
A newly designed benzodithiophene building block: tuning of the torsional barrier for non-halogenated and non-aromatic solvent-processible photovoltaic polymers†
Hye Won
Cho‡
a,
Sang Young
Jeong‡
b,
Ziang
Wu‡
b,
Hyojin
Lim
b,
Won-Woo
Park
c,
Woojin
Lee
a,
Jonnadula Venkata
Suman Krishna
b,
Oh-Hoon
Kwon
 c,
Jin Young
Kim
*ad and
Han Young
Woo
*b
c,
Jin Young
Kim
*ad and
Han Young
Woo
*b
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: jykim@unist.ac.kr
bDepartment of Chemistry, Korea University, Seoul 136-713, Republic of Korea. E-mail: hywoo@korea.ac.kr
cDepartment of Chemistry, College of Natural Sciences, Ulsan National Institute of Science and Technology, Ulsan 44919, Republic of Korea
dGraduate School of Carbon Neutrality, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 28th February 2023
Abstract
A new benzodithiophene (BDT) building block, 4,8-bis(5-(2-ethylhexyl)-3-fluoro-4-hexylthiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene (3-FBDT), was designed by tailoring the positions and number of alkyl and F substituents by adjusting the torsional energy barrier. The incorporation of 3-FBDT into a representative BDT-based polymer donor (PBDB-T-2F) yielded a new photovoltaic copolymer (PBDB-T-2F(3/4)) with a decreased valence band level and considerably improved solubility in non-halogenated and non-aromatic solvents such as tetrahydrofuran (THF). Side-chain engineering has been widely studied to control solution processability in eco-friendly solvents, but the torsional property control of conjugated main chains has rarely been attempted. Although PBDB-T-2F showed a significant drop in power conversion efficiencies (PCEs: 17.46 to 9.73%) owing to serious aggregation caused by replacing chloroform with THF as a processing solvent, the THF-processed PBDB-T-2F(3/4) device maintained a high PCE (14.3 to 13.86%) with little detrimental effect. Charge carrier dynamics and film morphology analyses suggested that the electronic and morphological properties of PBDB-T-2F(3/4) are mainly governed by the majority moiety of the PBDB-T-2F blocks. Owing to its high solubility and outstanding photoelectrical properties, PBDB-T-2F(3/4) was successfully employed to fabricate flexible, semi-transparent, and large-area THF-processed devices. The incorporation of the 3-FBDT building block into various BDT-based photovoltaic polymers can be an effective strategy to improve the solution processability and broaden the solvent selection for fabricating eco-friendly solar cells without significantly disrupting their photoelectrical properties.
Introduction
Organic solar cells (OSCs) have attracted growing attention because of their advantages such as facile tunability of chemical structures and their optical/photoelectrical properties, light weight, mechanical flexibility, semi-transparency, and solution processability on flexible plastic substrates. Subsequent to the development of nonfullerene acceptors, i.e., 2,2′-((2Z,2′Z)-((12,13-bis(2-ethylhexyl)-3,9-diundecyl-12,13-dihydro-[1,2,5]thiadiazolo[3,4-e]thieno[2′′,3'':4′,5']thieno[2′,3′:4,5]pyrrolo[3,2-g]thieno[2′,3′:4,5]thieno[3,2-b]indole-2,10 diyl)bis(methanylylidene))bis(5,6-difluoro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile (Y6), the power conversion efficiencies (PCEs) of solution-processed single-junction OSCs have been remarkably improved to over 19%.1–4 This demonstrates a great potential of OSCs for future applications as a green energy-harvesting source. To date, most studies on photovoltaic materials have mainly focused on the structural modification and optimization of nonfullerene acceptors,5–9 while counterpart donor materials have been relatively overlooked.10–12The benzodithiophene (BDT) moiety is considered the most attractive building block for donor materials, and various BDT-based photovoltaic polymers have been successfully developed.13–17 In 2012, Hou's group first reported the PBDB-T polymer, demonstrating a PCE of 6.67% with PC61BM as an acceptor.18 In 2016, the PCE of PBDB-T OSCs was further improved to 12% by incorporating a nonfullerene IT-M acceptor, which possesses a high-lying lowest unoccupied molecular orbital (LUMO) and suitable crystallinity.19 In 2015, an outstanding PCE of 13.7% was achieved by introducing an extra F atom onto the BDT moiety to produce a polymer, PBDB-T-2F.20 Compared to PBDB-T, PBDB-T-2F exhibited a stronger interchain packing (via intra-/interchain H bonding), superior hole transport, and a deeper highest occupied molecular orbital (HOMO) energy level, which has been extensively studied as a representative donor polymer. In 2017, Hou prepared a new BDT polymer (PBDB-T-SF) by inserting an S atom into the BDT core.21 The outstanding light-absorption property and crystalline morphology of PBDB-T-SF:IT-4F films yielded a PCE of 13%, together with a high tolerance towards the thickness of the active layer. In 2018, by replacing the F atom by a Cl atom in PBDB-T-2F, PBDB-T-2Cl was developed by Zhang's group.22 When blended with IT-4F as an acceptor, the higher dipole moment of the C–Cl bond in PBDB-T-Cl further decreased the HOMO energy level of the polymer, achieving a 13.1% PCE with an open-circuit voltage (Voc) of 0.88 V.23
Although the representative BDT-based polymer, PBDB-T-2F, performs well because of its structural rigidity and chain planarity with tight packing, the processing solvents are limited to toxic halogenated ones such as chloroform (CF), chlorobenzene, and 1,2-dichlorobenzene, which are detrimental to human health and the environment. Alternate non-halogenated solvents, including toluene, 2-methylanisole and o-xylene have also been considered for device fabrication, but these aromatic solvents still bear considerable toxicity (for example, most of them are carcinogenic) (Table S1†).24–26 Non-halogenated and non-aromatic solvents (such as tetrahydrofuran (THF), 2-methyltetrahydrofuran, and ethanol) can be ideal processing solvents for the mass production of OSCs via roll-to-roll processes.27–31 The successful demonstration of OSC devices using non-halogenated and non-aromatic solvents has been very limited.32 To overcome the solubility issue of organic semiconductors in eco-friendly solvents, previous studies mainly focused on the side-chain engineering, i.e., branched ones with shifting a branch point, asymmetric side chain strategy, swapping the position of alkyl side chains, and introduction of polarizable oligo(ethylene glycol) side chains.31,33–42 However, improving the solubility by incorporating insulating aliphatic side chains often deteriorates the electrical properties with suppressed carrier transport in the active layer. As an alternate strategy, a conjugated building block can be designed to improve solution processability by modulating the dihedral angle and torsional energy barrier.43–45 However, this strategy has not been extensively studied. Therefore, the development of a new BDT building block that enables a polymer to be processible in nonhalogenated and nonaromatic green solvents remains a challenge.
In this study, a new BDT-based building block, 4,8-bis(5-(2-ethylhexyl)-3-fluoro-4-hexylthiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene (3-FBDT), was designed and synthesized and incorporated into PBDB-T-2F to yield an eco-friendly (non-aromatic and non-halogenated) solvent-processible photovoltaic copolymer, PBDB-T-2F(3/4). For comparison, a 3-FBDT-based homopolymer (PBDB-T-2F(3)) was also prepared. Compared to the building block of PBDB-T-2F, in 4,8-bis(5-(2-ethylhexyl)-4-fluorothiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene (4-FBDT), an additional alkyl chain was introduced at the 4-position, and the F substituent was moved from the 4- to 3-position in the thiophene side chain. This chemical modification decreased the HOMO level (with improved Voc) and increased the torsional energy barrier between the thiophene and BDT moieties (with a tilted structure), resulting in a dramatic improvement in the solubility of the resulting polymer, even in non-aromatic/non-halogenated solvents (i.e., THF). Taking advantage of the outstanding processability in THF, PBDB-T-2F(3/4)-based devices maintained high PCEs when the solvent was changed from chloroform to THF, in stark contrast to PBDB-T-2F-based devices, which showed a dramatic decrease in the PCE. Charge recombination analysis, transient absorption (TA) spectroscopy tracking charge-carrier dynamics, and grazing incidence wide-angle X-ray scattering (GIWAXS) measurements revealed that the electronic and morphological properties of PBDB-T-2F(3/4) were mainly governed by the PBDB-T-2F blocks. The incorporation of a small amount of 3-FBDT into PBDB-T-2F resulted in a little damage to the optical and photoelectrical properties of the resulting copolymers. Finally, we conducted stability and feasibility tests by fabricating a flexible device, a large-area module, and a semi-transparent OSC device, demonstrating great potential for the eco-friendly fabrication of OSCs using non-aromatic/non-halogenated solvents. Furthermore, a new 3-FBDT building block can be considered as a co-monomer in various BDT-based photovoltaic polymers. This new building block would improve the processability in green solvents without disrupting their photoelectrical properties by incorporating a small number of 3-FBDT units.
Results and discussion
The synthesis route to the new BDT building block, 3-FBDT, is shown in Scheme 1. Using compound 1 as the starting material, [1,3-bis(diphenylphosphino)propane]dichloronickel(II) (NidpppCl2) catalyzed coupling reaction with 2-ethylhexylmagnesium bromide produced compound 2 with two alkyl substituents. Following bromination with N-bromosuccinimide (NBS), compound 4 was obtained in 78% yield via lithium diisopropylamide (LDA)-induced one-pot bromine isomerization and trimethylsilyl protection. Next, fluorination was performed in 80% yield using tert-butyllithium (t-BuLi) and N-fluorobenzenesulfonimide (NFSI). It is to be noted that this reaction should be controlled carefully at −78 °C because the 3-position lithiated thiophene can easily undergo a ring-opening reaction at an elevated temperature. Deprotection of the silyl group was performed to produce 6 using tetrabutylammonium fluoride (TBAF) in THF for 1 h (yield: 80%). Finally, compound 6 was reacted with benzo[1,2-b:4,5-b′]dithiophene-4,8-dione to yield 3-FBDT (56%) via lithiation and addition reactions. The chemical structure of each compound was confirmed by 1H, 13C, and 19F nuclear magnetic resonance (NMR) spectroscopy and matrix-assisted laser desorption time-of-flight mass spectrometry (MALDI-TOF). The detailed synthetic procedures, NMR, and MALDI-TOF data can be found in the ESI (Fig. S1–S19†).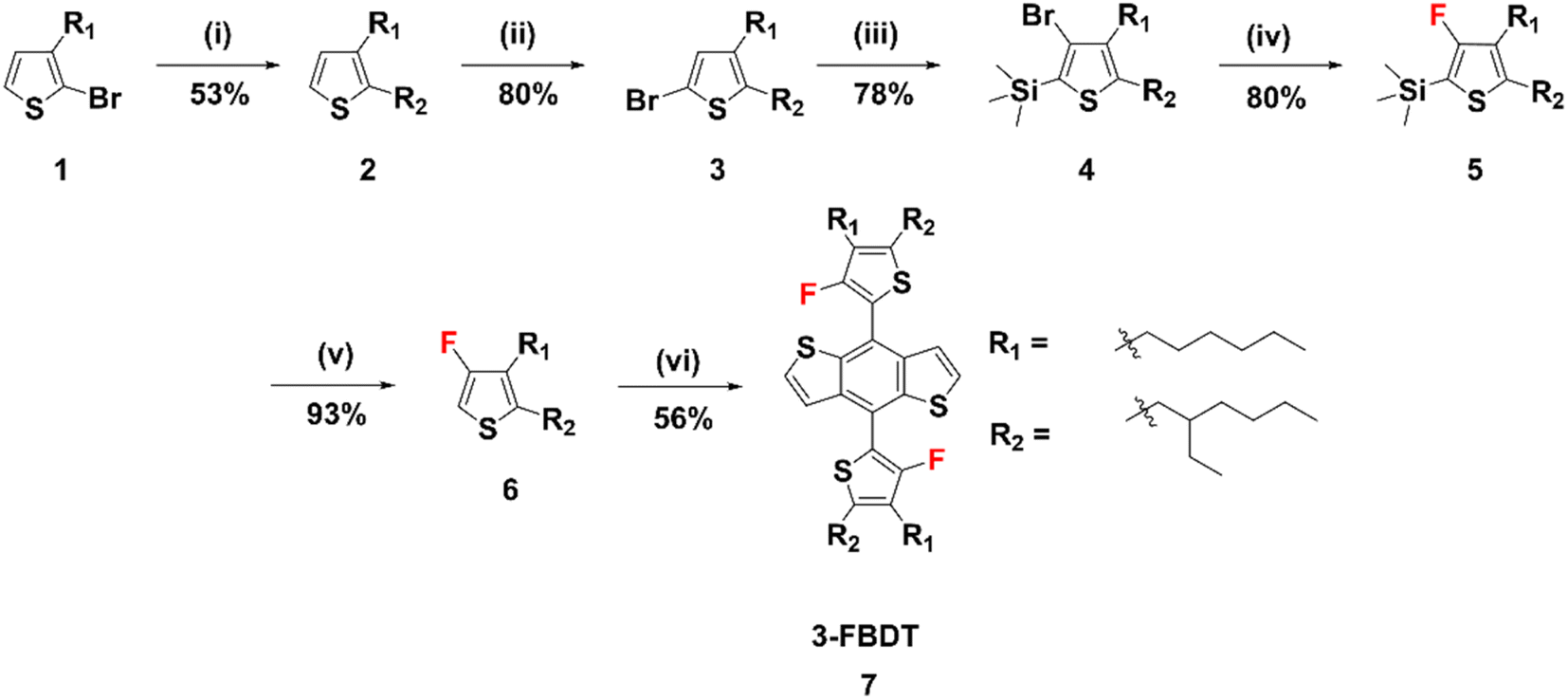 | ||
| Scheme 1 Synthetic scheme of 3-FBDT. Reagents and conditions: (i) 2-ethylhexylmagnesium bromide, NidpppCl2, Et2O, reflux, 12 h, (ii) NBS, DMF, room temp., 1 h, (iii) LDA, Me3SiCl, THF, 0 °C to room temp., 2 h, (iv) t-BuLi, NFSI, THF, −78 °C to room temp., 3 h, (v) TBAF, THF, 1 h, (vi) benzo[1,2-b:4,5-b′]dithiophene-4,8-dione, LDA, THF, 0 °C to room temp., 12 h. | ||
Density functional theory (DFT) calculations were performed using the B3LYP functional and 6-31G* as the basis set to investigate the energy-minimized 3-FBDT and 4-FBDT molecular geometries and frontier molecular orbital (FMO) structures (Fig. 1a). The alkyl chains of the two structures were truncated to methyl groups to reduce the computational burden. As shown in Fig. 1a, 3-FBDT exhibits a larger torsional angle (59.6°) between the BDT core and thiophene side chain than 4-FBDT (56°). Although the torsional angles of 3-FBDT and 4-FBDT are similar, the torsional energy barriers strongly depend on the fluorine position: 22.94 kJ mol−1 for 4-FBDT and 50.35 kJ mol−1 for 3-FBDT (Fig. 1b and c). The F atom at the 3-position of the thiophene side chains significantly increases the torsional barrier because of the additional steric hindrance originating from the fluorine (in thiophene) and hydrogen atoms (in BDT). For C–C bond rotation, steric hindrance needs to be overcome, which is related to the higher torsional energy barrier. Based on the calculated torsional profiles, we assume that the 3-FBDT building block is more likely to form a tilted nonplanar conformation, even in the solid film, which will be discussed in the following section. The different F positions also influence the FMO energy levels. The HOMO and LUMO levels were calculated to be −5.18/−1.34 eV for 3-FBDT and −5.34/−1.61 eV for 4-FBDT. In addition, different electrostatic potential (ESP) distributions were calculated depending on the F positions. The calculations revealed that the negative charges are more localized in the BDT core in 3-FBDT compared to that in 4-FBDT because of the F atoms near the BDT core in the 3-FBDT moiety.
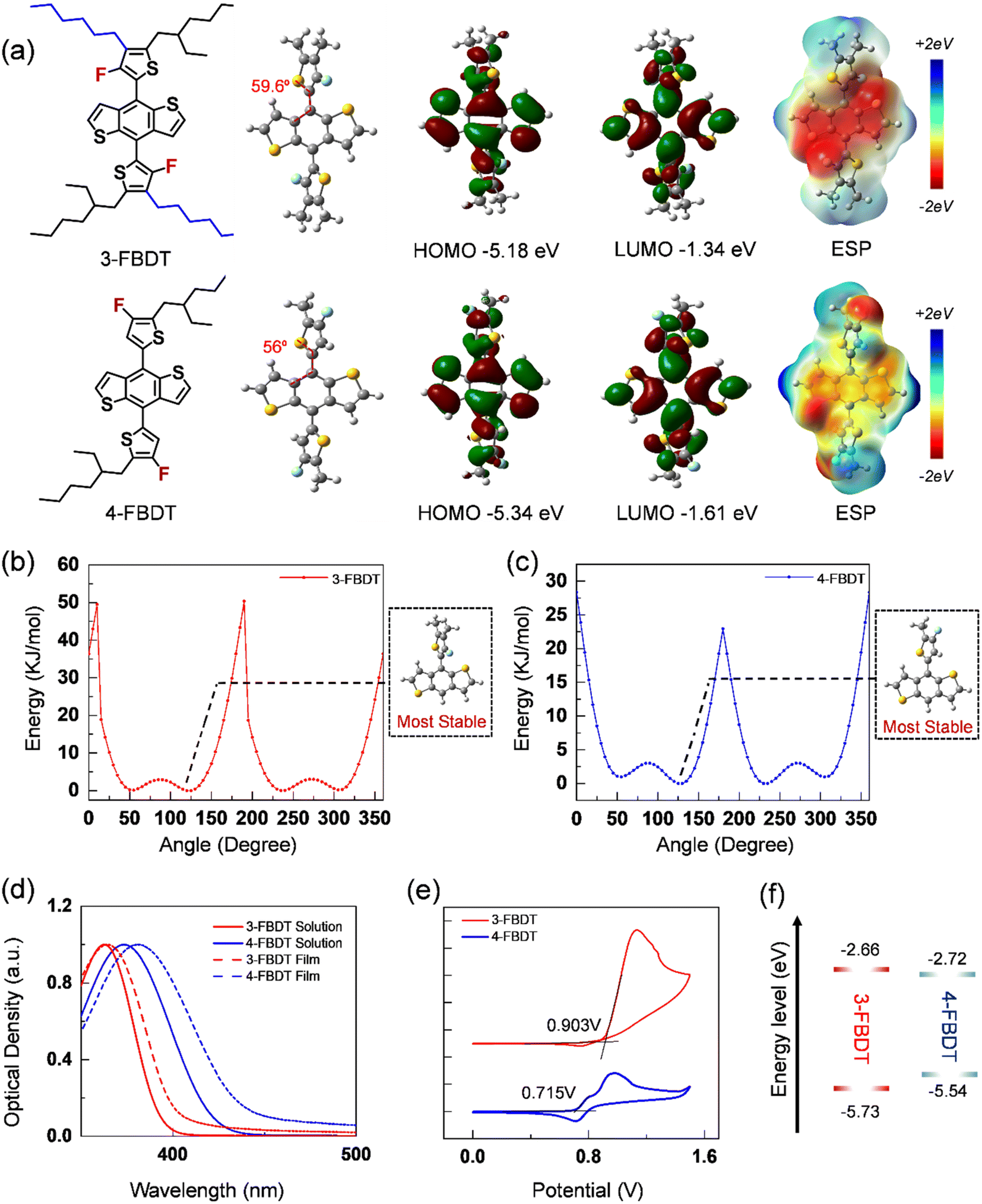 | ||
| Fig. 1 (a) Energy-minimized molecular geometries, FMOs, and electrostatic potential maps of 3-FBDT and 4-FBDT by DFT calculation (B3LYP/6-31G*). Torsional energy profiles of (b) 3-FBDT and (c) 4-FBDT. (d) UV-vis spectra of 3-FBDT and 4-FBDT in solution (chloroform) and in film. (e) Cyclic voltammograms, and (f) energy level diagram. | ||
To investigate the optical properties, we measured the ultraviolet-visible (UV-vis) absorption spectra of 3-FBDT and 4-FBDT in chloroform and in thin films (Fig. 1d). The maximum absorption was measured at λabs = 363 nm for 3-FBDT and λabs = 373 nm for 4-FBDT in the solution (Table 1). The substantially blue-shifted absorption of 3-FBDT compared with that of 4-FBDT may be related to its tilted molecular structure with a larger torsional angle. In the film, red-shifted absorption was measured for 4-FBDT (λabs = 373–381 nm), suggesting stronger intermolecular aggregation in 4-FBDT films with a planarized molecular structure in the solid state. In contrast, there was a negligible shift in the λabs values for 3-FBDT in solution (363 nm) and in the film (364 nm), indicating a tilted structure even in a solid film owing to the large torsional energy barrier. This finding is in good agreement with the aforementioned results. The optical band gaps (Eoptg) of 3-FBDT and 4-FBDT films were determined to be 3.07 and 2.82 eV, respectively, from the absorption edge in film.
| Material | λ abs_Sol. (nm) | λ abs_Film (nm) | E optg (eV) | E HOMO (eV) | E LUMO (eV) |
|---|---|---|---|---|---|
| a In chloroform. b Optical bandgap was estimated from the absorption onset in the film. c HOMO energy level was determined from the oxidation onset potential obtained from the CV measurements. d LUMO energy level was calculated from the HOMO energy level and corresponding optical bandgap. | |||||
| 3-FBDT | 363 | 364 | 3.07 | −5.73 | −2.66 |
| 4-FBDT | 373 | 381 | 2.82 | −5.54 | −2.72 |
| PBDB-T-2F | 615 | 623 | 1.80 | −5.55 | −3.71 |
| PBDB-T-2F(3/4) | 607 | 614 | 1.81 | −5.57 | −3.75 |
| PBDB-T-2F(3) | 528 | 589 | 1.90 | −5.64 | −3.74 |
Cyclic voltammetry (CV) was also carried out to examine the electrochemical properties of 3-FBDT and 4-FBDT using a ferrocene/ferrocenium (Fc/Fc+) redox couple as the internal standard in a 0.1 M Bu4NPF6 acetonitrile solution. Ag/AgNO3 was employed as the reference electrode, a platinum wire as the counter electrode, and a platinum electrode coated with a thin film as the working electrode (Fig. 1e and f). Based on the oxidation onset potential, the HOMO energy levels of 3-FBDT and 4-FBDT were determined to be −5.73 and −5.54 eV, respectively. The LUMO energy level was found to be −2.66 eV for 3-FBDT and −2.72 eV for 4-FBDT from the HOMO value and corresponding optical band gap using the equation: ELUMO = EHOMO + Eoptg. An evidently different trend was observed in the CV measurements compared with that in the DFT calculation, which can be understood by considering that the calculation was performed for the gas phase but CV was performed in a solid state (in film). The larger band gap of 3-FBDT (with a deeper HOMO and higher LUMO than those of 4-FBDT) may originate from its nonplanar chain conformation in the film. In contrast, a planar conformation was expected for the 4-FBDT in the film.
The synthetic schemes for the 3-FBDT-based homopolymer (PBDB-T-2F(3)) and copolymer (PBDB-T-2F(3/4)) are shown in Scheme 2. The 3-FBDT-based monomer (8) was prepared by lithiation of 3-FBDT with LDA at −78 °C for 1 h, followed by stannylation using trimethyltin chloride (yield: 64%). The other monomers, 2,6-bis(trimethytin)-4,8-bis(5-(2-ethylhexyl)-4-fluorothiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene (BDTT-F-Sn) and 1,3-bis(5-bromothiophen-2-yl)-5,7-bis(2-ethylhexyl)benzo[1,2-c:4,5-c′]dithiophene-4,8-dione (BDD), were purchased from Solarmer Materials Inc. The final polymers were obtained by Stille cross-coupling of compound 8, BDTT-F-Sn, and BDD by adjusting the feed ratios using Pd2(dba)3/P(o-tol)3 as a catalyst in toluene under microwave irradiation (80–150 °C) for PBDB-T-2F(3) (71% yield) or Pd(PPh3)4 in toluene at 80–110 °C in an oil bath for PBDB-T-2F(3/4) (yield: 55.1%). The detailed reaction procedures are described in the ESI.† The resulting polymers were purified by sequential Soxhlet extraction with hexane, dichloromethane, tetrahydrofuran, and chloroform. The number-average molecular weights (Mn) and dispersity of the two polymers were determined by gel permeation chromatography using o-dichlorobenzene as the eluent at 80 °C relative to a polystyrene standard. The Mn values of PBDB-T-2F(3) and PBDB-T-2F(3/4) were 26.9 (dispersity 2.31) and 22.6 kDa (3.06), respectively. PBDB-T-2F was purchased from Brilliant Matter (Mn: ∼39 kDa). The thermal behavior of the polymers was investigated using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) under nitrogen (Fig. S20†). All polymers showed high thermal stability with a decomposition temperature (at 5% weight loss) over 350 °C. No clear melting and crystallization transitions were observed in the DSC heating and cooling scans (in the temperature range of 30–310 °C), which may be because of the tilted 3-FBDT moiety, resulting in an amorphous morphology.
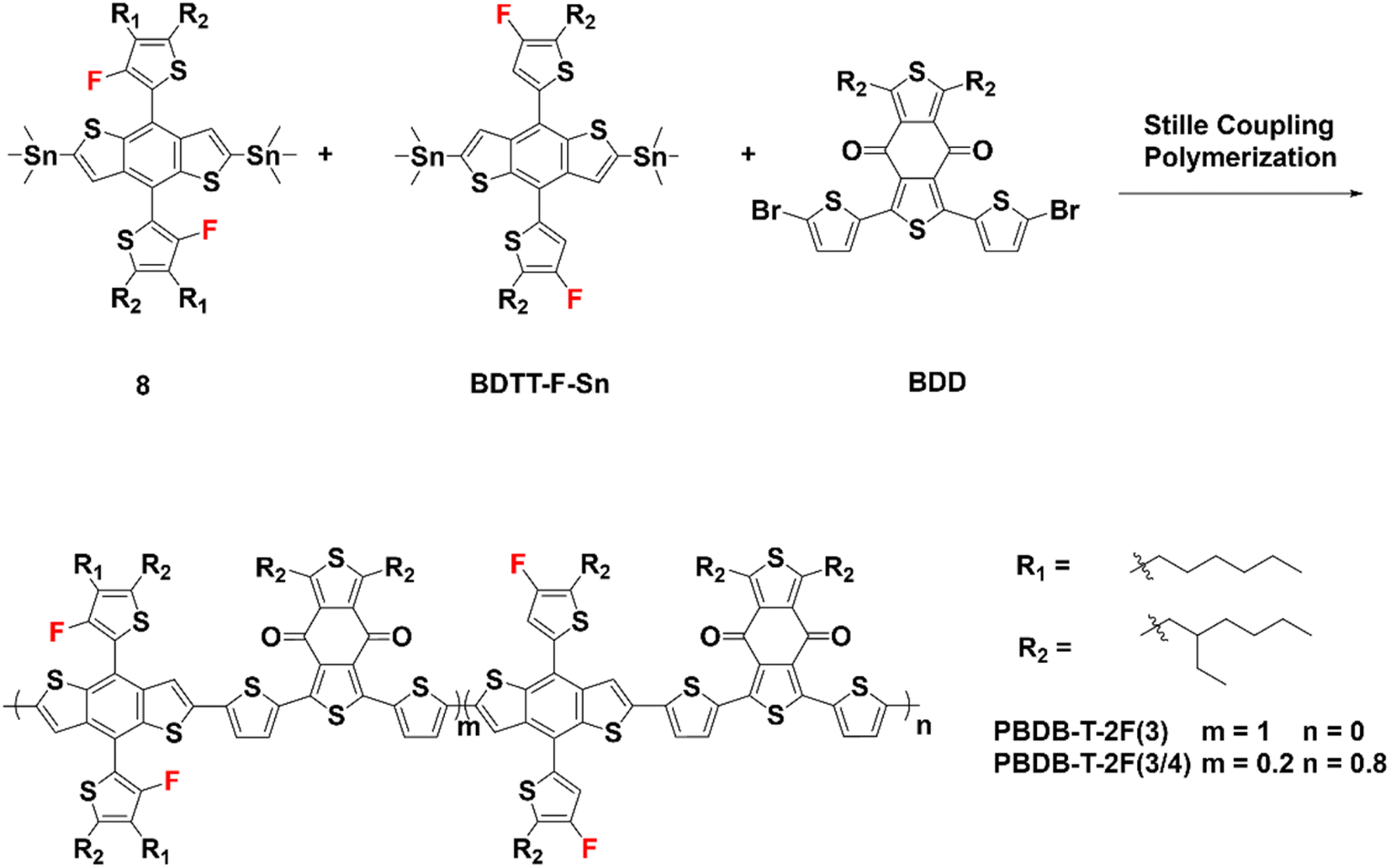 | ||
| Scheme 2 Synthetic scheme to PBDB-T-2F(3) and PBDB-T-2F(3/4). Reagents and conditions: Pd2dba3/P(o-tol)3, toluene, 80–150 °C under microwave irradiation for PBDB-T-2F(3); Pd(PPh3)4, toluene, 80–110 °C, 5.5 h in an oil bath for PBDB-T-2F(3/4). | ||
The normalized UV-vis spectra of PBDB-T-2F(3), PBDB-T-2F(3/4), PBDB-T-2F, and Y6-HU in chloroform and in the film are shown in Fig. 2b and c. The absorption maxima in solution were measured at λabs = 528, 607, and 615 nm for PBDB-T-2F(3), PBDB-T-2F(3/4), and PBDB-T-2F, respectively (Table 1). It is noteworthy to mention that PBDB-T-2F(3) shows a significantly blue-shifted absorption compared to PBDB-T-2F. The incorporation of 3-FBDT into PBDB-T-2F(3/4) also induced a blue shift in λabs relative to PBDB-T-2F. This can be understood in terms of a tilted 3-FBDT unit with a high torsional energy barrier that shows weak aggregation. We also measured the UV-vis spectra of the polymers in chlorobenzene by increasing the solution temperature to 100 °C (Fig. S21†). The resultant spectra indicated a similar aggregation tendency. In the film state, the spectra were substantially red-shifted compared to those in solution for all polymers; the maximum absorption was measured at λabs = 589, 614, and 623 nm for PBDB-T-2F(3), PBDB-T-2F(3/4), and PBDB-T-2F, respectively. The blue-shifted absorption of PBDB-T-2F(3) also suggested that the incorporation of the 3-FBDT moiety can suppress the interchain aggregation of polymers, even in the solid state. The optical band gap was determined to be 1.90, 1.81 and 1.80 eV for PBDB-T-2F(3), PBDB-T-2F(3/4), and PBDB-T-2F, respectively. All polymers showed complementary absorption with that of the Y6-HU acceptor, which is a requisite for the optimal light-harvesting ability of the blend films. We also studied the conformational optimization of PBDB-T-2F(3) and PBDB-T-2F (Fig. S22†). An energy-minimized conformation for the two polymers can be discussed similarly for 3-FBDT and 4-FBDT. The higher torsional energy barrier of 3-FBDT can influence the solubility and interchain aggregation, inducing a nonplanar conformation in PBDB-T-2F(3), even in solid films. The UV-vis spectra of the three polymers can be discussed in terms of the presence of tilted 3-FBDT building blocks in PBDB-T-2F(3) and PBDB-T-2F(3/4). Cyclic voltammetry (CV) was performed to investigate the electrochemical properties of the polymers in the films (Fig. S23†). Compared with PBDB-T-2F (−5.55 eV), lower HOMO energy levels were obtained for PBDB-T-2F(3/4) (−5.57 eV) and PBDB-T-2F(3) (−5.64 eV) (Fig. 2d). The LUMO level was also down-shifted owing to the incorporation of the 3-FBDT unit: −3.74 eV for PBDB-T-2F(3), −3.75 eV for PBDB-T-2F(3/4), and −3.71 eV for PBDB-T-2F. The low-lying HOMO energy level is beneficial for decreasing the sub-bandgap energy loss in OSCs and increasing the resulting Voc of the devices.
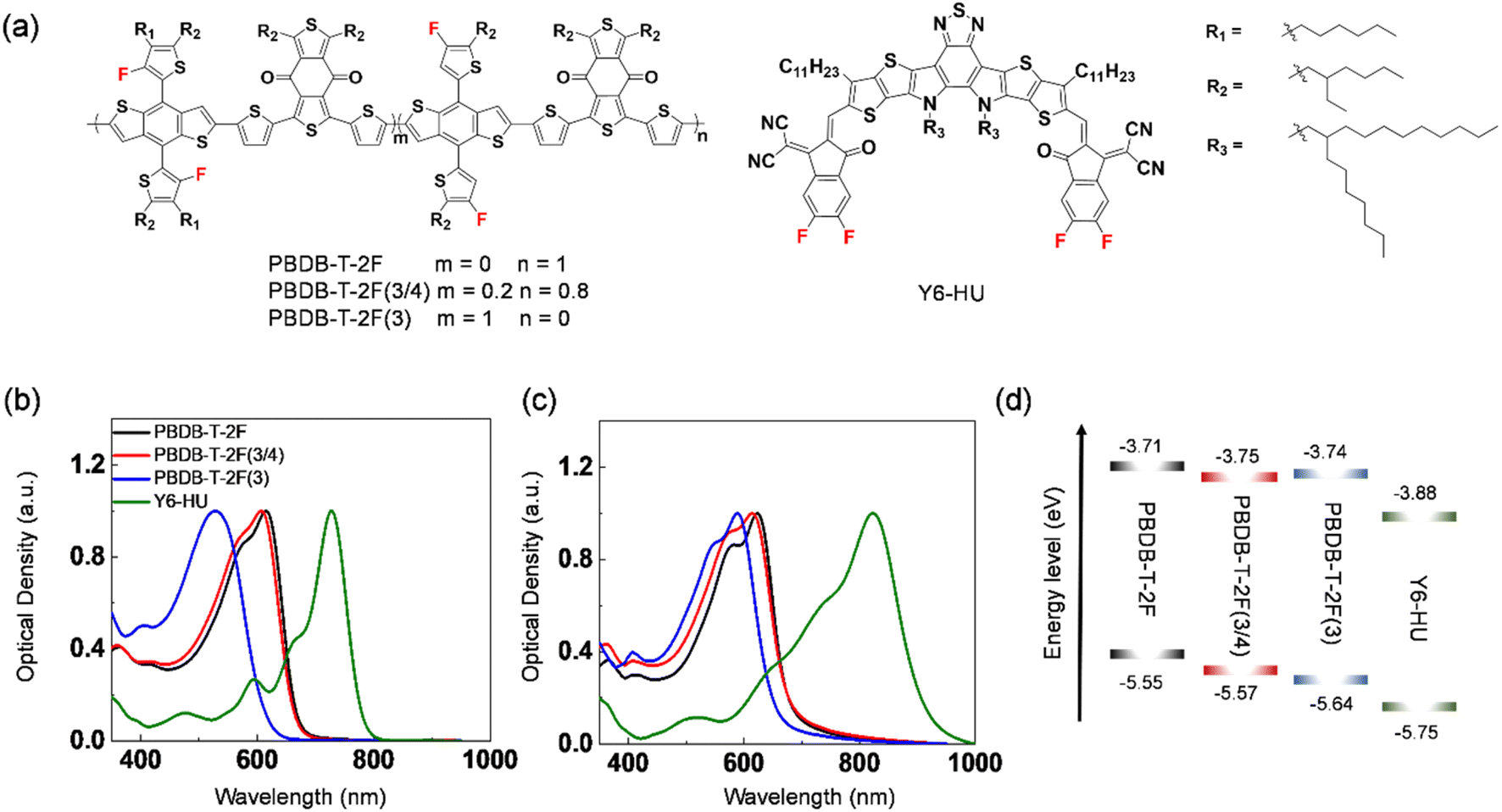 | ||
| Fig. 2 (a) Chemical structures of polymers and Y6-HU. UV-Vis absorption spectra of PBDB-T-2F(3), PBDB-T-2F(3/4), PBDB-T-2F, and Y6-HU (b) in solution and (c) in film. (d) Energy-level diagram. | ||
To investigate the effect of the different building blocks in the donor polymers on the photovoltaic performance, OSCs with a conventional device structure of ITO/PEDOT:PSS/polymer:Y6-HU/ZnO/Al were fabricated using CF as the processing solvent. The detailed procedures for device fabrication are described in the Experimental section (ESI†), and the corresponding photovoltaic parameters are summarized in Fig. 3a and Table 2. When CF was used as the processing solvent, the PBDB-T-2F:Y6-HU device delivered the best PCE of 17.46% with a higher fill factor (FF) and short-circuit current density (Jsc) than PBDB-T-2F(3/4):Y6-HU and PBDB-T-2F(3):Y6-HU. This is in good agreement with the external quantum efficiency (EQE) spectra, as shown in Fig. 3b. The optimal PCEs of the PBDB-T-2F(3/4):Y6-HU and PBDB-T-2F(3):Y6-HU devices were determined to be 14.30 and 9.39%, respectively. The higher Voc of the PBDB-T-2F(3)-based OSCs originated from the deeper HOMO of PBDB-T-2F(3). Despite having the highest Voc, the lowest PCE was measured for PBDB-T-2F(3):Y6-HU, with much smaller Jsc and FF values than the other two OSCs (PBDB-T-2F(3/4):Y6-HU and PBDB-T-2F:Y6-HU). To elucidate the difference in Jsc and FF characteristics, we measured the hole and electron mobilities (μh and μe) of pristine donor and blend films by the space-charge-limited-current (SCLC) method.46 As shown in Fig. 3c, the μh of pristine PBDB-T-2F, PBDB-T-2F(3/4) and PBDB-T-2F(3) is 8.30 × 10−4, 5.11 × 10−4 and 0.39 × 10−4 cm2 V−1 s−1, respectively. The lower hole mobility of PBDB-T-2F(3) must be related to the tilted chain conformation and reduced aggregation properties. As seen in Fig. S24 and Table S2,† the PBDB-T-2F(3):Y6-HU blend film also exhibits the lowest μh and μe values of 7.25 × 10−6 and 2.94 × 10−4 cm2 V−1 s−1, which leads to an unbalanced charge transport (μh/μe = 0.025). To further study the impact of the incorporation of new building moieties on the charge generation and transport behaviors, the photocurrent versus effective voltage (Jphvs. Veff) characteristics were studied (Fig. 3d–f and Table S3†). From the Jphvs. Veff measurements, the exciton dissociation efficiency (Pdiss) and charge collection efficiency (Pcoll) were determined.47Veff is Va − V0 where Va is the applied voltage and V0 is the voltage where Jph is zero. Pdiss is defined as Pdiss = Jph/Jsat under short-circuit conditions, where Jsat is the saturated photocurrent density. The Pdiss values were calculated to be 99.2, 95.3, and 90.3% for PBDB-T-2F-, PBDB-T-2F(3/4)-, and PBDB-T-2F(3)-based OSCs, respectively. Furthermore, the Pcoll value (defined by Pcoll = Jph/Jsat at the maximum power point) was the lowest for PBDB-T-2F(3):Y6-HU (65.2%), and the highest for PBDB-T-2F:Y6-HU (87.6%). The Pcoll value of PBDB-T-2F(3/4):Y6-HU was found to be 75.8%. To investigate the charge recombination behavior, the light intensity (Plight)-dependent Jsc and Voc values were measured. Jsc was proportional to Plightα. For all three blends, the power index α was near unity, indicating insignificant bimolecular recombination. From the plots of Voc as a function of ln(Plight), the slope (S) for PBDB-T-2F:Y6-HU was determined to be 1.03 kTq−1 (k is the Boltzmann constant, T the absolute temperature, and q the elementary charge). This was the smallest among those of the three samples, suggesting a suppressed monomolecular or trap-assisted recombination. The incorporation of 3-FBDT (into PBDB-T-2F(3/4) and PBDB-T-2F(3)) increased the S value, indicating enhanced trap-assisted charge recombination.
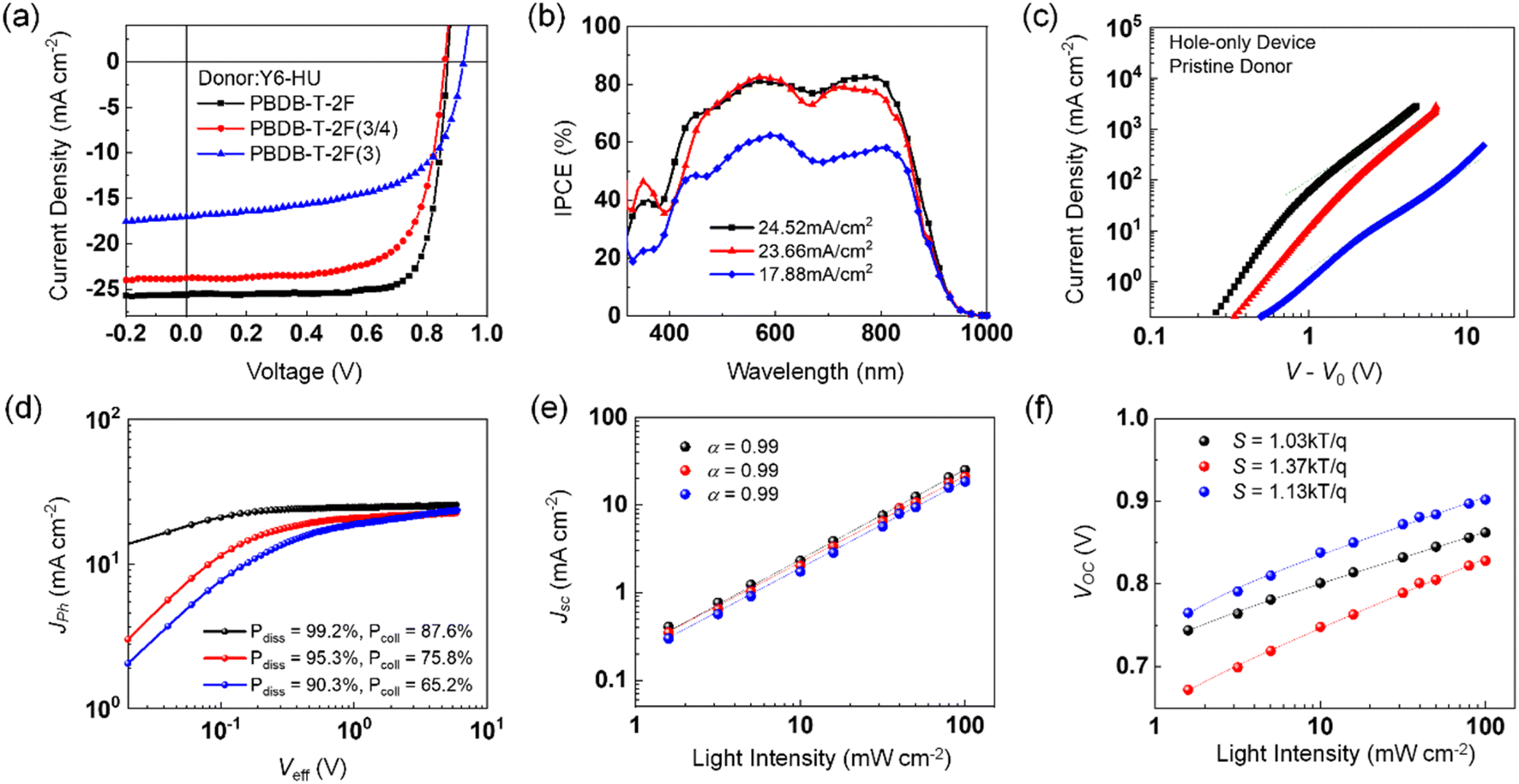 | ||
| Fig. 3 (a) J–V and (b) EQE curves of donor:Y6-HU-based OSCs fabricated with CF. (c) SCLC curves of hole-only devices based on donor polymer neat films. (d) Photocurrent density (Jph) versus effective voltage (Veff) characteristics. Light intensity dependent (e) Jsc and (f) Voc measurements. | ||
| Blend | Solvent | J sc [mA cm−2] | V oc [V] | FF | PCE [%] |
|---|---|---|---|---|---|
| a Average values and standard deviations were determined from 10 independent measurements. | |||||
| PBDB-T-2F:Y6-HU | CF | 25.91 (25.26 ± 0.53)a | 0.87 (0.864 ± 0.005) | 0.78 (0.77 ± 0.01) | 17.46 (16.88 ± 0.55) |
| THF | 15.15 (14.40 ± 0.57) | 0.86 (0.863 ± 0.002) | 0.75 (0.72 ± 0.02) | 9.73 (8.98 ± 0.54) | |
| PBDB-T-2F(3/4):Y6-HU | CF | 23.78 (23.25 ± 0.49) | 0.86 (0.855 ± 0.007) | 0.70 (0.69 ± 0.01) | 14.30 (13.62 ± 0.43) |
| THF | 23.28 (22.21 ± 0.51) | 0.86 (0.860 ± 0.002) | 0.69 (0.71 ± 0.01) | 13.86 (13.51 ± 0.15) | |
| PBDB-T-2F(3):Y6-HU | CF | 17.05 (17.46 ± 0.72) | 0.92 (0.919 ± 0.004) | 0.60 (0.58 ± 0.02) | 9.39 (9.26 ± 0.14) |
| THF | 17.58 (17.09 ± 1.05) | 0.90 (0.899 ± 0.002) | 0.57 (0.56 ± 0.02) | 9.03 (8.56 ± 0.40) | |
2D grazing-incidence wide angle X-ray scattering (GIWAXS) measurements were performed to study the interchain packing of pristine donor and blend films fabricated using CF as the processing solvent (Fig. 4 and Table S4†). As shown in 2D-GIWAXS images and line-cut profiles, the PBDB-T-2F, PBDB-T-2F(3/4) and PBDB-T-2F(3) pristine films display a face-on dominant orientation with a (100) lamellar peak at qxy = 0.29–0.30 Å−1 in the in-plane (IP) direction, together with a strong (010) π–π stacking peak at qz = 1.65 (dπ–π spacing = 3.81 Å), 1.64 (3.83 Å) and 1.52 Å−1 (4.13 Å) in the out-of-plane (OOP) direction, respectively. With an increase in the content of the 3-FBDT moiety in the polymeric backbone, dπ–π spacing gradually increases, which originates from the tilted 3-FBDT with a large torsional barrier, suppressing the cofacial packing interactions. The corresponding crystal coherence length (CCL) was measured based on the OOP (010) peak. A similar trend was observed: the CCL010 values were determined to be 20.50, 18.57, and 16.42 Å for PBDB-T-2F, PBDB-T-2F(3/4), and PBDB-T-2F(3), respectively.48 A pronounced face-on packing was also observed for all three polymers:Y6-HU blend films with an OOP (010) scattering at qz = 1.66–1.67 Å−1 and an IP (100) peak at qxy = 0.29–0.30 Å−1. In the GIWAXS spectra of the blend films, a similar (010) peak (in the OOP direction) was observed for the three blends, indicating a strong contribution to the (010) π–π stacking signal from the Y6-HU domains. Overall, the tilted (with a higher torsional energy barrier) 3-FBDT building block hindered the interchain packing and resulting charge transport, however the effect on the film morphology and photoelectrical properties of PBDB-T-2F(3/4) was not significant.
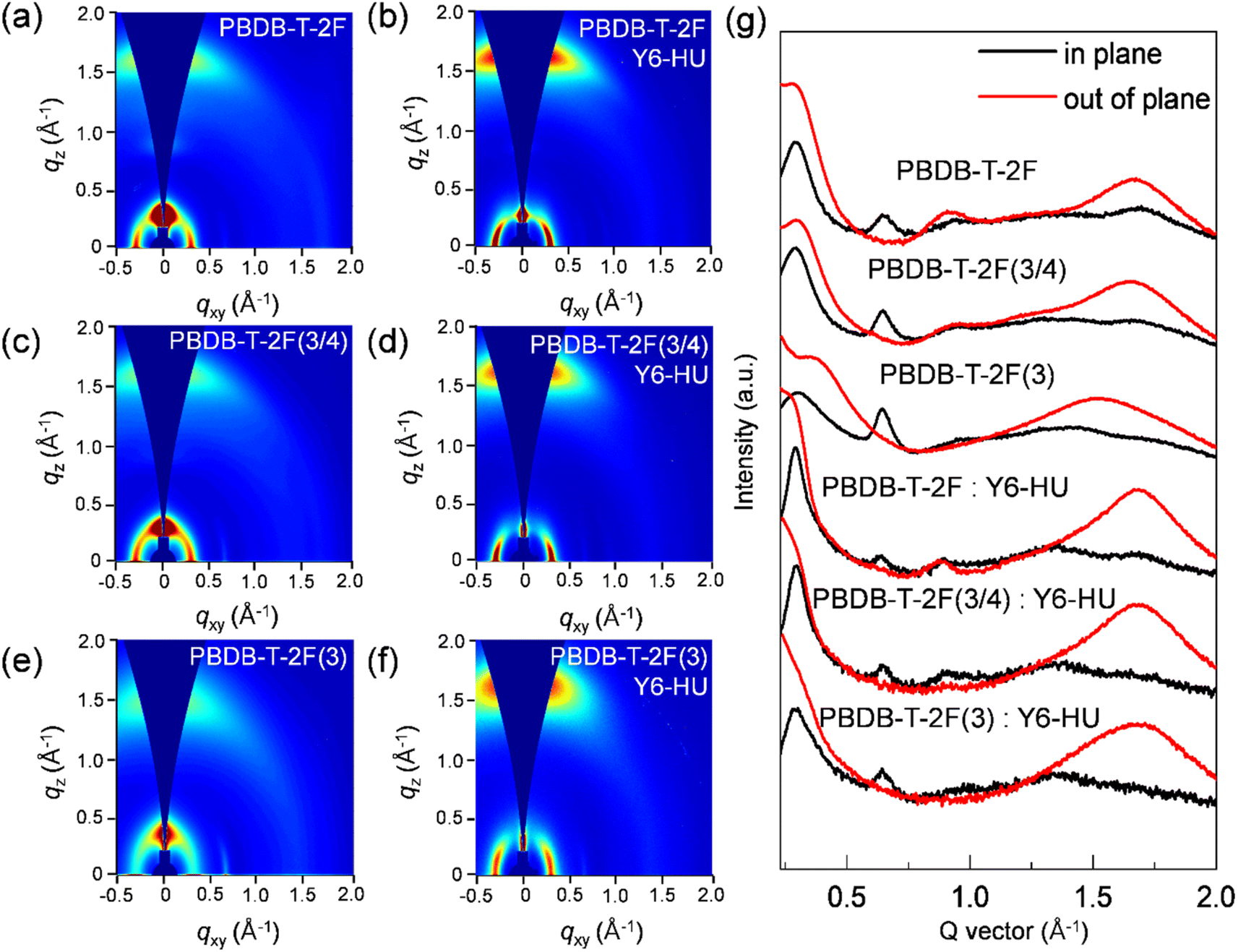 | ||
| Fig. 4 2D GIWAXS images of pristine and blend films of (a and b) PBDB-T-2F, (c and d) PBDB-T-2F(3/4), and (e and f) PBDB-T-2F(3). (g) Corresponding in-plane and out-of-plane line-cuts. | ||
To verify the influence of different building blocks on the charge separation (CS) dynamics of the donor polymer:Y6-HU blends, we conducted the transient absorption (TA) spectroscopy measurements. Hole transfer in the pristine acceptor and its blends was monitored by selectively exciting Y6-HU at 750 nm (Fig. 5a, b, S25 and S26†). Upon excitation, the TA spectra of neat Y6-HU and its blend films showed a negative absorption band centered at approximately 840 nm, which corresponds to the ground-state bleach (GSB) of Y6-HU. Similar to previous studies, in neat Y6-HU, the majority of excited species (excitons or carriers) are relaxed within a nanosecond.49,50 For the blends, in addition to the long-lived GSB of Y6-HU, an additional negative absorption band emerged centered at approximately 630 nm, which corresponds to the GSB of the donor polymer. The emergence of the GSB of the donor polymer is indicative of hole transfer that occurs to form the CS state.49–51 To gain insight into the hole-transfer efficiency, we constructed lifetime-associated spectra in the full spectral and temporal ranges by global-lifetime analysis.51,52Fig. 5c and d show the lifetime-associated spectra of Y6-HU and PBDB-T-2F(3/4):Y6-HU, respectively. For satisfactory global-lifetime analysis, at least three lifetime components were introduced for Y6-HU and the blends. The three lifetime-associated spectra of Y6-HU showed similar spectral features, indicating that the three different conformers relaxed on different timescales. The two fast components (∼0.8 and 140 ps) may originate from relaxation via conical intersection.53
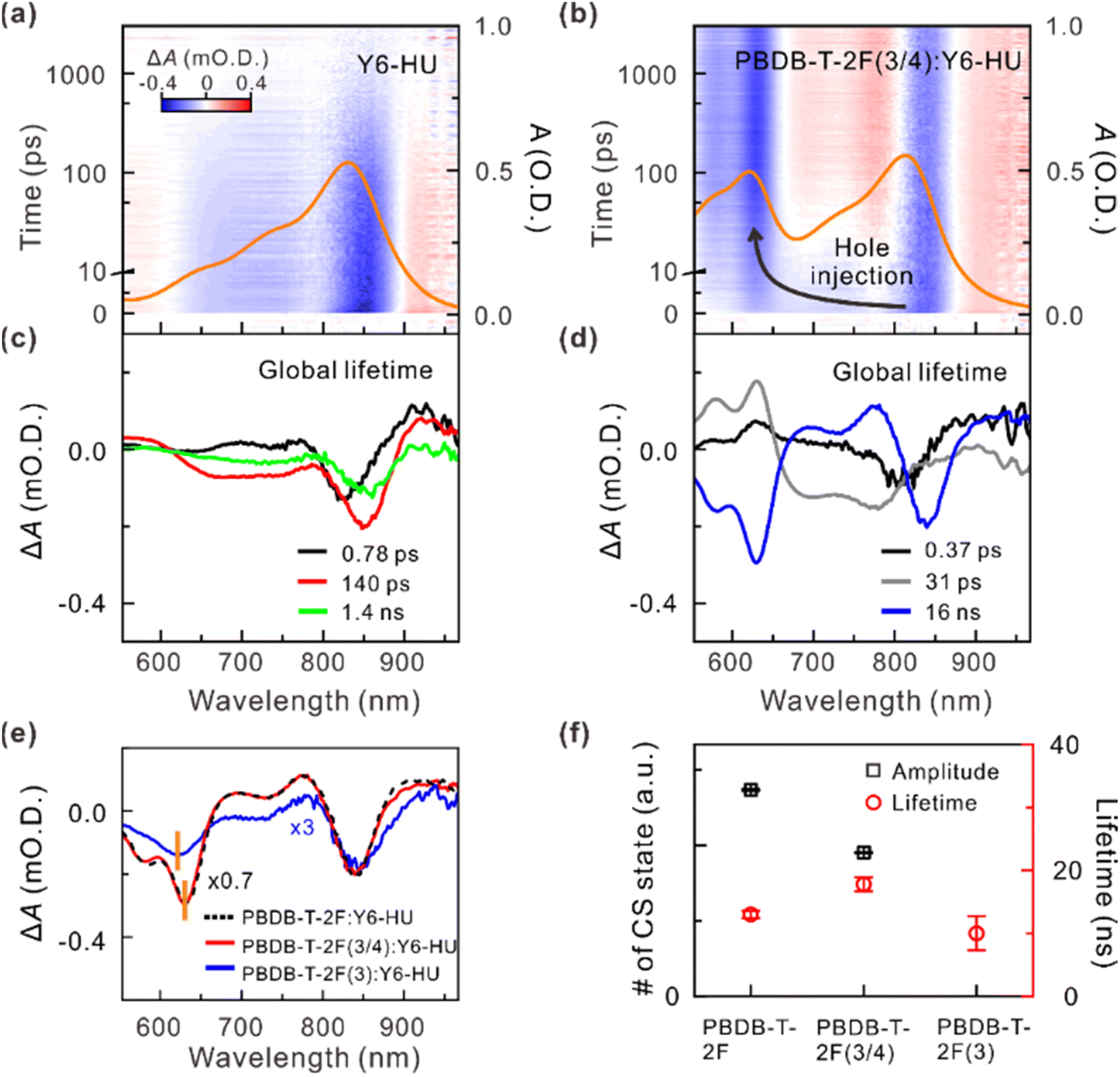 | ||
| Fig. 5 Hole-transfer and charge-separation dynamics. Two-dimensional TA maps of (a) neat Y6-HU and (b) PBDB-T-2F(3/4):Y6-HU films upon excitation at 750 nm. The steady-state absorption spectrum of each sample is also presented (orange solid line). Lifetime-associated spectra of (c) Y6-HU and (d) PBDB-T-2F(3/4):Y6-HU by the global-lifetime analysis. Global lifetimes are given in the panels. (e) Spectra of the long-lived global-lifetime components (CS states) of PBDB-T-2F:Y6-HU (black, dashed line), PBDB-T-2F(3/4):Y6-HU (red, solid line), and PBDB-T-2F(3):Y6-HU (blue, solid line). The lifetime-associated spectra are scaled for comparison. (f) Observed amplitude (black, square) and lifetime (red, circle) of the CS state in polymer:Y6-HU blends. | ||
The lifetime-associated spectra of the blends clearly showed the characteristic spectral features of the CS state.49–51 The spectral characteristic feature of neat Y6-HU disappeared entirely for PBDB-T-2F(3/4):Y6-HU and PBDB-T-2F:Y6-HU. In the CS state, the holes and electrons reside in the valence band of the donor polymer and the conduction band of Y6-HU, respectively. After a hole in Y6-HU is transferred to the donor polymer to form the CS state, each carrier leads to GSB at each domain, which recovers on the same timescale (a few tens of nanoseconds). In the lifetime-associated spectra of PBDB-T-2F(3/4):Y6-HU, the mirror features of the 0.37 and 31 ps components to the spectrum of the CS state (16 ns) below 700 nm indicate that the hole transfer process occurs on those timescales. For PBDB-T-2F(3):Y6-HU, the lifetime-associated spectra of the 0.53 ps, 15 ps, and 220 ps components show the characteristic spectral features of Y6-HU (Fig. S26†). Thus, it was inferred that the carriers were not fully transferred to PBDB-T-2F(3). In contrast, the lifetime-associated spectrum of the CS state of PBDB-T-2F(3/4):Y6-HU matched well with that of PBDB-T-2F:Y6-HU (Fig. 5e). This suggests that exciton dissociation and hole transfer are mainly governed by the PBDB-T-2F blocks in the PBDB-T-2F(3/4) copolymer. Finally, we quantitatively compared the amount and lifetime of the CS state by analyzing the kinetic profiles at the peak of the GSB of PBDB-T-2F (Fig. 5f and S27, Table S5†). The extent of the formation of the CS state was observed to be slightly lesser for PBDB-T-2F(3/4):Y6-HU than for PBDB-T-2F:Y6-HU. Interestingly, the TAS data showed that relaxation of the CS state was significantly retarded for PBDB-T-2F(3/4):Y6-HU compared to PBDB-T-2F:Y6-HU.
Next, we fabricated conventional OSCs by replacing the processing solvent CF by THF, which is non-halogenated and non-aromatic. As shown in Fig. S28,† PBDB-T-2F has poor solubility in THF, showing large aggregates in both the solution and film of PBDB-T-2F:Y6-HU. In contrast to PBDB-T-2F, PBDB-T-2F(3/4) and PBDB-T-2F(3) showed dramatically improved solubility in THF upon incorporation of the 3-FBDT moiety into the polymer backbone. To elucidate the relationship between polymer structures and their solubility, Hansen solubility parameters of PBDB-T-2F and PBDB-T-2F(3) were calculated using the group contribution method.54 Hansen solubility parameters (HSPs) considered three kinds of intermolecular interactions: dispersion interaction (δD), permanent dipolar interaction (δP), and hydrogen bonding interaction (δH). As summarized in Table S8,† the calculated δD, δP, and δH parameters are 24.85, 2.50, and 2.93 MPa1/2 for PBDB-T-2F and 23.18, 1.94, and 2.59 MPa1/2 for PBDB-T-2F(3). In addition, the relative distance in Hansen space (Ra) refers to the similarity in solubility between two substances, and can be used to roughly estimate the solubility of materials in a certain solvent. The detailed calculation method and parameters are described in the ESI (Tables S6–S9†). Compared to PBDB-T-2F, PBDB-T-2F(3) showed lower Ra values in different solvents, including both polar and non-polar solvents, indicating that PBDB-T-2F(3) is more likely to dissolve in solvents than PBDB-T-2F.54,55 For example, the Ra value between the donor polymer and THF was calculated to be 17.18 and 14.35 MPa1/2 for PBDB-T-2F and PBDB-T-2F(3), respectively. This result suggests that structural modification by incorporation of 3-FBDT improves the solution processability of the resulting polymers and broadens the choice of device fabrication solvents for eco-friendly PSCs.
It is noteworthy that the incorporation of 3-FBDT enhanced the solvent processability dramatically, which enables the successful fabrication of OSCs using non-halogenated and non-aromatic processing solvents (i.e., THF). The detailed device fabrication conditions using THF as the processing solvent are described in the ESI.† The optimal J–V characteristics are displayed in Fig. 6a, and the corresponding photovoltaic parameters are summarized in Tables 2 and S10–S12.† PBDB-T-2F:Y6-HU showed a significant drop in PCE (9.73%) compared to that of the CF-processed sample (17.46%). In contrast, owing to their superior solubility in THF, the PBDB-T-2F(3/4) and PBDB-T-2F(3) OSCs showed PCEs of 13.86 and 9.03%, respectively, showing a negligible PCE drop. The PCE ratios of the THF-/CF-processed OSCs (PCETHF/PCECF) were 55.7, 96.9, and 96.2% for PBDB-T-2F, PBDB-T-2F(3/4), and PBDB-T-2F(3), respectively. Similar results have been reported by designing copolymeric structures to realize efficient OSCs using nonhalogenated and/or nonaromatic solvents. For example, Cui et al. synthesized a series of copolymers by incorporating an ester group-substituted thiophene (EST) unit into PM6, showing a PCE over 15% using a low-toxic nonhalogen/nonaromatic mixture of THF and N-methyl pyrrolidone.43 A modulation of intermolecular aggregation and solution processibility was successfully demonstrated by preparing terpolymer acceptors based on indacenodithiophene dicyanindenone (IDTIC), showing PCEs up to 11.94% by blending with PM6 using o-xylene as a processing solvent.44 Wu et al. reported a non-halogenated or nonaromatic solvent-processible single component block copolymer named PBDB-T-b-PYT, and similar PCEs of ∼11% were obtained using THF or toluene (as a processing solvent), compared to the device processed using CF.45 However, the copolymer strategy for improving solution processibility may significantly change the electronic structure and photoelectrical properties of the homopolymer by combining a differently structured comonomer. Unlike previous studies, we incorporated a BDT building block (3-FBDT by tailoring the positions and number of alkyl and F substituents and adjusting the torsional energy barrier) into a BDT-based polymer, improving the processability in green solvents with negligibly disrupting its photoelectrical properties.
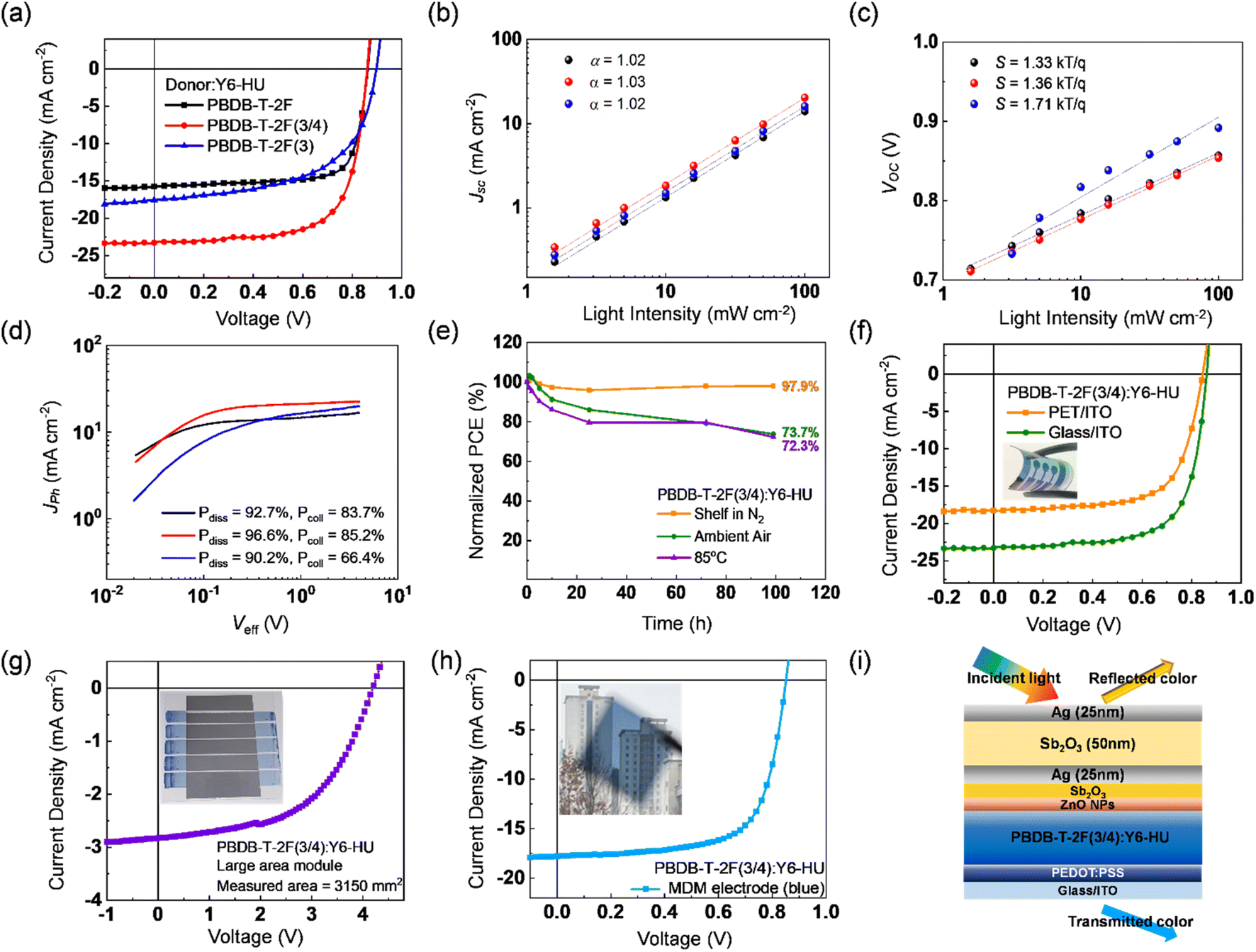 | ||
| Fig. 6 (a) J–V curves of donor:Y6-HU-based OSCs fabricated with THF. Light intensity dependent (b) Jsc and (c) Voc characteristics. (d) Jphvs. Veff curves and (e) device stability (including shelf-storage stability, air stability, and thermal stability). J–V characteristics of (f) flexible, (g) large area module, and (h) blue-colored semi-transparent PBDB-T-2F(3/4):Y6-HU OSCs. (i) Device structure of a blue-colored semi-transparent OSC with an MDM top electrode. | ||
To investigate the charge generation, transport, and recombination behaviors of OSCs processed by THF, Jphvs. Veff and the light intensity-dependent Jsc and Voc characteristics were measured (Fig. 6b–d and Table S13†). The PBDB-T-2F(3/4)-based OSC exhibited the highest Pdiss of 96.6% and Pcoll of 85.2%, showing a different trend from that of the CF-processed devices. This result emphasizes the importance of the solution processability and the consequent blend morphology. Both negligible bimolecular recombination (with the power index α of ∼1) and suppressed trap-assisted recombination (S value of 1.36 kTq−1) were also confirmed for PBDB-T-2F(3/4):Y6-HU by its Jscvs. Plight and Vocvs. ln(Plight) characteristics.
The THF-processed blend film morphology was characterized using atomic force microscopy (AFM) and GIWAXS measurements. The height images show that the root-mean-square averaged surface roughness (Rq) gradually decreases from 2.27 nm for PBDB-T-2F to 0.56 nm for PBDB-T-2F(3) as the solubility improves (Fig. S29†). In the phase and 3D images, the PBDB-T-2F blend films showed significant aggregation, whereas a smooth surface was observed for the PBDB-T-2F(3/4) and PBDB-T-2F(3) blend films. The GIWAXS data (Fig. S30 and Table S14†) demonstrates a dominant face-on orientation for all three blends, exhibiting lamellar scattering in the IP direction and a (010) π–π stacking peak along the OOP direction. By incorporating the 3-FBDT units in the PBDB-T-2F backbone, the solution processibility in THF improved and optimal film morphology was obtained for PBDB-T-2F(3/4):Y6-HU to show the higher CCL value of 25.24 Å compared to that of PBDB-T-2F (21.64 Å) and PBDB-T-2F(3) (15.76 Å) blends based on the OOP (010) scattering peak. Consequently, the favorable film morphology of PBDB-T-2F(3/4):Y6-HU with a face-on ordered smooth surface and a larger CCL facilitates charge generation and transport. The combination of the 3-FBDT and 4-FBDT units in the same polymeric backbone successfully yielded both pronounced solution processability (in THF) and photoelectrical properties.
To demonstrate the feasibility of eco-friendly solvent-processed PBDB-T-2F(3/4), stability tests were conducted under various conditions. Furthermore, flexible devices, large-area modules, and colorful semi-transparent OSCs were fabricated. First, we measured the shelf storage stability, ambient air stability, and thermal stability of PBDB-T-2F(3/4):Y6-HU, as shown in Fig. 6e. The unencapsulated device was kept in a glove box filled with N2 for the shelf storage test and it retained 97.9% of the initial PCE after 100 h. Ambient air stability was also tested at 25 °C and 25% (relative humidity) without encapsulation. The PBDB-T-2F(3/4)-based device showed 73.7% of the initial performance after 100 h. Similarly, the device under continuous thermal treatment at 85 °C for 100 h maintained 72.3% of the initial efficiency. The good stability characteristics may be related to the suppressed excessive phase separation owing to the weak interchain aggregation caused by the tilted 3-FBDT moiety.56 Next, a flexible device with a structure of PET/ITO/PEDOT:PSS/PBDB-T-2F(3/4):Y6-HU/ZnO/Al was fabricated (Fig. 6f and Table S15†). The flexible device showed a Jsc of 18.26 mA cm−2, Voc of 0.85 V, FF of 0.67, and PCE of 10.39%. Compared with the device with a glass substrate, Jsc decreased, which is related to the low transmittance of ITO/PET substrates. Besides, the Voc and FF are comparable to that of the rigid devices (0.86 V and 0.69, respectively), which means the formation of a smooth surface helps to properly contact with the charge transporting layer. Despite the Jsc loss, it retained 75% of its PCE compared to the OSC on glass.
As illustrated in Fig. S31a,† a large-area module was successfully fabricated with a device architecture of ITO/ZnO/PBDB-T-2F(3/4):Y6-HU/MoO3/Ag. The bar coating method was used to coat the active layer at a blade speed of 10 mm s−1. The module (with an active area of 38.43 cm2) consists of series-connected 4 sub-cells (8.19 cm2) and one sub-cell (5.67 cm2), as presented in Fig. S31b.†Fig. 6g shows the J–V characteristics measured using a mask with an area of 31.50 cm2. The large-area PBDB-T-2F(3/4):Y6-HU module exhibited a PCE of 6.26% with a Voc of 4.21, Jsc of 2.83 mA cm−2, and FF of 0.53 (Table S16†). The Voc value of 4.21 V, close to five times the Voc of the single cell (0.86 V) was obtained, indicating that 5 sub-cells are successfully connected in series. It is because the thickness gradient between the start and end points of the coating is overcome by acquiring the film uniformity suppressing excessive molecular aggregation.57 Afterward, to demonstrate the aesthetic property, the blue-colored semi-transparent OSC was fabricated with incorporation of a Fabry–Pérot etalon-type metal/dielectric/metal (MDM) electrode.58,59 The fine-modulation of transmitted light using the MDM electrodes has been reported by us previously.59 As depicted in Fig. 6i, Sb2O3 (2 nm)/Ag (25 nm)/Sb2O3 (50 nm)/Ag (25 nm) was employed as the cathode. The blue transparent electrode was chosen because the absorption of PBDB-T-2F(3/4):Y6-HU is inherently low in the range of 400–500 nm. Therefore, we can minimize the absorption loss by adopting the blue MDM electrode and sufficiently utilize light absorption in the 500–900 nm range. The blue colored semi-transparent OSC exhibited 10.31% PCE with a Jsc of 17.79 mA cm−2, Voc of 0.85 V, and FF of 0.68 (Table S17†). The Voc and FF values were almost similar to those of the device with the Al electrode, but only Jsc slightly decreased. As shown in Fig. S32,† the EQE difference between the OSCs of MDM and Al electrodes was clearly measured in the 400–500 nm region, which originates from the absorption loss because of the light transmitted via the MDM electrodes. The picture in Fig. 6h suggests the potential of semi-transparent OSCs for aesthetic and colorful solar windows.60–62 Overall, non-halogenated and non-aromatic THF-processed PBDB-T-2F(3/4) successfully demonstrated its great potential as an eco-friendly photoactive donor for real applications of OSCs as a clean energy source.
Conclusions
We synthesized a new BDT building block (3-FBDT) and its copolymer PBDB-T-2F(3/4) to fabricate eco-friendly solvent-processed OSCs. To control the solution processability in eco-friendly solvents, side-chain engineering has been widely studied, but little attention has been paid to the torsional property control of conjugated main chains. Compared with the building block (4-FBDT) of PBDB-T-2F, swapping the F position and incorporating an additional hexyl substituent produced a tilted structure with an increased torsional energy barrier. The incorporation of 3-FBDT into a representative donor polymer, PBDB-T-2F, produced a highly soluble copolymer, PBDB-T-2F(3/4), in THF, which showed a comparably high PCE of 13.86% with the CF-processed device (14.3%) by fabricating OSCs using a non-halogenated and non-aromatic processing solvent. In contrast, PBDB-T-2F showed a significant PCE drop (17.46 to 9.73%) because of the serious aggregation that occurred from replacing CF with THF as a processing solvent. Charge recombination analysis, charge-carrier dynamics by TA analysis, and GIWAXS measurements suggested that the electronic and morphological properties of PBDB-T-2F(3/4) were mainly governed by the majority moiety of the PBDB-T-2F blocks. Furthermore, combining the excellent electronic properties and unique solubility, the PBDB-T-2F(3/4):Y6-HU blend was successfully employed to fabricate a flexible device, large-area module, and blue-colored semi-transparent solar cell using THF as the processing solvent. In conclusion, a new 3-FBDT building block and its copolymer PBDB-T-2F(3/4) may be a useful and effective strategy to improve the solution processability while maintaining its photoelectrical properties. The fine adjustment of interchain aggregation by modulating the 3-FBDT content can control the solvent processability and broaden the selection of solvents. Our approach can be extended to various high-performance photovoltaic polymers to advance the fabrication of eco-friendly OSCs for mass production.Experimental
Full details of experimental procedures including the material synthesis, device fabrication and characterization can be found in the ESI.†Author contributions
Z. W. and H. L. synthesized the material. S. Y. J. and J. V. S. K. analyzed the optical and electronic properties of the materials. H. W. C. and W. L. performed the fabrication and characterization of organic solar cell devices. O. H. K. and W. W. P. conducted the TAS experiments. J. Y. K. and H. Y. W. supervised this research project and revised the manuscripts. All authors discussed the results and participated in preparing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT (NRF-2022R1A4A1033247, 2019R1A2C2085290, 2020M3H4A3081814, 2019R1A6A1A11044070).References
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed.
- W. Gao, F. Qi, Z. Peng, F. R. Lin, K. Jiang, C. Zhong, W. Kaminsky, Z. Guan, C.-S. Lee, T. J. Marks, H. Ade and A. K. Y. Jen, Adv. Mater., 2022, 34, 2202089 CrossRef CAS PubMed.
- Y. Wei, Z. Chen, G. Lu, N. Yu, C. Li, J. Gao, X. Gu, X. Hao, G. Lu, Z. Tang, J. Zhang, Z. Wei, X. Zhang and H. Huang, Adv. Mater., 2022, 34, 2204718 CrossRef CAS PubMed.
- H. Chen, H. Liang, Z. Guo, Y. Zhu, Z. Zhang, Z. Li, X. Cao, H. Wang, W. Feng, Y. Zou, L. Meng, X. Xu, B. Kan, C. Li, Z. Yao, X. Wan, Z. Ma and Y. Chen, Angew. Chem., Int. Ed., 2022, 61, e202209580 CAS.
- Y. Shi, Y. Chang, K. Lu, Z. Chen, J. Zhang, Y. Yan, D. Qiu, Y. Liu, M. A. Adil, W. Ma, X. Hao, L. Zhu and Z. Wei, Nat. Commun., 2022, 13, 3256 CrossRef CAS PubMed.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- X. Kong, C. Zhu, J. Zhang, L. Meng, S. Qin, J. Zhang, J. Li, Z. Wei and Y. Li, Energy Environ. Sci., 2022, 15, 2011–2020 RSC.
- R. Sun, W. Wang, H. Yu, Z. Chen, X. Xia, H. Shen, J. Guo, M. Shi, Y. Zheng, Y. Wu, W. Yang, T. Wang, Q. Wu, Y. Yang, X. Lu, J. Xia, C. J. Brabec, H. Yan, Y. Li and J. Min, Joule, 2021, 5, 1548–1565 CrossRef CAS.
- Z. Liao, D. Hu, H. Tang, P. Huang, R. Singh, S. Chung, K. Cho, M. Kumar, L. Hou, Q. Chen, W. Yu, H. Chen, K. Yang, Z. Kan, F. Liu, Z. Xiao, G. Li and S. Lu, J. Mater. Chem. A, 2022, 10, 7878–7887 RSC.
- J. Wang, Y. Cui, Y. Xu, K. Xian, P. Bi, Z. Chen, K. Zhou, L. Ma, T. Zhang, Y. Yang, Y. Zu, H. Yao, X. Hao, L. Ye and J. Hou, Adv. Mater., 2022, 34, 2205009 CrossRef CAS PubMed.
- B. Huang, X. Deng, H. Jin, K. Liu, S. Chen, Z. Ma, J. Oh, C. Yang, J. Liu and L. Chen, J. Mater. Chem. A, 2022, 10, 18714–18722 RSC.
- P. Liu, K. Zhang, F. Liu, Y. Jin, S. Liu, T. P. Russell, H.-L. Yip, F. Huang and Y. Cao, Chem. Mater., 2014, 26, 3009–3017 CrossRef.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Adv. Mater., 2014, 26, 1118–1123 CrossRef CAS PubMed.
- L. Dou, J. Gao, E. Richard, J. You, C.-C. Chen, K. C. Cha, Y. He, G. Li and Y. Yang, J. Am. Chem. Soc., 2012, 134, 10071–10079 CrossRef CAS PubMed.
- Y. Huang, F. Liu, X. Guo, W. Zhang, Y. Gu, J. Zhang, C. C. Han, T. P. Russell and J. Hou, Adv. Energy Mater., 2013, 3, 930–937 CrossRef CAS.
- X. Guo, M. Zhang, L. Huo, F. Xu, Y. Wu and J. Hou, J. Mater. Chem., 2012, 22, 21024–21031 RSC.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. Tan and J. Hou, Macromolecules, 2012, 45, 9611–9617 CrossRef CAS.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- Q. Fan, Q. Zhu, Z. Xu, W. Su, J. Chen, J. Wu, X. Guo, W. Ma, M. Zhang and Y. Li, Nano Energy, 2018, 48, 413–420 CrossRef CAS.
- Q. Wu, W. Wang, Z. Chen, X. Xia, M. Gao, H. Shen, H. Zhu, X. Lu, L. Ye, J. Xia and J. Min, J. Mater. Chem. C, 2022, 10, 1850–1861 RSC.
- H. Tan, W. Zhang, P. Zhang, X. Lv, A. Hexig, J. Huang, B. Li and C. Zhan, Sol. RRL, 2022, 6, 2200147 CrossRef CAS.
- J. Lee, J. W. Kim, S. A. Park, S. Y. Son, K. Choi, W. Lee, M. Kim, J. Y. Kim and T. Park, Adv. Energy Mater., 2019, 9, 1901829 CrossRef.
- D. Wang, G. Zhou, Y. Li, K. Yan, L. Zhan, H. Zhu, X. Lu, H. Chen and C.-Z. Li, Adv. Funct. Mater., 2022, 32, 2107827 CrossRef CAS.
- L. Hong, H. Yao, Z. Wu, Y. Cui, T. Zhang, Y. Xu, R. Yu, Q. Liao, B. Gao, K. Xian, H. Y. Woo, Z. Ge and J. Hou, Adv. Mater., 2019, 31, 1903441 CrossRef PubMed.
- L. Ye, Y. Xiong, Z. Chen, Q. Zhang, Z. Fei, R. Henry, M. Heeney, B. T. O'Connor, W. You and H. Ade, Adv. Mater., 2019, 31, 1808153 CrossRef PubMed.
- C. Xie, S. Liang, G. Zhang and S. Li, Polymers, 2022, 14, 4229 CrossRef CAS PubMed.
- C. Kim, H. Kang, N. Choi, S. Lee, Y. Kim, J. Kim, Z. Wu, H. Y. Woo and B. J. Kim, J. Mater. Chem. C, 2020, 8, 15224–15233 RSC.
- S. Lee, Y. Kim, Z. Wu, C. Lee, S. J. Oh, N. T. Luan, J. Lee, D. Jeong, K. Zhang, F. Huang, T.-S. Kim, H. Y. Woo and B. J. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 45038–45047 CrossRef CAS PubMed.
- S. Li, H. Zhang, S. Yue, X. Yu and H. Zhou, Nanotechnology, 2022, 33, 072002 CrossRef CAS PubMed.
- B. Kang, Z. Wu, M. J. Kim, H. Y. Woo and J. H. Cho, Chem. Mater., 2020, 32, 1111–1119 CrossRef CAS.
- X. Chen, X. Liu, M. A. Burgers, Y. Huang and G. C. Bazan, Angew. Chem., Int. Ed., 2014, 53, 14378–14381 CrossRef CAS PubMed.
- B. Fan, L. Ying, Z. Wang, B. He, X.-F. Jiang, F. Huang and Y. Cao, Energy Environ. Sci., 2017, 10, 1243–1251 RSC.
- Z. Li, L. Ying, P. Zhu, W. Zhong, N. Li, F. Liu, F. Huang and Y. Cao, Energy Environ. Sci., 2019, 12, 157–163 RSC.
- Y. Qin, L. Ye, S. Zhang, J. Zhu, B. Yang, H. Ade and J. Hou, J. Mater. Chem. A, 2018, 6, 4324–4330 RSC.
- Y. Chen, Y. Cui, S. Zhang and J. Hou, Polym. Chem., 2015, 6, 4089–4095 RSC.
- C. Liao, M. Zhang, X. Xu, F. Liu, Y. Li and Q. Peng, J. Mater. Chem. A, 2019, 7, 716–726 RSC.
- Y. Do, H. Park, T. Gokulnath, K. Sung, H.-Y. Park and S.-H. Jin, Macromol. Res., 2022, 30, 183–189 CrossRef CAS.
- Z. Zheng, O. M. Awartani, B. Gautam, D. Liu, Y. Qin, W. Li, A. Bataller, K. Gundogdu, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1604241 CrossRef PubMed.
- W. Zhao, S. Zhang, Y. Zhang, S. Li, X. Liu, C. He, Z. Zheng and J. Hou, Adv. Mater., 2018, 30, 1704837 CrossRef PubMed.
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Xu, K. Xian, B. Gao, J. Qin, J. Zhang, Z. Wei and J. Hou, Adv. Mater., 2019, 31, 1808356 CrossRef PubMed.
- H. Liu, L. Wang, H. Liu, M. Guan, C.-J. Su, U. S. Jeng, B. Zhao, C. Weng, K. You and X. Lu, Chem. Eng. J., 2022, 429, 132407 CrossRef CAS.
- Y. Wu, J. Guo, W. Wang, Z. Chen, Z. Chen, R. Sun, Q. Wu, T. Wang, X. Hao, H. Zhu and J. Min, Joule, 2021, 5, 1800–1815 CrossRef CAS.
- P. W. M. Blom, M. J. M. de Jong and M. G. van Munster, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, R656–R659 CrossRef CAS.
- V. D. Mihailetchi, L. J. A. Koster, J. C. Hummelen and P. W. M. Blom, Phys. Rev. Lett., 2004, 93, 216601 CrossRef CAS PubMed.
- T. Liu, Z. Luo, Q. Fan, G. Zhang, L. Zhang, W. Gao, X. Guo, W. Ma, M. Zhang, C. Yang, Y. Li and H. Yan, Energy Environ. Sci., 2018, 11, 3275–3282 RSC.
- Y. W. Lee, J. Yeop, J. Y. Kim and H. Y. Woo, Macromol. Res., 2021, 29, 871–881 CrossRef CAS.
- R. Wang, C. Zhang, Q. Li, Z. Zhang, X. Wang and M. Xiao, J. Am. Chem. Soc., 2020, 142, 12751–12759 CrossRef CAS PubMed.
- B. Lee, S. Kim, H.-W. Nho, J. Oh, G. Park, M. Jeong, Y. Cho, S. M. Lee, O.-H. Kwon and C. Yang, Adv. Energy Mater., 2021, 11, 2102594 CrossRef CAS.
- H.-W. Nho, W.-W. Park, B. Lee, S. Kim, C. Yang and O.-H. Kwon, Phys. Chem. Chem. Phys., 2022, 24, 1982–1992 RSC.
- V. C. Stephanie, A. C. Sue and R. Garry, Proc. SPIE, 2005, 5938, 59380J Search PubMed.
- D. W. Van Krevelen and K. Te Nijenhuis, in Properties of Polymers, Elsevier, Amsterdam, 4th edn, 2009, pp. 189–227 Search PubMed.
- C. Lee, H. R. Lee, J. Choi, Y. Kim, T. L. Nguyen, W. Lee, B. Gautam, X. Liu, K. Zhang, F. Huang, J. H. Oh, H. Y. Woo and B. J. Kim, Adv. Energy Mater., 2018, 8, 1802674 CrossRef.
- J. Xin, J. Feng, B. Lin, H. B. Naveed, J. Xue, N. Zheng and W. Ma, Small, 2022, 18, 2200608 CrossRef CAS PubMed.
- X. F. Liao, Q. Xie, Y. X. Guo, Q. N. He, Z. Chen, N. Yu, P. P. Zhu, Y. J. Cui, Z. F. Ma, X. B. Xu, H. M. Zhu and Y. W. Chen, Energy Environ. Sci., 2022, 15, 384 RSC.
- S. Song, H. W. Cho, J. Jeong, Y. J. Yoon, S. Y. Park, S. Song, B. H. Woo, Y. C. Jun, B. Walker and J. Y. Kim, Sol. RRL, 2020, 4, 2000201 CrossRef CAS.
- H. R. Yeom, S. Song, S. Y. Park, H. S. Ryu, J. W. Kim, J. Heo, H. W. Cho, B. Walker, S.-J. Ko, H. Y. Woo and J. Y. Kim, Nano Energy, 2020, 77, 105146 CrossRef CAS.
- T. Liu, Y. Zheng, Y. Xu, X. Liu, C. Wang, L. Yu, M. Fahlman, X. Li, P. Murto, J. Chen and X. Xu, Nano Energy, 2022, 103, 107776 CrossRef CAS.
- B. Lee, L. Lahann, Y. Li and S. R. Forrest, Sustainable Energy Fuels, 2020, 4, 5765–5772 RSC.
- S. Guan, Y. Li, K. Yan, W. Fu, L. Zuo and H. Chen, Adv. Mater., 2022, 34, 2205844 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta10030d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |