
Organic–inorganic hybrid catalysts containing new Schiff base for environment friendly cyclohexane oxidation†
R. Antonya,
S. Theodore David Manickam*a,
K. Karuppasamya,
Pratap Kollub,
P. V. Chandrasekarc and
S. Balakumara
aCentre for Scientific and Applied Research, PSN College of Engineering and Technology (Autonomous), Tirunelveli 627152, Tamil Nadu, India. E-mail: S.theodore.david@gmail.com; Fax: +91 4634 279680
bNewton International Fellow, Thin Film Magnetism Group, Department of Physics, Cavendish Laboratory, University of Cambridge, JJ Thomson Avenue, Cambridge, CB3 0HE, UK
cCollege of Physics and Information Engineering, Institute of Optoelectronic Display, Fuzhou University, Fuzhou 350002, PR China
First published on 9th September 2014
Abstract
Organic and inorganic entities have been hybridized using 3-aminopropyltriethoxysilane (APTES) linker for the synthesis of three novel organic–inorganic hybrid catalysts [Cu(II), Co(II) and Ni(II)]. During the course of the synthesis, the static inorganic moiety was functionalized with versatile imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) groups. The prepared catalysts were characterized by spectral techniques (EDS, FT-IR, DR UV-Vis., 29Si CP/MAS NMR, powder-XRD and ESR), thermal study (TG-DTG) and surface studies (SEM and AFM). The reported catalysts are significant because of their geometrical and dispersive surface properties. The synthesised catalysts were tested with cyclohexane oxidation reactions using H2O2, to make the catalytic system eco-friendly. Compared to the previously reported catalysts, the present catalysts have shown better cyclohexane conversion and selectivity, and are also cost effective. Among the three catalysts studied, Cu(II) catalyst showed the maximum conversion efficiency (44%) with product selectivity of 29% cyclohexanol and 71% cyclohexanone. Because of the combination of interesting structural properties and acceptable catalytic property with better selectivity and environment benign character along with a low cost, these hybrid catalysts could be the best future catalysts for cyclohexane oxidation.
N) groups. The prepared catalysts were characterized by spectral techniques (EDS, FT-IR, DR UV-Vis., 29Si CP/MAS NMR, powder-XRD and ESR), thermal study (TG-DTG) and surface studies (SEM and AFM). The reported catalysts are significant because of their geometrical and dispersive surface properties. The synthesised catalysts were tested with cyclohexane oxidation reactions using H2O2, to make the catalytic system eco-friendly. Compared to the previously reported catalysts, the present catalysts have shown better cyclohexane conversion and selectivity, and are also cost effective. Among the three catalysts studied, Cu(II) catalyst showed the maximum conversion efficiency (44%) with product selectivity of 29% cyclohexanol and 71% cyclohexanone. Because of the combination of interesting structural properties and acceptable catalytic property with better selectivity and environment benign character along with a low cost, these hybrid catalysts could be the best future catalysts for cyclohexane oxidation.
Introduction
Schiff bases formed by the reaction of primary amines with aldehydes/ketones display borderline character between hard and soft Lewis bases, and also exhibit stability under diverse conditions.1,2 In coordination chemistry, the role of Schiff bases as ligands is remarkable and widespread.3–5 The fascinating fact is that the formation of Schiff base complexes with metal ions increases their applications, predominantly as catalysts for many reactions such as oxidation, cyclopropanation, polymerization, coupling, hydrogenation, etc.6–9 Many Schiff base complexes belong to a typical class of homogeneous catalysts. However, quite often homogenous catalysts are not environment friendly due to the numerous problems associated with them, such as difficulties in catalyst recovery and product separation. Consequently, it is essential to find a new and effective route to eliminate these problems related with the homogeneous catalysts. The best way to overcome the aforementioned problems is providing a solid support for the homogeneous complexes.10,11 Though many supports like alumina,12 synthetic and bio-polymers,13,14 zeolite and montmorillonite clay15 have been used to make an immobilized Schiff base metal complexes, SiO2 has emerged as the pre-eminent and familiar solid support because of its high surface area, good accessibility, good mechanical and thermal stability, and flexible surface silanol groups.16 SiO2-containing Schiff base complexes are also termed as distinctive organic–inorganic hybrid catalysts, where the inorganic moiety is static and organic entity is flexible. These hybrid catalysts are very exceptional due to their mixed properties of both homogeneous (mobile reactive centre) and heterogeneous catalysts (catalytic recovery and recyclability). 3-Aminopropyltriethoxysilane (APTES) is an extensively employed chemical agent to embed the organic entity on to the inorganic SiO2 support via the functionalization of surface hydroxyl groups of SiO2 gel. The addition of amino groups on the surface of SiO2 gel can offer an opportunity to install a range of functional groups, often imine groups, on SiO2 gel's surface.17–19Oxidation of cyclohexane to cyclohexanol and cyclohexanone through the inert C–H bond activation is the most desired organic transformation because it's products are applied as intermediates for the synthesis of commercially essential products such as nylon-6 and nylon-6,6′. Cyclohexanol and cyclohexanone are also used as solvents for lacquers, shellacs and varnishes; stabilizers and homogenizers for soaps and emulsions; and starting materials for manufacturing insecticides, herbicides and pharmaceuticals.20–22 In industries, the current cyclohexane oxidation process in the presence of homogeneous boric cobalt salts requires a high temperature (150–170 °C) and pressure (115–175 psi) and yields only 4% conversion and poor product selectivity ratio (cyclohexanone–cyclohexanol). This industrial process is risky and environmentally hazardous reaction too.22–24 These issues eventually made different research groups to search for novel catalysts for the oxidation of cyclohexane under milder conditions. In quest of environment friendly oxidant, H2O2 and molecular O2 appear to be the preferable oxidants because they generate water as their by-product. However, high activation energy is required to break O–O double bond of molecular O2 during the addition of an organic substrate, whereas the spin mismatch between the ground state of O2 (triplet) and organic substrate (singlet) suppresses the use of O2 as the oxidant. Moreover, O2 involving organic reactions may also cause explosions.25,26 Therefore, H2O2 would be the better oxidant than the molecular O2.
In search of an improved catalytic system for the cyclohexane oxidation in presence of H2O2, variety of heterogeneous catalysts has been investigated. A few cases are Co(II) and Cr(VI) on poly(4-vinylpyridine-co-divinylbenzene) (4.64% conversion);27 Fe(III) scorpionate complex on carbon materials (20.8% conversion)28 and desilicated MOR zeolite (37.2% conversion)29 in the presence of co-catalyst and co-oxidant; Au nanoparticles on carbon materials (3.6% conversion);30 and chitosan supported Co(II) catalyst (26% conversion even after electron beam irradiation).31 To date, relatively superior cyclohexane conversion has been observed with SiO2 supported catalysts. For instance, though Urus et al. have achieved 99.9% cyclohexane conversion under microwave reaction conditions, the poor selectivity reduces its usability.32 Whereas, the catalysts involving mesoporous SiO2 supports such as MCM-41 (45.5% conversion with 51.9% cyclohexanol and 48.1% cyclohexanone selectivity)33 and SBA-15 (71.1% conversion with 26% cyclohexanol and 74% cyclohexanone selectivity)34 have shown sufficient cyclohexane conversion and better selectivity. However, tedious procedures, more chemicals and relatively high cost are required for the synthesis of mesoporous SiO2 materials. These limitations may suppress the effective use of SiO2 supported catalysts for cyclohexane oxidation. Thus, synthesizing a simple, low cost and effectual SiO2 supported catalysts for cyclohexane oxidation with enhanced selectivity appears to be very essential. We have already reported a simple and low cost SiO2 supported catalyst for adequate cyclohexane oxidation.35
By considering the abovementioned facts and in continuation with our work, we have attempted to develop a new catalytic system with adequate catalytic ability and selectivity under mild and environmentally tolerable conditions for the cyclohexane oxidation. Therefore, a novel SiO2-supported Schiff base ligand with its Cu(II), Co(II) and Ni(II) complexes were synthesized and their structural, thermal and morphological characterizations were also studied. Very importantly, the reported complexes were used as catalysts in the cyclohexane oxidation using H2O2 as an oxidant, and the stability of the catalysts were assessed in the successive catalytic run.
Experimental methods
Materials
All the chemicals were of AnalaR grade and used without further purification. APTES and 1,2-diphenylethanedione (DPED) were obtained from Himedia. SiO2 gel, cyclohexane, acetonitrile, toluene, H2O2 and metal salts were purchased from E. Merck, India.Synthesis of SiO2 supported APTES (Si–NH2)
The synthesis protocol for installing APTES on to SiO2 was adapted from the earlier reported procedure.32 Typically, 50 g SiO2 gel and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hydrochloric acid solution were mixed and refluxed for 6 h for the pre-treatment. This pre-treated SiO2 gel was filtered off and rinsed with a sufficient amount of deionized water till the pH of the filtrate reached 7. It was then dried at 120 °C for 12 h under vacuum. In 100 ml toluene, a mixture of 20 g pre-treated SiO2 gel and 20 ml APTES were mixed and refluxed for approximately 3 days. The resulting suspension was filtered off, washed with excess amount of toluene, ethanol and diethyl ether and dried at 100 °C under vacuum.
:1 hydrochloric acid solution were mixed and refluxed for 6 h for the pre-treatment. This pre-treated SiO2 gel was filtered off and rinsed with a sufficient amount of deionized water till the pH of the filtrate reached 7. It was then dried at 120 °C for 12 h under vacuum. In 100 ml toluene, a mixture of 20 g pre-treated SiO2 gel and 20 ml APTES were mixed and refluxed for approximately 3 days. The resulting suspension was filtered off, washed with excess amount of toluene, ethanol and diethyl ether and dried at 100 °C under vacuum.
Synthesis of SiO2 supported Schiff base (Si–NH2–DPED)
A mixture of 5 g SiO2–NH2 and 5 mmol DPED was stirred in 100 ml water at 60 °C for 24 h. The obtained solid product was filtered, washed with excess amount of water and dried at 100 °C.Synthesis of organic–inorganic hybrids [M(Si–NH2–DPED)Cl2]
2 g SiO2–NH2–DPED and 5 mmol metal chloride salt were mixed in 50 ml water. This mixture was then magnetically stirred at ambient temperature for 24 h. The final product was filtered, washed with excess amount of water and dried at 90 °C under vacuum.Cyclohexane oxidation
Cyclohexane oxidation was carried out in a 25 ml flask equipped with magnetic stirrer using the following procedure: 0.05 g catalyst ([Cu(SiO2–NH2–DPED)Cl2]/[Co(SiO2–NH2–DPED)Cl2]/[Ni(SiO2–NH2–DPED)Cl2]) was taken in 10 ml acetonitrile. Subsequently, 10 mmol of 30% H2O2 solution and 5 mmol of cyclohexane were added to the flask. This catalytic mixture was stirred at 70 °C under atmospheric pressure conditions for 12 h. Aliquots from the reaction mixture were taken at intervals of every 2 h up to 12 h for the product analysis. By the same reaction procedure, two separate blank catalytic experiments were also run, one without catalyst and another without oxidant. The product samples were analysed using Hewlett-Packard gas chromatograph (HP 6890) annexed with FID detector, a capillary column (HP-5), programmed oven with temperature range from 50 to 200 °C using nitrogen as the carrier gas with 0.5 cm3 min−1 flow rate.Instruments
FT-IR spectra of the compounds were recorded on a Jasco FT-IR 4100 spectrophotometer in the wave number region of 4000–400 cm−1 with 2 cm−1 resolution using KBr disks having 1% sample. DR UV-Vis. spectra were performed within 200–800 nm wavelength range on a Shimadzu UV-2600 double beam spectrophotometer, containing an integrating sphere attachment for solid samples. The solid-state 29Si cross polarized magic angle spinning nuclear magnetic resonance (29Si CP MAS NMR) spectral studies were performed for Si–NH2–DPED and [Cu(Si–NH2–DPED)Cl2] by a Mercury Plus 300 MHz NMR spectrometer (VARIAN, USA) annexed with a 5 mm dual broad band probe. The thermal properties of the compounds were investigated up to 800 °C at a heating rate of 10 °C min−1 under dynamic nitrogen atmosphere using Mettler Toledo star system. Powder X-ray diffraction (powder XRD) measurements were performed on an X-ray diffractometer (XPERT PRO PANalytical, Netherlands) for phase identification. The powder XRD patterns were obtained using CuKα radiation with a secondary monochromator (λ = 0.1540 nm) at 40 kV and 30 mA. X-band electron spin resonance (ESR) experiment of Cu(II) complex was performed on a JES-FA200 ESR spectrometer (JEOL, Japan) at 9.65 GHz micro wave frequency. Surface morphology of the compounds was assessed using a scanning electron microscope of JSM-SEM 6400 model at 15 kV accelerating voltage with 2 k× magnification range under liquid N2 atmosphere. Atomic force microscopic (AFM) study of [Cu(SiO2–NH2–DPED)Cl2] was performed on AFM XE 70, Park systems to analyse the particle distribution.Results and discussion
Characterization of hybrid catalysts
The presence of expected elements in each synthesized compound of this study was identified and confirmed by their energy peaks. The EDS spectra of all the reported compounds are provided (see ESI†). The occurrence of metal ion peaks in the respective EDS spectrum suggests the successful formation of organic–inorganic hybrid catalysts.Vibrational spectroscopy is the best scientific tool to thoroughly study the changes in SiO2 during the chemical modifications. Fig. 1 shows representative FT-IR spectra of SiO2 gel, Si–NH2, Si–NH2–DPED and the catalysts. In FT-IR spectrum of pure SiO2 gel (Fig. 1(a)), the bands observed at 1094 and 798 cm−1 were attributed to the characteristic anti-symmetric and symmetric stretches of Si–O–Si bond, respectively. The band at 462 cm−1 could be due to the bending vibrations of O–Si–O units. A small shoulder at 978 cm−1 is allocated to Si–OH stretching mode. The significant stretch of surface silanol groups (Si–O–H) is found at 3548 cm−1 as a broad band.36 All these characteristic bands were also present in the FT-IR spectra of all the modified forms of SiO2, which indicates that SiO2 framework is not affected even after the chemical modifications. In Fig. 1(b), a new continuous band attributed to –NH2 stretch has emerged in the region of 3290–3000 cm−1, confirming aminopropylation of SiO2. It was further supported by the occurrence of stretching and bending vibrations of aliphatic –CH2 groups at 2938 and 1495 cm−1, respectively. The –OH stretch of silanol was shifted to a lower wave number region, which shows the grafting of APTES on to surface silanol groups.37 In Fig. 1(c), the characteristic band of –NH2 groups were found to be disappeared. It can be attributed to the reaction between Si–NH2 and DPED. The new band at 1629 cm−1 for a distinctive stretch of –CN group, further confirms the reaction between Si–NH2 and DPED. It was also confirmed by the significant aromatic –CC– stretch, which emerges at 1474 cm−1 as a medium band. In addition, an additional band is found at 1720 cm−1 suggesting that one of the –CO groups of DPED did not undergo Schiff base reaction. The FT-IR spectra of all the complexes are similar to that of Si–NH2–DPED, except for the few changes. Considerably, such changes were the shifts in the stretching vibrations of –CN and –CO to a lower wave number. This proves the coordination of –CN and –CO groups with metal ions.38–40 However, the significant M − O and M − N bonds were not visible in the FT-IR spectra of complexes because they might be hidden by the bending vibrations of O–Si–O units.
 | ||
| Fig. 1 FT-IR spectra of (a) SiO2 gel, (b) Si–NH2, (c) Si–NH2–DPED, (d) [Cu(Si–NH2–DPED)Cl2], (e) [Co(Si–NH2–DPED)Cl2] and (f) [Ni(Si–NH2–DPED)Cl2]. | ||
The electronic absorption spectra of Si–NH2–DPED and its complexes are depicted in Fig. 2. The UV-Vis. spectra of SiO2 gel and Si–NH2 are also individually provided (ESI†). Unlike for the SiO2, the UV-Vis. pattern of Si–NH2 showed a significant band below 400 nm, which may be due to the characteristic n–π* transition of the non-bonded electrons of –NH2 groups. The electronic spectrum of the Schiff base, formed by the reaction between SiO2–NH2 and DPED (Fig. 2(a)), exhibited an additional band below 400 nm, which can be allotted to π–π* transition of the aromatic π-electrons. Emergence of this extra band apparently confirmed the formation of the Schiff base, Si–NH2–DPED. However, the n–π* transition band was due to the existence of non-bonded electrons of CN and CO groups, and not due to the NH2 groups. Moreover, the UV-Vis. spectra of complexes displayed the characteristic n–π* and π–π* transitions bands below 400 nm, but they slightly differ either in their position or intensity, which may be due to the coordination of the metals with Si–NH2–DPED. The charge transfer transition (metal to ligand π-back bonding) may also contribute to these absorption bands (below 450 nm) observed in the electronic spectra of metal complexes. However, a new band appeared in the UV-Vis. spectra of all the complexes, which can be the characteristic of the d–d transition of the central metal ions. This band is very significant because it provides information about the geometry of the metal complexes. In Fig. 2(b), it is obvious that the [Cu(Si–NH2–DPED)Cl2] shows its d–d transition band at around 600 nm which is an indication of 2B1g → 2A1g transition, characteristic of square planar geometry. In the UV-Vis. spectra of [Co(Si–NH2–DPED)Cl2] (Fig. 2(c)) and [Ni(Si–NH2–DPED)Cl2] (Fig. 2(d)) this band appeared at 600 and 460 nm, respectively, because of 1A1g → 1B1g transition, which confirms the square planar geometry of the complexes.41–43
 | ||
| Fig. 2 DR UV-Vis. spectra of (a) Si–NH2–DPED, (b) [Cu(Si–NH2–DPED)Cl2], (c) [Co(Si–NH2–DPED)Cl2] and (d) [Ni(Si–NH2–DPED)Cl2]. | ||
29Si CP MAS NMR spectroscopy was successfully employed in the characterization of SiO2 based materials by studying the silicon environment in the materials. In general, two characteristic peaks were expected for the unmodified SiO2 gel at ∼110 and ∼105 ppm which are the evidences of ((SiO)4Si) sites and ((SiO)3SiOH) silanol sites, respectively. The abundance of ((SiO)4Si) sites in SiO2 gel might make the peak at 110 ppm predominant;44 29Si NMR spectra of Si–NH2–DPED and its Cu(II) complex are separately shown (ESI†). The two expected characteristic peaks of SiO2 gel are observed in the 29Si NMR spectra of both Si–NH2–DPED and its Cu(II) complex. However, the intensity of the peak at 105 ppm (characteristic of ((SiO)3SiOH) sites) appears to be significantly low, which may be due to the successful modification of surface –OH groups with organic functional groups.45 The appearance of a new significant peak noted at ∼65 ppm in the 29Si NMR spectrum of Si–NH2–DPED can be due to the formation of ((SiO)3SiC) sites as the consequence of organic modification of surface –OH groups.46–48 This peak is also found in 29Si NMR spectrum of Cu(II) complex but with some changes in intensity and peak area. These changes may be ascribed to the coordination of Si–NH2–DPED with Cu(II). Thus, the 29Si NMR spectral data support the results obtained from FT-IR and UV-Vis spectral studies.
Powder XRD patterns of all the compounds were taken in a wide angle region (between the 2θ region of 10° and 80°). The XRD patterns of SiO2, Si–NH2, Si–NH2–DPED and the complexes are presented in Fig. 3. SiO2 exhibited a broad band centered at 2θ = 22°, characteristic of topological structure of SiO2.49 After the modification with APTES and DPED, this peak was found to have lost its intensity and sharpness. This may be due to the slight disorder in the topological structure of SiO2 in its modified forms because of the immobilization of DPED and entry of metal ions on to the porous wall of SiO2 or reduction in X-ray scattering contrast between the channel wall of silicate frame work and ligand/complex.16,50 Some new intense peaks also appeared in the XRD pattern of Si–NH2–DPED, and significantly these new peaks appear to be analogous with the characteristic peaks of DPED. This further supports the organo-functionalization of Si–NH2 with DPED. Co(II) and Ni(II) complexes show similar XRD pattern like Si–NH2–DPED, while Cu(II) complex does not. This may be due to the higher crystallinity of Cu(II) complex compared to the other two.
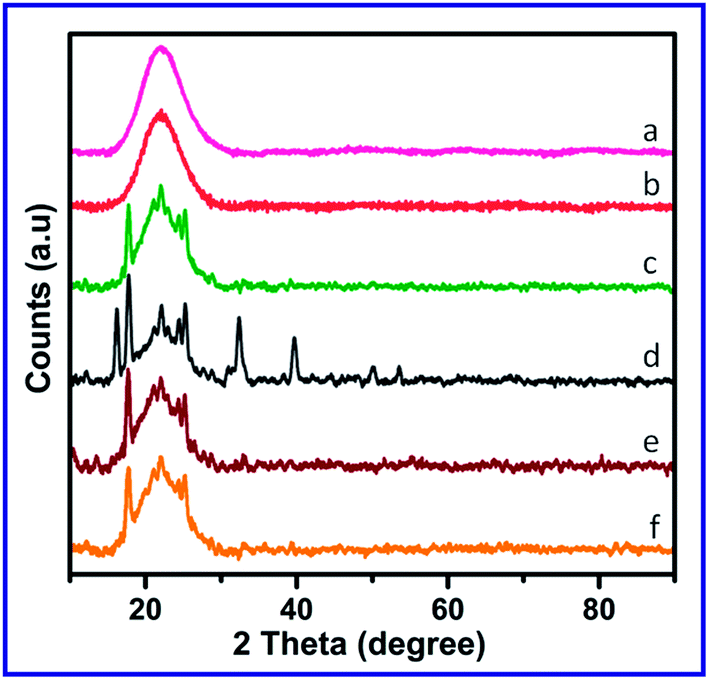 | ||
| Fig. 3 Powder XRD patterns of (a) SiO2 gel, (b) Si–NH2, (c) Si–NH2–DPED, (d) [Cu(Si–NH2–DPED)Cl2], (e) [Co(Si–NH2–DPED)Cl2] and (f) [Ni(Si–NH2–DPED)Cl2]. | ||
ESR spectroscopy has been established as the most excellent technique to explore the geometry of metal complexes, in particular Cu(II) complexes.51–53 In the present study, ESR spectral study of Cu(II) complex was performed. The recorded ESR spectrum of Cu(II) complex is shown in Fig. 4. It reveals two g factors, g‖ and g⊥. The obtained spectrum provides evidence about the axial nature of Cu(II) complex. After comparison, the calculated g factors are found to have the order of g‖ (2.223) > g⊥ (2.094) > 2.0023, and this indicates the presence of unpaired electron in dx2 − dy2 orbital. This investigation suggest that the geometry was square planar, which is in agreement with the result obtained from UV-Vis. spectroscopy.54 The lesser value of g‖ as compared to 2.3 is a good evidence for the covalent character of Cu–ligand interaction, which was recommended by Kivelson and Niemon.55
 | ||
| Fig. 4 X-band ESR spectrum of [Cu(Si–NH2–DPED)Cl2]. | ||
The following expression can be employed to compute the exchange coupling factor (G):
| G = (g‖ − 2)/(g⊥ − 2) |
It has already been proved that parallely aligned or slightly misaligned local axes have G value greater than 4.0, whereas, the G value will be less than 4.0 for remarkably misaligned local axes with considerable exchange coupling. In this case, the G value of Cu(II) complex is 4.9, thus the exchange coupling could be negligible.56,57
The TG-DTG patterns of SiO2, Si–NH2, Si–NH2–DPED and the complexes are illustrated in Fig. 5 and in particular, TG-DTG curves of SiO2 and Si–NH2 are provided separately (see ESI†). TG-DTG pattern of SiO2 gel exhibited two decomposition peaks corresponding to the loss of physically adsorbed water molecules (0–100 °C, 2.94%) and dehydroxylation of surface silanol groups (370–800 °C, 1.4%). Though Si–NH2 showed two similar decomposition peaks, the second one is higher (345–645 °C, 5.67%), and this may be due to the decay of APTES entity. TG-DTG curves (Fig. 6(a)) of Si–NH2–DPED revealed a different outline compared to that of both SiO2 and Si–NH2; there was no loss of physically adsorbed water molecules, however two thermal decays are found. They are attributed to the loss of DPED moiety (100–355 °C, 22.90%) and the loss of APTES along with the dehydroxylation of surface silanols (355–655 °C, 16.52%). The massive weight loss between 100–355 °C clearly indicates the loading of organic entity (DPED) onto Si–NH2. TG-DTG models of all the complexes are found to be similar to each other and show three mass loss steps. The initial step can be assigned to the desorption of physically adsorbed water molecules, second step is due to the loss of DPED and third step may be collectively due to the removal of APTES and dehydroxylation of surface silanols. In addition to this similarity, only Cu(II) complex shows weight gain above 580 °C which may be due to the oxidation reaction and this behaviour can be assigned to the crystalline character of Cu(II) complex.
 | ||
| Fig. 5 TG-DTG patterns of (a) Si–NH2–DPED, (b) [Cu(Si–NH2–DPED)Cl2], (c) [Co(Si–NH2–DPED)Cl2] and (d) [Ni(Si–NH2–DPED)Cl2]. | ||
 | ||
| Fig. 6 SEM images of (a) Si–NH2–DPED, (b) [Cu(Si–NH2–DPED)Cl2], (c) [Co(Si–NH2–DPED)Cl2] and (d) [Ni(Si–NH2–DPED)Cl2]. | ||
Changes in the surface properties of SiO2 gel because of the series of chemical modifications were examined by SEM analysis. SEM images of SiO2 and Si–NH2 are displayed in ESI,† whereas that of Si–NH2–DPED and its complexes are collectively portrayed in Fig. 6. The SEM photograph of SiO2 gel exposes the lumpy shaped apparent surface. It is considerably evident that the particle size of the modified forms is smaller than that of the unmodified SiO2 gel. Therefore, it is apparent that the modification of SiO2 with APTES, DPED and metal ions can provide more efficient catalysts. In addition, the chemical modifications of SiO2 with functional groups generally assist and enhance the adsorption of metal ions.58,59 Because of the availability of the functional groups, metal ions can be adsorbed on SiO2's surface through coordination reaction. This coordination of the functional groups on silica would enhance the life span of metal ions on the surface of SiO2. Consequently, this may increase the stability of the complexes in successive catalytic runs. The surface of the complexes is rougher than that of Si–NH2–DPED as a consequence of the presence of metal ions. SEM images of all the complexes show well-dispersed metal ions, which means that adsorbed particles are not agglomerated. This surface behaviour of the organic–inorganic hybrids would help to achieve better catalytic ability.
2D and 3D AFM images of [Cu(Si–NH2–DPED)Cl2] were obtained, given as Fig. 7, to verify the dispersion and distribution of particles. They confirm that particles are properly arranged and dispersed; the particles appear like cones. They are not agglomerated and agree with the surface information obtained from the SEM images.
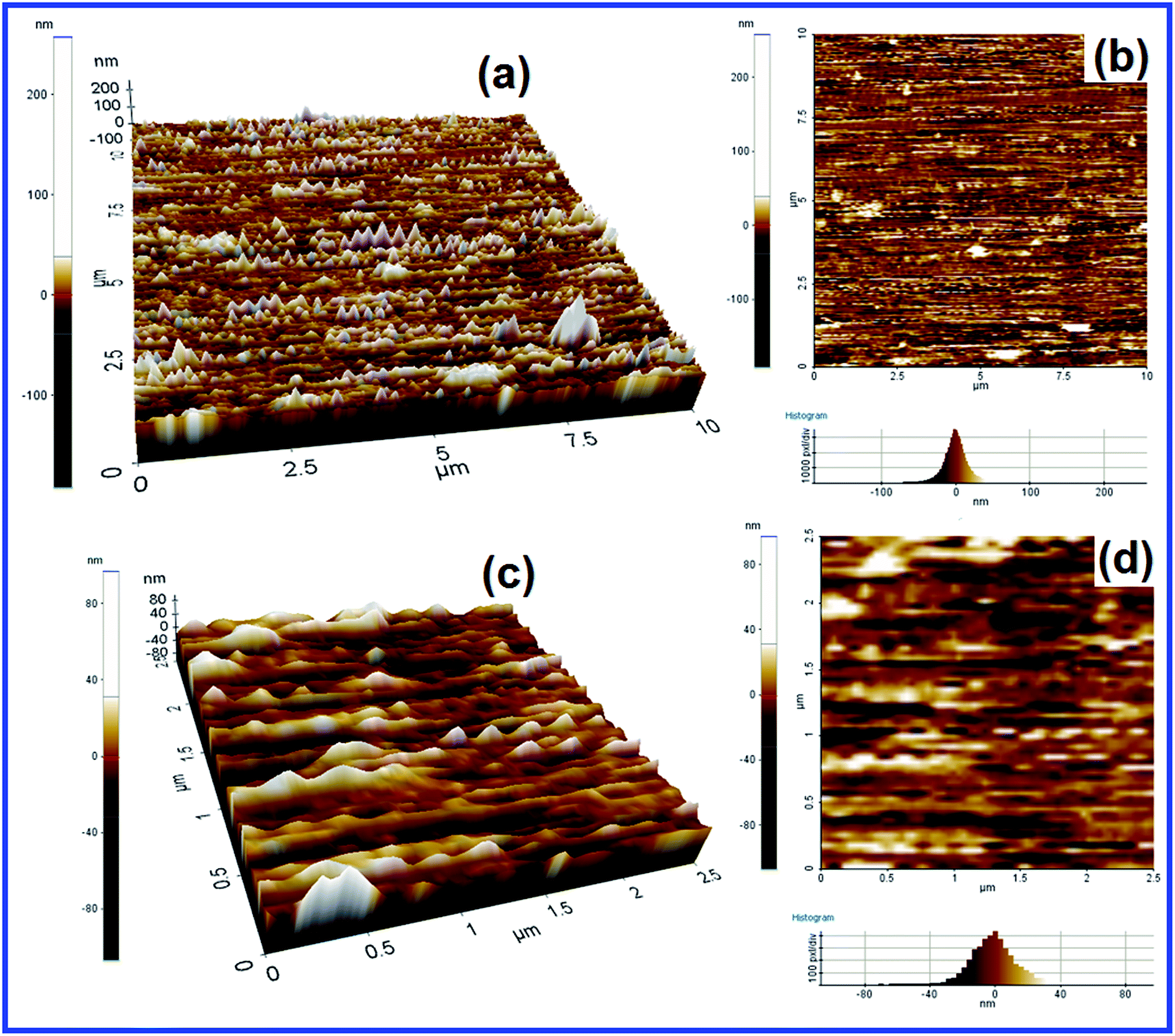 | ||
| Fig. 7 2D (a) and 3D (b) AFM images (10 × 10 μm); 2D (c) and 3D (d) AFM images (2.5 × 2.5 μm) of [Cu(Si–NH2–DPED)Cl2]. | ||
Catalytic study
Catalytic activities of the reported catalysts ([Cu(Si–NH2–DPED)Cl2]/[Co(Si–NH2–DPED)Cl2]/[Ni(Si–NH2–DPED)Cl2]) were investigated by carrying out a model catalysis reaction, where cyclohexane was oxidized into cyclohexanol and cyclohexanone in the presence of H2O2. This model reaction catalysed by organic–inorganic hybrid catalysts with H2O2 is promising and desirable due to its industrial importance and environment friendly character. The product analysis was performed by gas chromatography technique. During product analysis, there was no cyclohexane conversion identified either in the absence of catalyst or oxidant. This proves the significance of catalyst and H2O2 in the cyclohexane oxidation. Similar to the catalyst, the role of solvents in catalysis were also found to be crucial because of their ability to convert different phases into a uniform and upholding mass transportation. The significance of solvents also includes their ability to change the reaction mechanism by disturbing reaction intermediates and amend the surface properties of catalysts and reaction path-ways.60 In the literature, acetonitrile was established as a widely used and effective solvent.31,34,61 Therefore, in our present study, all the catalytic runs were executed in acetonitrile. In all the experiments, the observed products were only cyclohexanone and cyclohexanol. No white precipitate was observed on cooling the product mixture and the absence of acid in the product mixture was confirmed by titrating against NaOH. These results clearly demonstrated the absence of adipic acid in the product mixture. | ||
| Fig. 8 Effect of temperature on cyclohexane oxidation catalysed by Cu(II), Co(II) and Ni(II) hybrid catalysts. | ||
 | ||
| Fig. 9 Effect of time on selectivity of products obtained from cyclohexane oxidation catalysed by Cu(II), Co(II) and Ni(II) hybrid catalysts. | ||
Among the three catalysts, [Cu(Si–NH2–DPED)Cl2] was found to show a higher catalytic efficacy compared to the other two. The cyclohexane conversion efficiencies of [Cu(Si–NH2–DPED)Cl2], [Co(Si–NH2–DPED)Cl2] and [Ni(Si–NH2–DPED)Cl2] were 44, 38 and 35%, respectively, at 70 °C for 12 h (see Table S1 in ESI†). From Fig. 9, we can conclude that [Cu(Si–NH2–DPED)Cl2] would be the better choice for the formation of cyclohexanol and [Ni(Si–NH2–DPED)Cl2] can be preferred for cyclohexanone. This study shows that better selectivity for cyclohexanol by [Cu(Si–NH2–DPED)Cl2] can be achieved only below 6 h of the reaction. This problem can be possibly eliminated by concurrently removing cyclohexanol from catalytic reaction mixture. The variation in product selectivity may depend on the surface morphology of the catalysts.
 | ||
| Fig. 10 Catalytic reusability of [Cu(Si–NH2–DPED)Cl2]. Reaction conditions: 5 mmol cyclohexane, 10 mmol 30% H2O2, 0.05 g catalyst, 10 ml CH3CN, 70 °C and 12 h. | ||
We have already reported the similar kind of catalysts for cyclohexane oxidation but the support used was chitosan, not SiO2. Our current study suggests that the SiO2 based organic–inorganic hybrid catalysts are more effective than the chitosan based catalysts for the oxidation of cyclohexane.62 The reason may possibly be the well dispersive character of SiO2 based catalysts than their chitosan supported analogues. These SiO2 supported catalysts also appeared as good reusable catalysts than the corresponding chitosan supported catalysts.
Similar to the blank catalytic experiments without oxidant and catalyst, two other experiments were carried out to check the catalytic activity of SiO2 gel and Si–NH2–DPED under the identical conditions; no cyclohexane conversion was detected. It proves the inability of SiO2 and Si–NH2–DPED to oxidize cyclohexane. To study the mechanism of cyclohexane oxidation, UV-Vis. spectroscopy was used. For this mechanistic study, a mixture of H2O2, nitric acid and catalyst in acetonitrile was prepared and its electronic spectrum was recorded. A shoulder peak was identified between 410–420 nm in the UV-Vis. spectra of all the complexes. This can be attributed to the metal-peroxo species63 and it further suggests that cyclohexane oxidation followed the peroxidative mechanism, as given in the ESI.†
Conclusions
Three organic–inorganic hybrid catalysts (Cu(II), Co(II) and Ni(II) complexes) were synthesized by embedding imine (CN) functional groups on the SiO2 gel surface with the help of APTES linker. The studied hybrid catalysts were characterized by spectral, thermal and surface analytical techniques. The catalytic activities of catalysts were studied in generic, facile and mild cyclohexane oxidation using H2O2 oxidant for an eco-friendly approach. The salient features of the study are that all the catalysts showed adequate catalytic activity as well as good selectivity at reasonable temperature and atmospheric pressure. Importantly, except cyclohexanone and cyclohexanol (KA oil), no other products were obtained at the end of the cyclohexane oxidation. Upon increasing the reaction time, cyclohexanone % was found to be increasing while the cyclohexanol % was decreasing. It is found that the cyclohexane is first oxidised to cyclohexanol which is subsequently oxidised to cyclohexanone. Therefore, by lengthening the reaction time, our present catalytic system would become more selective. Cu(II) hybrid catalyst exhibited superior catalytic efficiency than the Co(II) and Ni(II) catalysts. The reusability study confirmed the stability of organic–inorganic hybrid catalysts inside the catalytic reactions, and therefore our catalysts can be repeatedly used in the catalytic run without any major decrease in their catalytic efficacy. With the merits of H2O2 oxidant, better product selectivity, dispersive nature and reusability of catalysts, the present hybrid catalysts for cyclohexane oxidation would be effective and preferable for future catalysis. Currently, the electron beam impact on the structural and catalytic properties of these organic–inorganic hybrid catalysts is currently under study in our laboratory.
Acknowledgements
STDM is very grateful to the Department of Atomic Energy-Board of Research in Nuclear Sciences (DAE-BRNS), Mumbai, India for providing financial aid to successfully carry out this work. RA sincerely thanks DAE-BRNS for giving Junior and Senior Research Fellowships. PK is very happy to thank the SAIF, IIT Bombay for providing characterization facilities (SEM with EDS, 29Si NMR, powder XRD and ESR). There are insufficient words to thank the valuable contribution of Mr K. Saravanan, CSIR, CSCMRI, Bhavnagar, India for the discussion of catalytic activities.References
- A. D. Garnovskii, A. L. Nivorozhkin and V. I. Minkin, Coord. Chem. Rev., 1993, 126, 1–69 CrossRef CAS.
- R. Ziessel, Coord. Chem. Rev., 2001, 216, 195–223 CrossRef.
- Y.-B. Dong, M. D. Smith and H.-C. zur Loye, Inorg. Chem., 2000, 39, 4927–4935 CrossRef CAS.
- L. Sacconi and I. Bertini, J. Am. Chem. Soc., 1966, 88, 5180–5185 CrossRef CAS.
- J.-C. Wu, S.-X. Liu, T. D. Keene, A. Neels, V. Mereacre, A. K. Powell and S. Decurtins, Inorg. Chem., 2008, 47, 3452–3459 CrossRef CAS PubMed.
- S. Adsule, V. Barve, D. Chen, F. Ahmed, Q. P. Dou, S. Padhye and F. H. Sarkar, J. Med. Chem., 2006, 49, 7242–7246 CrossRef CAS PubMed.
- J. A. Miller, W. Jin and S. T. Nguyen, Angew. Chem., 2002, 114, 3077–3080 CrossRef.
- N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS PubMed.
- N. Debono, M. Iglesias and F. Sanchez, Adv. Synth. Catal., 2007, 349, 2470–2476 CrossRef CAS PubMed.
- C. W. Jones, M. W. McKittrick, J. V. Nguyen and K. Yu, Top. Catal., 2005, 34, 67–76 CrossRef CAS PubMed.
- F. R. Hartley, Supported Metal Complexes: A New Generation of Catalysts, Springer, 1985 Search PubMed.
- M. Salavati-Niasari, M. Hassani-Kabutarkhani and F. Davar, Catal. Commun., 2006, 7, 955–962 CrossRef CAS PubMed.
- R. Antony, G. Tembe, M. Ravindranathan and R. Ram, J. Mol. Catal. A: Chem., 2001, 171, 159–168 CrossRef CAS.
- R. Antony, S. Theodore David, K. Saravanan, K. Karuppasamy and S. Balakumar, Spectrochim. Acta, Part A, 2013, 103, 423–430 CrossRef CAS PubMed.
- F. Bedioui, Coord. Chem. Rev., 1995, 144, 39–68 CrossRef CAS.
- R. Sharma, A. Pandey and S. Gulati, Appl. Catal., A, 2012, 431, 33–41 CrossRef PubMed.
- K. Dhara, K. Sarkar, D. Srimani, S. K. Saha, P. Chattopadhyay and A. Bhaumik, Dalton Trans., 2010, 39, 6395–6402 RSC.
- M. Nandi, P. Roy, H. Uyama and A. Bhaumik, Dalton Trans., 2011, 40, 12510–12518 RSC.
- S. Jana, B. Dutta, R. Bera and S. Koner, Langmuir, 2007, 23, 2492–2496 CrossRef CAS PubMed.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- R. Mokaya and M. Poliakoff, Nature, 2005, 437, 1243–1244 CrossRef CAS PubMed.
- T. F. Silva, E. C. Alegria, L. M. Martins and A. J. Pombeiro, Adv. Synth. Catal., 2008, 350, 706–716 CrossRef CAS PubMed.
- T. F. Silva, G. S. Mishra, M. F. G. da Silva, R. Wanke, L. M. Martins and A. J. Pombeiro, Dalton Trans., 2009, 9207–9215 RSC.
- R. Whyman, Applied organometallic chemistry and catalysis, Oxford University Press, 2001 Search PubMed.
- H. H. Thorp, Science, 2000, 289, 882–883 CrossRef CAS PubMed.
- X. Wang, G. Wu, W. Wei and Y. Sun, Transition Met. Chem., 2010, 35, 213–220 CrossRef CAS.
- D. Lončarević, J. Krstić, J. Dostanić, D. Manojlović, Ž. Čupić and D. Jovanović, Chem. Eng. J., 2010, 157, 181–188 CrossRef PubMed.
- L. Martins, M. P. de Almeida, S. Carabineiro, J. Figueiredo and A. Pombeiro, ChemCatChem, 2013, 5, 3847–3856 CrossRef CAS PubMed.
- L. Martins, A. Martins, E. Alegria, A. Carvalho and A. Pombeiro, Appl. Catal., A, 2013, 464, 43–50 CrossRef PubMed.
- S. Carabineiro, L. Martins, M. Avalos-Borja, J. Buijnsters, A. Pombeiro and J. Figueiredo, Appl. Catal., A, 2013, 467, 279–290 CrossRef CAS PubMed.
- R. Antony, S. Theodore David, K. Karuppasamy, G. Sanjeev and S. Balakumar, Spectrochim. Acta, Part A, 2014, 124, 178–186 CrossRef CAS PubMed.
- S. Uruş, M. Dolaz and M. Tümer, J. Inorg. Organomet. Polym. Mater., 2010, 20, 706–713 CrossRef.
- J. Zhao, W. Wang and Y. Zhang, J. Inorg. Organomet. Polym., 2008, 18, 441–447 CrossRef CAS.
- S. Alavi, H. Hosseini-Monfared and M. Siczek, J. Mol. Catal. A: Chem., 2013, 377, 16–28 CrossRef CAS PubMed.
- R. Antony, T. D. Selvanayagam, P. Kollu, P. Chandrasekar, K. Karuppasamy and S. Balakumar, RSC Adv., 2014, 4, 24820–24830 RSC.
- G. Dıaz, R. Perez-Hernandez, A. Gomez-Cortes, M. Benaissa, R. Mariscal and J. Fierro, J. Catal., 1999, 187, 1–14 CrossRef.
- P. Sharma, A. Lazar and A. Singh, Appl. Catal., A, 2012, 439, 101–110 CrossRef PubMed.
- E. F. Murphy, L. Schmid, T. Bürgi, M. Maciejewski, A. Baiker, D. Günther and M. Schneider, Chem. Mater., 2001, 13, 1296–1304 CrossRef CAS.
- T. R. Arun and N. Raman, Spectrochim. Acta, Part A, 2014, 127, 292–302 CrossRef CAS PubMed.
- A. Magro, L. Crociani, C. Prinzivalli, P. A. Vigato, P. L. Zanonato and S. Tamburini, Inorg. Chim. Acta, 2014, 410, 29–38 CrossRef CAS PubMed.
- W.-J. Zhou, B. Albela, P. Perriat, M.-Y. He and L. Bonneviot, Langmuir, 2010, 26, 13493–13501 CrossRef CAS PubMed.
- C. Brückner, V. Karunaratne, S. J. Rettig and D. Dolphin, Can. J. Chem., 1996, 74, 2182–2193 CrossRef.
- F. Feigl, Chemistry of specific, selective and sensitive reactions, Academic Press, 1949 Search PubMed.
- P. Yin, Y. Tian, Z. Wang, R. Qu, X. Liu, Q. Xu and Q. Tang, Mater. Chem. Phys, 2011, 129, 168–175 CrossRef CAS PubMed.
- D. Pérez-Quintanilla, I. del Hierro, M. Fajardo and I. Sierra, J. Mater. Chem., 2006, 16, 1757–1764 RSC.
- S. Singha, K. Parida and A. C. Dash, J. Porous Mater., 2011, 18, 707–714 CrossRef CAS.
- W. A. Carvalho, M. Wallau and U. Schuchardt, J. Mol. Catal. A: Chem., 1999, 144, 91–99 CrossRef CAS.
- X. Wang, G. Wu, W. Wei and Y. Sun, Catal. Lett., 2010, 136, 96–105 CrossRef CAS PubMed.
- N. An, W. Zhang, X. Yuan, B. Pan, G. Liu, M. Jia, W. Yan and W. Zhang, Chem. Eng. J., 2013, 215, 1–6 CrossRef PubMed.
- R. Qu, M. Wang, C. Sun, Y. Zhang, C. Ji, H. Chen, Y. Meng and P. Yin, Appl. Surf. Sci., 2008, 255, 3361–3370 CrossRef CAS PubMed.
- J. Evans, A. Zaki, M. El-Sheikh and S. El-Safty, J. Phys. Chem. B, 2000, 104, 10271–10281 CrossRef CAS.
- D. Zois, C. Vartzouma, Y. Deligiannakis, N. Hadjiliadis, L. Casella, E. Monzani and M. Louloudi, J. Mol. Catal. A: Chem., 2007, 261, 306–317 CrossRef CAS PubMed.
- A. R. Silva, M. M. A. Freitas, C. Freire, B. de Castro and J. L. Figueiredo, Langmuir, 2002, 18, 8017–8024 CrossRef CAS.
- S. Deshpande, D. Srinivas and P. Ratnasamy, J. Catal., 1999, 188, 261–269 CrossRef CAS.
- D. Kivelson and R. Neiman, J. Chem. Phys., 2004, 35, 149–155 CrossRef PubMed.
- B. Hathaway and D. Billing, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS.
- R. Dudley and B. Hathaway, J. Chem. Soc. A, 1970, 1725–1728 RSC.
- S. Uruş, S. Purtaş, G. Ceyhan and T. Erkenez, Sep. Purif. Technol., 2013, 118, 432–447 CrossRef PubMed.
- S. Uruş, S. Purtaş, G. Ceyhan and F. Tümer, Chem. Eng. J., 2013, 220, 420–430 CrossRef PubMed.
- A. Corma, M. Camblor, P. Esteve, A. Martinez and J. Perezpariente, J. Catal., 1994, 145, 151–158 CrossRef CAS.
- C. K. Modi and P. M. Trivedi, Arabian J. Chem., 2013 DOI:10.1016/j.arabjc.2013.03.016.
- R. Antony, S. Theodore David Manickam, K. Saravanan, K. Karuppasamy and S. Balakumar, J. Mol. Struct., 2013, 1050, 53–60 CrossRef CAS PubMed.
- P. Roy and M. Manassero, Dalton Trans., 2010, 39, 1539–1545 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08303b |
| This journal is © The Royal Society of Chemistry 2014 |