collected tear sample to the sampling area. In contrast to the ELISA method requiring several hours of multiple pipetting, incubation, and washing procedures, the simplicity of the μPAD makes it a widely applicable sensing tool for rapid diagnosis. Finally, the detection system proposed here is expected to be adaptable to the sensing of other metal binding proteins by changing the probe deposited on the sensing area.
Acknowledgements
This work was supported by JST (Japan Science and Technology Agency) SENTAN.Notes and references
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318–1320 CrossRef CAS PubMed.
- A. K. Yetisen, M. S. Akram and C. R. Lowe, Lab Chip, 2013, 13, 2210–2251 RSC.
- S. M. Z. Hossain and J. D. Brennan, Anal. Chem., 2011, 83, 8772–8778 CrossRef CAS PubMed.
- S. Hossain, C. Ozimok, C. Sicard, S. Aguirre, M. Ali, Y. Li and J. Brennan, Anal. Bioanal. Chem., 2012, 403, 1567–1576 CrossRef CAS PubMed.
- J. C. Jokerst, J. A. Adkins, B. Bisha, M. M. Mentele, L. D. Goodridge and C. S. Henry, Anal. Chem., 2012, 84, 2900–2907 CrossRef CAS PubMed.
- M. M. Mentele, J. Cunningham, K. Koehler, J. Volckens and C. S. Henry, Anal. Chem., 2012, 84, 4474–4480 CrossRef CAS PubMed.
- S. J. Vella, P. D. Beattie, R. Cademartiri, A. Laromaine, A. W. Martinez, S. T. Phillips, K. A. Mirica and G. M. Whitesides, Anal. Chem., 2012, 84, 2883–2891 CrossRef CAS PubMed.
- K. Abe, K. Suzuki and D. Citterio, Anal. Chem., 2008, 80, 6928–6934 CrossRef CAS PubMed.
- D. A. Bruzewicz, M. Reches and G. M. Whitesides, Anal. Chem., 2008, 80, 3387–3392 CrossRef CAS PubMed.
- Y. Lu, W. Shi, L. Jiang, J. Qin and B. Lin, Electrophoresis, 2009, 30, 1497–1500 CrossRef CAS PubMed.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091–7095 CrossRef CAS PubMed.
- J. Olkkonen, K. Lehtinen and T. Erho, Anal. Chem., 2010, 82, 10246–10250 CrossRef CAS PubMed.
- W. Dungchai, O. Chailapakul and C. S. Henry, Analyst, 2011, 136, 77–82 RSC.
- X. Li, J. Tian, G. Garnier and W. Shen, Colloids Surf., B, 2010, 76, 564–570 CrossRef CAS PubMed.
- K. Maejima, S. Tomikawa, K. Suzuki and D. Citterio, RSC Adv., 2013, 3, 9258–9263 RSC.
- N. Li, N. Wang, J. Zheng, X. M. Liu, O. W. Lever, P. M. Erickson and L. Li, J. Proteome Res., 2005, 4, 2052–2061 CrossRef CAS PubMed.
- K. Karns and A. E. Herr, Anal. Chem., 2011, 83, 8115–8122 CrossRef CAS PubMed.
- F. H. Grus, P. Sabuncuo and A. J. Augustin, Electrophoresis, 2001, 22, 1845–1850 CrossRef CAS.
- Y. Danjo, M. Lee, K. Horimoto and T. Hamano, Acta Ophthalmol., 1994, 72, 433–437 CrossRef CAS.
- S. V. Hetherington, J. K. Spitznagel and P. G. Quie, J. Immunol. Methods, 1983, 65, 183–190 CrossRef CAS.
- T. Abe, A. Nakajima, M. Matsunaga, S. Sakuragi and M. Komatsu, Br. J. Ophthalmol., 1999, 83, 684–687 CrossRef CAS.
- U. Sridhar and A. K. Gupta, in Clinical Ophthalmology: Contemporary Perspectives, ed. A. K. Gupta and V. Krishna, Elsevier Health Sciences, 9th edn, 2009 Search PubMed.
- L. Zhou, R. W. Beuerman, C. M. Chan, S. Z. Zhao, X. R. Li, H. Yang, L. Tong, S. P. Liu, M. E. Stern and D. Tan, J. Proteome Res., 2009, 8, 4889–4905 CrossRef CAS PubMed.
- N. Tomosugi, K. Kitagawa, N. Takahashi, S. Sugai and I. Ishikawa, J. Proteome Res., 2005, 4, 820–825 CrossRef CAS PubMed.
- P. F. Levay and M. Viljoen, Haematologica, 1995, 80, 252–267 CAS.
- E. N. Baker, B. F. Anderson, H. M. Baker, M. Haridas, G. E. Norris, S. V. Rumball and C. A. Smith, Pure Appl. Chem., 1990, 62, 1067–1070 CrossRef CAS.
- S. Sueda, S. Tanaka, S. Inoue and H. Komatsu, Anal. Biochem., 2012, 422, 52–54 CrossRef CAS PubMed.
- A. M. Lipchik and L. L. Parker, Anal. Chem., 2013, 85, 2582–2588 CrossRef CAS PubMed.
- Y. Kataoka, S. Shinoda and H. Tsukube, J. Nanosci. Nanotechnol., 2009, 9, 655–657 CrossRef CAS PubMed.
- S. Alila, S. Boufi, M. N. Belgacem and D. Beneventi, Langmuir, 2005, 21, 8106–8113 CrossRef CAS PubMed.
- J. L. Flanagan and M. D. P. Willcox, Biochimie, 2009, 91, 35–43 CrossRef CAS PubMed.
- R. Sariri and H. Ghafoori, Biochemistry, 2008, 73, 381–392 CAS.
- D. Meillet, P. L. Hoang, F. Unanue, N. Kapel, M. C. Diemert, F. Rousselie, A. Galli and J. Galli, Eur. J. Clin. Chem. Clin. Biochem., 1992, 30, 319–323 CAS.
- J. U. Prause, Acta Ophthalmol., 1983, 61, 272–282 CrossRef CAS.
- D. K. Sen and G. S. Sarin, Br. J. Ophthalmol., 1980, 64, 693–695 CrossRef CAS.
- E. D. Weinberg, J. Pharm. Pharmacol., 2001, 53, 1303–1310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c3an01926h |
| This journal is © The Royal Society of Chemistry 2014 |