Calmodulin–CaMKII mediated alteration of oxidative stress: interplay of the cAMP/PKA–ERK 1/2-NF-κB–NO axis on arsenic-induced head kidney macrophage apoptosis
Chaitali Banerjeea, Ambika Singhb, Rajagopal Ramanb and Shibnath Mazumder*a
aImmunobiology Laboratory, Department of Zoology, University of Delhi, Delhi 110 007, India. E-mail: shibnath1@yahoo.co.in; Tel: +91-11-27667212, Ext 203
bGut Biology Laboratory, Department of Zoology, University of Delhi, Delhi 110 007, India
First published on 31st July 2013
Abstract
Using the head kidney macrophages (HKM) from catfish (Clarias batrachus) we earlier demonstrated the role of calcium (Ca2+) and its dependent neutral protease calpain in arsenic-induced apoptosis. Here, we report the role of the CaM–CaMKII axis as an initiator of the process. With the help of specific assay kits and inhibitors we document the pro-apoptotic role of CaM and CaMKII in arsenic-induced HKM apoptosis. CaM-induced CaMKII activity influenced superoxide ion production in exposed cells with a consequent increase in intracellular cAMP levels. Using H-89, the specific inhibitor for PKA, we show for the first time the pro-apoptotic role of the cAMP/PKA pathway in arsenic-induced HKM apoptosis. We report the cAMP/PKA pathway to be critical for initiating downstream activation of MAPKs, namely ERK 1/2. The superoxide ions generated due to arsenic-stress also induce NF-κB activation in HKM. Inducible NOS activity and consequent NO production were evident in the exposed HKM and our study implicates the involvement of ERK 1/2 and NF-κB in the process. Arsenic exposure alters mitochondrial membrane potential, releases cytochrome C and activates caspase-9 leading to caspase-3 mediated apoptosis of HKM. Our findings, thus, provide insight into the underlying mechanism of arsenic toxicity and indicate that HKM could serve as an important in vitro model for immunotoxicity assays.
Introduction
Arsenic, a well known environmental toxicant, ranks first in the priority list of the Agency for Toxic Substances and Disease Registry. Inorganic arsenicals are more toxic and exist as trivalent arsenite and pentavalent arsenate in nature. Both forms are soluble over a wide pH range; arsenate predominates under aerobic conditions while under anaerobic conditions the more toxic arsenite predominates.1Arsenic has been implicated in multi-systemic health effects besides its role as a carcinogen.2 Being immunotoxic, arsenic interferes with macrophagic differentiation and functioning,3 and suppresses T-4 and B-cell proliferation5 posing important immunological concerns. The pro-apoptotic role of arsenic has also been documented suggesting its chemo-therapeutic potential.1
Arsenic is toxic to fish. It affects hatching in Oryzias latipes and causes DNA damage in Channa punctatus,6 and induces histological and ultra-structural changes in liver in C. batrachus.7 It has been suggested that exposure to non-lethal concentrations of arsenic can alter different haematological parameters in fish inducing time-dependent and tissue-specific changes in fish B- and T-cell functioning, rendering them immune-compromised and susceptible to pathogenic infections;8 establishing fish as a successful model to explore arsenic immunotoxicity.9 In recent years, we have demonstrated head kidney macrophages (HKM) from C. batrachus as an alternative model system to study arsenic-toxicity.7,10
Calcium, a ubiquitous second messenger, controls a broad range of cellular functions including growth, differentiation and death. Arsenic-induced alteration in intracellular Ca2+ levels has been reported to induce the apoptosis of several cell types, although the mechanisms are not well understood. The frequency and magnitude of Ca2+ flux elicited by various signalling ligands are sensed by cytoplasmic sensors and specifically directed to different signalling pathways by activating different Ca2+-dependent enzymes. Calmodulin (CaM) senses increases in the intracellular Ca2+ concentration and undergoes conformational changes on Ca2+-binding. Ca2+–CaM is a ubiquitous signal that regulates diverse cellular responses including activation of CaM-dependent kinases (CaMK), calcineurin, calpains and transcription factors such as NFAT.11 Recent reports have shown that the number of enzymes instrumental to apoptosis induction is CaM-dependent.12 Although the role of Ca2+ and its dependent protease calpain in arsenic-induced HKM apoptosis has been observed,13 many of the potential Ca2+-dependent mechanisms are yet to be studied in detail in arsenic-pathology, especially in fish.
The redox state of a cell is important in determining its susceptibility to different stimuli. Arsenic disturbs cellular oxidation and reduction equilibria through complex redox reactions with endogenous oxidants and cellular antioxidant systems.14 Superoxide ions, generated in response to arsenic exposure, have been implicated in a multitude of cellular functions including apoptosis.15 A crucial intracellular signalling event is the release of cyclic adenosine monophosphate (cAMP). The role of cAMP in ROS generation16 and vice versa has been observed.17 Elevated cAMP activates PKA which has been implicated in initiating a cascade of signalling molecules with anti- and pro-apoptotic effects in different cell types.18 In view of the arsenic-effect, it has been suggested that cAMP/PKA is critical in modulating the apoptosis of several cancer cell lines, although the mechanisms are less well understood.19
Arsenic-induced changes in cellular redox status have a profound impact on several transcription factors including NF-κB, which consequently deranges cell signaling and alters gene expression systems. From the available literature NF-κB appears to be a stress response transcription factor, which regulates the expression of a variety of downstream target genes, including those involved in pro- and anti-apoptosis.20
Nitric oxide (NO) is a free radical synthesized from L-arginine during its conversion to citrulline where NO is released as a by-product via the action of NO synthases (NOS). In macrophages NO has been identified as a pleiotropic messenger molecule regulating a variety of diverse cellular functions including apoptosis.21 Arsenicals have been reported to exert their toxicity by modulating NO production.22 In addition, ROS inhibitors also reduce NO levels by preventing iNOS expression through blockade of NF-κB activation.23 Inhibitors to different MAPKs can also block iNOS expression to different extents in macrophages, on stimulation with different stimuli.24
The activation of the mitochondria dependent or intrinsic pathway of apoptosis is an important signalling pathway in arsenic-induced cell death.25 The process involves alteration of mitochondrial membrane potential (ψm) and release of pro-apoptotic factors like cytochrome C (cyt C) in the cytosol leading to the formation of apoptosome to initiate the activation of the intrinsic caspase, caspase-9, which in turn recruits and activates effector caspases-3/7 to execute apoptotic death.26 It was observed that arsenic-exposure led to mitochondrial aggregation, leading to the suggestion that arsenic can either directly or via generated ROS affect the mitochondrial inner transmembrane potential promoting apoptosis.
Here, we investigated the role of the CaM–CaMKII axis in arsenic-induced HKM apoptosis and its relation to superoxide ion generation; looked for the possible mechanism of cAMP/PKA activation and defined the role of downstream effector molecules like ERK 1/2, iNOS, NF-κB and mitochondria mediated activation of the caspase-9 pathway in instigating the death pathway.
Results
Arsenic-induced CaM–CaMKII activation contributes to HKM apoptosis
We earlier demonstrated that arsenic-induced alterations in intracellular Ca2+ levels are an early event leading to HKM apoptosis.13 CaM is an important downstream target of Ca2+ and we were interested in investigating its role in the arsenic-induced apoptosis of HKM. At the onset, we looked for arsenic-induced changes in CaM expression in HKM. The HKM were incubated with arsenic and the changes in CaM concentrations measured at different time points using a specific CaM-assay kit. We noted maximum CaM activity at 4 h of exposure and thereafter it started declining and reached basal levels following 16 h of exposure (Fig. 1A). | ||
| Fig. 1 Arsenic exposure leads to CaM-CaMKII activation. (A) HKM were exposed to arsenic and the amount of CaM in the cell lysates measured at indicated time interval using the EIA assay kit. (B) HKM were pre-treated with CMZ and then exposed to arsenic, and CaM levels measured in the cell lysates 4 h post incubation. (C) HKM were pre-treated separately with or without CMZ, KN-93, KN-92 and STO-609 and then exposed to arsenic, and apoptosis studied 24 h post incubation using Hoechst 33342 staining and measuring the caspase-3 activity. (D) HKM were pre-treated separately with or without CMZ, KN-93 and KN-92 and then exposed to arsenic, and CaMKII levels measured in the cell lysates 24 h post incubation. *P < 0.05 vs. HKM; #P < 0.05 vs. HKM + As. HKM, control head kidney macrophage; HKM + As, HKM exposed to arsenic; HKM + CMZ + As, HKM pre-treated with CMZ for 1 h prior to arsenic exposure; HKM + KN-93 + As, HKM pre-treated with KN-93 for 1 h prior to arsenic exposure; HKM + KN-92 + As, HKM pre-treated with KN-92 for 1 h prior to arsenic exposure; HKM + STO-609 + As, HKM pre-treated with STO-609 for 2 h prior to arsenic exposure. CMZ, CaM antagonist; KN-93, CaMKII antagonist; KN-92, inactive analogue of KN-93; STO-609, CaMKK antagonist. The concentration of chemicals used was as mentioned in the Materials and methods section. | ||
Next, to determine the significance of CaM in the process, HKM were pre-treated with CaM-specific inhibitor CMZ then exposed to arsenic and CaM concentration and apoptosis checked in the exposed cells. It was observed that pre-treatment with CMZ significantly inhibited CaM activity (P < 0.05; Fig. 1B) and HKM apoptosis as evident from Hoechst 33342 and AV-PI staining and caspase-3 activity (Fig. 1C and 2), suggesting arsenic-induced CaM activation to be pro-apoptotic in HKM.
 | ||
| Fig. 2 Arsenic-induced apoptosis of HKM. The HKM were pre-incubated for definite time intervals with the indicated inhibitors and then exposed to arsenic, and apoptosis measured 24 h post incubation using Annexin V-FITC-propidium iodide staining. The early apoptotic cells stain only with AV and not with PI (AV+PI−). The late apoptotic cells undergo a gradual loss in their membrane integrity and stain with PI (AV+PI+), while the necrotic cells stain only with PI (AV−PI+). *P < 0.05 vs. HKM; #P < 0.05 vs. HKM + As. HKM, control head kidney macrophage; HKM + As, HKM exposed to arsenic; HKM + CMZ + As, HKM pre-treated with CMZ for 1 h prior to arsenic exposure; HKM + KN-93 + As, HKM pre-treated with KN-93 for 1 h prior to arsenic exposure; HKM + KN-92 + As, HKM pre-treated with KN-92 for 1 h prior to arsenic exposure; HKM + STO-609 + As, HKM pre-treated with STO-609 for 2 h prior to arsenic exposure; HKM + H-89 + As, HKM pre-treated with H-89 for 1 h prior to arsenic exposure; HKM + L-Nil + As, HKM pre-treated with L-Nil for 1 h prior to arsenic exposure; HKM + NF-κBi + As, HKM pre-treated with NF-κBi for 1 h prior to arsenic exposure; HKM + Z-LEHD-FMK + As, HKM pre-treated with Z-LEHD-FMK for 1 h prior to arsenic exposure. CMZ, CaM antagonist; KN-93, CaMKII antagonist; KN-92, inactive analogue of KN-93; STO-609, CaMKK antagonist; H-89, PKA antagonist; L-Nil, iNOS specific inhibitor; NF-κBi, NF-κB activation inhibitor; Z-LEHD-FMK, caspase-9 inhibitor. The concentration of chemicals used was as mentioned in the Materials and methods section. | ||
The relative involvement of two CaM dependent kinases, CaMKK and CaMKII, on arsenic-induced HKM apoptosis was studied. CaMKK and CaMKII were selected as both are conserved, well characterised, have wide tissue distribution including macrophages and work through distinct pathways regulating diverse biological functions.27 The HKM were pre-treated separately with KN-93, the CaMKII specific inhibitor, and STO-609, specific for CaMKK, and then incubated with arsenic and apoptosis was studied following 24 h of incubation. It was observed that pre-treatment with KN-93 significantly reduced (P < 0.05) arsenic-induced HKM apoptosis as evident from Hoechst 33342 and AV-PI staining and caspase-3 activity (Fig. 1C and 2). Pre-treatment with KN-92, the inactive analogue of KN-93, and STO-609 failed to inhibit apoptosis of exposed HKM (Fig. 1C). Our data suggest the importance of the CaMKII pathway on HKM exposed to arsenic.
To garner further evidence in support of our observation, the HKM were incubated in the presence of arsenic and CaMKII levels assayed following 24 h of incubation. We observed that CaMKII levels were elevated (P < 0.05) in the arsenic-treated HKM (P < 0.05) and pre-incubation with CMZ and KN-93 led to significant reduction in the enzyme activity (Fig. 1D). The inactive analogue KN-92 failed to inhibit CaMKII activity in the arsenic-treated HKM. Taken together, our observations clearly imply that CaM–CaMKII activation is an early event initiating arsenic-induced HKM apoptosis.
CaMKII activation induces superoxide ion generation in arsenic-treated HKM
We earlier reported that arsenic-induced release of superoxide ions instigates HKM apoptosis.7 Thus, after recognizing the importance of the CaMKII axis in arsenic stress we investigated its role in superoxide ion generation. To look into this, the HKM were pre-treated with KN-93, exposed to arsenic and superoxide ion production checked by NBT assay. We noted that pre-treatment of HKM with KN-93 significantly (P < 0.05) attenuated superoxide ion production. The inactive analogue KN-92 had no inhibitory effect on superoxide ion generation (Fig. 3A). Next we studied whether superoxide ions in turn had a role in CaMKII activation in our model. The HKM were pre-treated separately with Apo and DPI and then checked for arsenic-induced CaMKII levels. We observed that Apo and DPI had no inhibitory effect on CaMKII activity in the arsenic-treated HKM (Fig. 3B). Based on these results we suggest that CaMKII activation is upstream of and critical for superoxide ion generation in arsenic-treated HKM. | ||
| Fig. 3 Arsenic-induced superoxide ion production is downstream to CaMKII activation. (A) HKM were pre-treated separately with or without KN-93 and KN-92 and then exposed to arsenic, and superoxide ion production measured following 2 h of incubation. (B) HKM were pre-treated separately with or without Apo and DPI and then exposed to arsenic, and CaMKII levels measured in the cell lysates 24 h post incubation. *P < 0.05 vs. HKM; #P < 0.05 vs. HKM + As. HKM, control head kidney macrophage; HKM + As, HKM exposed to arsenic; HKM + KN-93 + As, HKM pre-treated with KN-93 for 1 h prior to arsenic exposure; HKM + KN-92 + As, HKM pre-treated with KN-92 for 1 h prior to arsenic exposure; HKM + Apo + As, HKM pre-treated with Apo for 1 h prior to arsenic exposure; HKM + DPI + As, HKM pre-treated with DPI for 2 h prior to arsenic exposure. KN-93, CaMKII antagonist; KN-92, inactive analogue of KN-93; Apo and DPI, NADPH oxidase inhibitor. The concentration of chemicals used was as mentioned in the Materials and methods section. | ||
Superoxide ions activate the cAMP/PKA pathway in arsenic-treated HKM
The cross-talk between ROS and the cAMP/PKA pathway has been reported earlier.16,17 We were interested in documenting a link between cellular superoxide ion generation and activation of the cAMP/PKA pathway in our model. To look into this, we initially studied the changes in intracellular cAMP levels in arsenic-treated HKM at indicated time intervals. It is evident from Fig. 4A that on arsenic treatment, the intracellular cAMP levels increased till 12 h and thereafter declined gradually reaching basal levels at 24 h. We also observed that cAMP release was significantly inhibited in the presence of Apo and DPI (Fig. 4B). The results of this study demonstrated that superoxide ion generation had a significant stimulatory effect on intracellular cAMP production in arsenic-treated HKM. | ||
| Fig. 4 cAMP/PKA is pro-apoptotic in the exposed HKM. (A) Changes in intracellular cAMP levels in cell lysates of arsenic-exposed and unexposed HKM were measured at indicated time intervals. (B) HKM were pre-incubated separately with or without Apo, DPI, U0126, and then exposed to arsenic, and changes in cAMP levels measured 12 h post incubation. (C) HKM were pre-incubated with or without H-89 and then exposed to arsenic, and apoptosis studied 24 h post incubation using Hoechst 33342 staining and measuring the caspase-3 activity. Vertical bars represent mean ± SE (n = 6). *P < 0.05 vs. HKM; #P < 0.05 vs. HKM + As. HKM, control head kidney macrophage; HKM + As, HKM exposed to arsenic; HKM + Apo + As, HKM pre-treated with Apo for 1 h prior to arsenic exposure; HKM + DPI + As, HKM pre-treated with DPI for 2 h prior to arsenic exposure; HKM + U0126 + As, HKM pre-treated with U0126 for 2 h prior to arsenic exposure; HKM + H-89 + As, HKM pre-treated with H-89 for 1 h prior to arsenic exposure. Apo and DPI, NADPH oxidase inhibitor; U0126, ERK 1/2 inhibitor; H-89, PKA antagonist. The concentration of chemicals used was as mentioned in the Materials and methods section. | ||
Protein kinase A (PKA) is the major regulator in the cAMP signal transduction pathway, so we used H-89, the specific PKA inhibitor, to examine the possible role of the PKA pathway in HKM apoptosis induced by arsenic. The HKM were pre-treated with H-89 and then incubated in the presence of arsenic in complete RPMI and apoptosis studied following 24 h of incubation. From Hoechst 33342 and AV-PI staining and caspase-3 activity (Fig. 2 and 4C) it is indeed evident that HKM apoptosis was significantly reduced (P < 0.05) in the presence of H-89, suggesting the essentially pro-apoptotic role of cAMP/PKA in arsenic-induced HKM apoptosis. However, pre-treatment of HKM with H-89 had no effect on superoxide ion production (data not shown). Thus we conclude that superoxide ion-induced cAMP led to pro-apoptotic activation of PKA in the arsenic-treated HKM.
cAMP/PKA is critical for ERK 1/2 activation in arsenic-treated HKM
The critical role of ERK 1/2 in arsenic-induced HKM apoptosis was earlier reported by us.13 In the present study we investigated the role of the cAMP/PKA pathway in ERK 1/2 activation. To look into this, the HKM were pre-treated with or without H-89 then incubated with arsenic and the changes in total and pERK 1/2 levels measured following 24 h of incubation. It is evident from Fig. 5B that pre-treatment with H-89 led to a significant decline (P < 0.05) in the level of phospho ERK 1/2 in arsenic-treated HKM. No change was noticed in the total ERK 1/2 levels in H-89 pre-treated HKM on exposure to arsenic. On the other hand pre-treatment with U0126 did not interfere with cAMP levels in HKM exposed to arsenic (Fig. 4B). Our results imply the role of the cAMP/PKA pathway in ERK 1/2 activation in arsenic-treated HKM. | ||
| Fig. 5 Superoxide ions induce pro-apoptotic activation of NF-κB and trigger phosphorylation of ERK 1/2 in the exposed HKM. (A) HKM were pre-treated separately with NF-κBi and then exposed to arsenic, and apoptosis studied 24 h post incubation using Hoechst 33342 staining and measuring the caspase-3 activity. (B) HKM were pre-treated separately with or without H-89, L-Nil, respectively, and then exposed to arsenic, and the changes in total and phosphorylated ERK 1/2 levels measured 24 h post incubation from the cell lysates using EIA kits. (C) HKM were pre-treated separately with or without Apo, DPI, NF-κBi respectively and then exposed to arsenic, and changes in total and phosphorylated NF-κB-p65 measured 24 h post incubation from the cell lysates using EIA kits. Vertical bars represent mean ± SE (n = 6). *P < 0.05 vs. HKM; #P < 0.05 vs. HKM + As. HKM, control head kidney macrophage; HKM + As, HKM exposed to arsenic; HKM + H-89 + As, HKM pre-treated with H-89 for 1 h prior to arsenic exposure; HKM + L-Nil + As, HKM pre-treated with L-Nil for 1 h prior to arsenic exposure; HKM + Apo + As, HKM pre-treated with Apo for 1 h prior to arsenic exposure; HKM + DPI + As, HKM pre-treated with DPI for 2 h prior to arsenic exposure; HKM + NF-κBi + As, HKM pre-treated with NF-κBi for 1 h prior to arsenic exposure. H-89, PKA antagonist; L-Nil, iNOS specific inhibitor; Apo and DPI, NADPH oxidase inhibitor; NF-κBi, NF-κB activation inhibitor. The concentration of chemicals used was as mentioned in the Materials and methods section. | ||
ERK 1/2 instigates NO production in arsenic-treated HKM
After establishing the link between cAMP/PKA and ERK 1/2 we looked for the likely downstream signalling molecules instigating arsenic-induced HKM apoptosis and NO appeared to be an attractive candidate. We first investigated whether exposure to arsenic induces NO production in the HKM. The HKM were treated with arsenic and NO production assayed by Griess’ reagent at indicated time intervals. It is evident from Fig. 6A that arsenic-induced NO production was maximum in 24 h exposed HKM. Hence, this time point was taken for the subsequent studies on NO. | ||
| Fig. 6 Arsenic-induced HKM apoptosis involves the pro-apoptotic activation of inducible NOS. (A) HKM were exposed to arsenic and NO release measured using Griess’ reagent at the indicated time intervals. (B) HKM were pre-treated separately with or without L-Nil, NF-κBi, U0126 and then exposed to arsenic, and NO release measured 24 h post incubation using Griess’ reagent. (C) HKM were pre-treated separately with or without L-Nil prior to arsenic exposure, and apoptosis studied 24 h post incubation using Hoechst 33342 staining and measuring the caspase-3 activity. (D) HKM were pre-treated separately with or without L-Nil, NF-κBi, U0126 prior to arsenic exposure and the iNOS activity studied by immunofluorescence using anti-iNOS antibody (×40). The image represents the best of three replicates. Vertical bars represent mean ± SE (n = 6). *P < 0.05 vs. HKM; #P < 0.05 vs. HKM + As. HKM, control head kidney macrophage; HKM + As, HKM exposed to arsenic; HKM + L-Nil + As, HKM pre-treated with L-Nil for 1 h prior to arsenic exposure; HKM + NF-κBi + As, HKM pre-treated with NF-κBi for 1 h prior to arsenic exposure; HKM + U0126 + As, HKM pre-treated with U0126 for 2 h prior to arsenic exposure. L-Nil, iNOS specific inhibitor; NF-κBi, NF-κB activation inhibitor; U0126, ERK 1/2 inhibitor. The concentration of chemicals used was as mentioned in the Materials and methods section. | ||
Our next step was to establish the role of NO in arsenic-treated HKM apoptosis. The HKM were pre-treated with or without L-Nil, the iNOS specific inhibitor, and then exposed to arsenic for 24 h following which NO production and iNOS expression were studied. The results clearly documented arsenic-induced NO production and iNOS activity in the HKM which underwent marked inhibition in the presence of L-Nil (Fig. 6B and 6D). It was also observed that pre-incubation with L-Nil significantly inhibited HKM apoptosis as evident from Hoechst 33342 and AV-PI staining and caspase-3 activity (Fig. 2 and 6C).
It has been suggested that MAPKs have a role in NO production in different cell types. Since ERK 1/2 was implicated in the arsenic-induced HKM apoptosis pathway we hypothesised its role in NO production. To test this, the HKM were pre-treated with U0126 prior to arsenic exposure and NO production and iNOS expression studied. It is evident from Fig. 6B and 6D that pre-treatment with U0126 significantly inhibited NO production and iNOS expression in the arsenic-treated HKM. The role of NO in ERK 1/2 activation was also studied and we observed that pre-treatment with L-Nil had no inhibitory effect on pERK 1/2 expression in arsenic-treated HKM (Fig. 5B).
Together, our results suggest that the signals from the ERK 1/2 pathway due to cAMP/PKA activation instigate iNOS activity and the release of pro-apoptotic NO in exposed HKM.
Superoxide ion-induced NF-κB activation is also critical in iNOS activation
It has been reported that ROS can activate NF-κB to initiate a cascade of events.28 NF-κB consists of five Rel-domain-containing proteins including p65/RelA. In our study, p65 activity was assayed as phosphorylation of this domain is considered to be a critical index for measuring NF-κB activity.28 The HKM were pre-treated separately with or without Apo and DPI prior to arsenic exposure and the expression of total and phosphorylated p65 of NF-κB studied at indicated time intervals using a specific EIA kit. It was noted that the levels of total p65 remained unchanged in control and arsenic-treated HKM. However, a time dependent increase in phosphorylated p65 levels (P < 0.05) was noted with maximum changes recorded in 24 h arsenic-treated HKM (data not shown). The peak phosphorylating time of 24 h was selected for subsequent study. The HKM were treated with Apo or DPI prior to arsenic exposure and the changes in phosphorylated p65 levels measured following 24 h of incubation (Fig. 5C). This clearly suggests the role of superoxide ions in NF-κB activation in arsenic-treated HKM.It has been reported that NF-κB has both pro- and anti-apoptotic effects on cells.20 To look into this, the HKM were pre-treated with or without the NF-κB activation inhibitor (NF-κBi) prior to arsenic exposure and apoptosis studied following 24 h of incubation. The NF-κB inhibitor used for the study is 6-amino-4-(4-phenoxyphenylethylamino) quinazoline which has been reported to specifically block NF-κB-dependent transcription (http://www.merckmillipore.com/calbiochem) by “unknown mechanisms”.29 Our inhibitor studies clearly indicate the pro-apoptotic role of NF-κB in arsenic-induced HKM apoptosis (Fig. 5A).
Our next step was to identify the likely downstream target for NF-κB that initiated the apoptosis of HKM and iNOS was an attractive candidate.23 The HKM were pre-treated with NF-κBi then incubated with arsenic for 24 h and NO production and iNOS expression studied by Griess’ reagent and immunofluorescence respectively. We observed that the inhibition of NF-κB activation by its specific activation inhibitor led to a significant decline in NO release (P < 0.05, Fig. 6B) and iNOS expression (Fig. 6D), suggesting the involvement of NF-κB in NO production and iNOS expression.
Mitochondrial signalling has a critical role in arsenic-induced HKM apoptosis
In the present study we sought to determine the role of mitochondria in arsenic-induced apoptosis. The HKM were incubated with arsenic and the changes in mitochondrial membrane potential (ψm) checked at indicated time periods using JC-1 dye under a confocal microscope. Under normal conditions, JC-1 forms intracellular aggregates, the J aggregates, emitting red fluorescence at 590 nm. At low membrane potentials, JC-1 continues to exist as a monomer and produces green fluorescence (emission at 527 nm). Thus, JC-1 exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (529 nm) to red (590 nm), and mitochondrial depolarization indicated by a decrease in the red/green fluorescence intensity ratio. As seen in Fig. 7, the red fluorescence suggests the presence of distinct J-aggregates in the unexposed HKM. In arsenic-treated HKM we recorded a time dependent decrease in red fluorescence due to JC-1 monomer which is suggestive of loss in ψm (Fig. 7). From the above results it is evident that exposure to arsenic alters the ψm in HKM.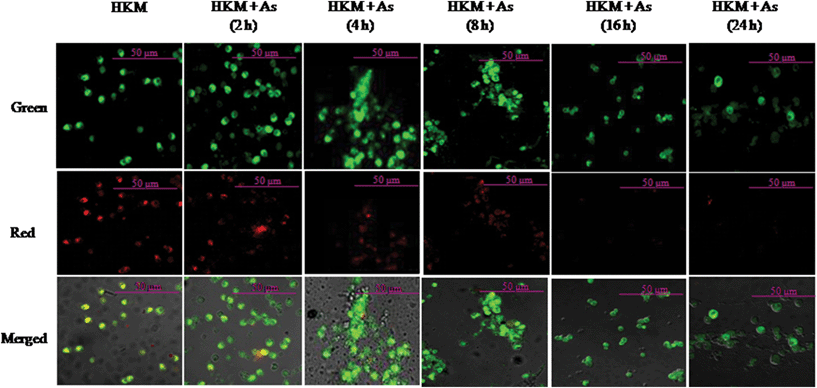 | ||
| Fig. 7 Arsenic exposure leads to time dependent loss of ψm. HKM were exposed to arsenic and the ψm loss studied at indicated time points using JC-1 dye (×40). Red images indicate the JC-1 aggregate, while green images indicate JC-1 monomers. Merged images indicate the co-localization of JC-1 aggregates and monomers. The image represents the best of three replicates. HKM, control head kidney macrophage; HKM + As, HKM exposed to arsenic. | ||
ROS have been implicated in ψm alterations in several studies.30 As we observed arsenic-induced superoxide ion generation in HKM we were keen to see its effects on ψm. To look into this, the HKM were pre-treated with Apo and DPI separately prior to arsenic exposure and alterations in ψm studied following 24 h of incubation. This particular time interval was selected because at 24 h of incubation maximum apoptosis was recorded7 and most of the exposed HKM were also found to have lost their ψm along with evident detection of JC-1 monomer (Fig. 7). We observed that pre-treatment with Apo and DPI effectively inhibited arsenic-induced loss of ψm in the HKM (Fig. 8A). This indicates that a positive correlation exists between superoxide ions and mitochondrial events leading to HKM apoptosis induced by arsenic.
 | ||
| Fig. 8 Superoxide ion-induced ψm loss leads to MPTP opening and cytosolic release of cyt C. (A) HKM were pre-treated with or without Apo, DPI, CsA separately and the ψm loss studied following 24 h of incubation using JC-1 dye (×40). Red images indicate the JC-1 aggregate, while green images indicate JC-1 monomers. Merged images indicate the co-localization of JC-1 aggregates and monomers. Images shown are representative observations from three independent experiments. (B) HKM were pre-treated with or without CsA and the release of cyt C in the cytosol studied 24 h post incubation by immunoblotting using anti-cyt C antibody. β-Actin served as the loading control. The image represents the best of three replicates. HKM, control head kidney macrophage; HKM + As, HKM exposed to arsenic; HKM + Apo + As, HKM pre-treated with Apo for 1 h prior to arsenic exposure; HKM + DPI + As, HKM pre-treated with DPI for 2 h prior to arsenic exposure; HKM + CsA + As, HKM pre-treated with CsA for 1 h prior to arsenic exposure. Apo and DPI, NADPH oxidase inhibitor; CsA, cyclosporin A, inhibits ψm loss and prevents MPTP formation. The concentration of chemicals used was as mentioned in the Materials and methods section. | ||
It is reported that CsA at sub-micromolar concentration could effectively inhibit reduction in the ψm and MPTP formation and inhibit cytosolic release of cyt C.31 To evaluate this, HKM were pre-treated with CsA prior to arsenic exposure and alterations in ψm studied. We observed that CsA pre-treatment could prevent loss of ψm (Fig. 8A). Also, it is evident from Fig. 8B that cyt C released in the cytosol due to arsenic exposure was significantly inhibited following pre-incubation of HKM with CsA. Together our results suggest that arsenic-induced reduction in the ψm and subsequent MPTP formation leads to the release of cyt C from mitochondria into the cytosol of exposed HKM.
Caspase-9 initiates arsenic-induced HKM apoptosis
It has been reported that cyt C induces caspase-9 activity inducing apoptosis.26 On observing cyt C activity in arsenic-treated HKM we hypothesised the involvement of caspase-9 as an initiator caspase in the process. Caspase-9 activation was checked from its ability to cleave pNA from LEHD-pNA. The HKM were incubated with or without arsenic and the levels of pNA in the cell lysates measured and results plotted as relative increase over basal activity. We observed a significant increase (P < 0.05) in pNA levels in arsenic-treated HKM, compared to control HKM (Fig. 9A). We also evaluated the effect of CsA on caspase-9 activity and observed that pre-treatment of HKM with CsA prior to arsenic exposure could effectively inhibit caspase-9 activity in the exposed HKM (Fig. 9A). | ||
| Fig. 9 Arsenic exposure leads to activation of caspase-9. (A) HKM were pre-treated with or without L-Nil, CsA, Z-LEHD-FMK respectively and then exposed to arsenic, and caspase-9 activity measured in the cell lysate following 24 h of incubation using the activity assay kit. Results are plotted over increase in basal activity. (B) HKM were pre-treated separately with or without Z-LEHD-FMK prior to arsenic exposure and apoptosis study done using Hoechst 33342 staining and measuring the caspase-3 activity following 24 h of incubation. *P < 0.05 vs. HKM; #P < 0.05 vs. HKM + As. HKM, control head kidney macrophage; HKM + As, HKM exposed to arsenic; HKM + L-Nil + As, HKM pre-treated with L-Nil for 1 h prior to arsenic exposure; HKM + CsA + As, HKM pre-treated with CsA for 1 h prior to arsenic exposure; HKM + Z-LEHD-FMK + As, HKM pre-treated with Z-LEHD-FMK for 1 h prior to arsenic exposure. L-Nil, iNOS specific inhibitor; CsA, Cyclosporin A, inhibits ψm loss and prevents MPTP formation; Z-LEHD-FMK, caspase-9 inhibitor. The concentration of chemicals used was as mentioned in the Materials and methods section. | ||
For further evidence of the involvement of caspase-9 the HKM were pre-treated with or without the caspase-9 inhibitor Z-LEHD-FMK prior to arsenic exposure and apoptosis checked. It was noted that pre-treatment of HKM with Z-LEHD-FMK effectively blocked caspase-9 activity (P < 0.05; Fig. 9A), and inhibited caspase-3 activity and apoptosis (P < 0.05; Fig. 2 and 9B). Modulation of caspase-9 activity with respect to iNOS expression is well studied.32 To establish this relationship, the HKM were pre-treated with Z-LEHD-FMK prior to arsenic exposure and iNOS expression and NO production studied. Similarly, in another set of experiments, HKM were pre-treated with L-Nil, exposed to arsenic and caspase-9 activity checked. We observed that pre-treatment of HKM with L-Nil effectively decreased caspase-9 activity (Fig. 9A) though Z-LEHD-FMK pre-treatment to the arsenic-treated HKM exerted no inhibitory effect on NO production and iNOS expression (data not shown), indicating that the iNOS-activated mitochondrial pathway plays an important role in arsenic-induced apoptosis.
Discussion
While elucidating the molecular mechanism of apoptosis induced by arsenic we observed a significant increase in CaM levels in exposed HKM. The presence of CaM is well documented in fish33 and to the best of our knowledge CaM involvement in arsenic-toxicity in fish macrophages has not been reported. We observed that the CaM antagonist CMZ effectively reduced CaM levels and inhibited HKM apoptosis. This is the first report on the involvement of CaM in arsenic-induced apoptosis in fish cells. Our results also concur with earlier reports suggesting the pro-apoptotic role of CaM in the arsenic-induced apoptosis of several cancer cell lines, although the mechanisms are not well understood.34 Comparing our results with those reporting anti-apoptotic roles of CaM,35 we hypothesize that the amount of CaM available affects downstream CaM-dependent pathways determining the cellular fate and the eventual outcome of arsenic stress.How CaM induces HKM apoptosis was not clear in our model. However, in the context of arsenic-toxicity, the involvement of CaM-dependent kinases has been reported.36 We hypothesised that downstream activation of CaM-dependent kinases could be important for initiating death pathways. Inhibiting the enzyme activity would rescue the cells from apoptotic death and therefore prove the hypothesis. Our inhibitor studies with the CaMKK inhibitor STO-609 clearly ruled out the involvement of CaMKK in the process. This observation also ruled out the involvement of CaMKI and CaMKIV on arsenic-induced apoptosis as both the kinases require upstream phosphorylation of CaMKK for activation.12 The pro-apoptotic role of CaMKII in apoptosis has also been documented in cardiomyocytes,37 hepatocytes,38 nerve cells,39 and in apoptosis induced by tributyltin,40 UV light and TNF-α.41 We noted that KN-93, the specific inhibitor for CaMKII, interfered with CaMKII activation and significantly blocked arsenic-induced apoptosis. The structural analogue for KN-93 was neither able to inhibit CaMKII activation nor affect apoptotic death in the exposed HKM. Together, our results clearly suggested CaMKII to be the kinase translating Ca2+–CaM mediated death signalling in arsenic-treated HKM.
On establishing the importance of the CaM–CaMKII axis in arsenic-induced apoptosis we investigated how it regulates the death pathway. Arsenicals induce ROS generation and it has been observed that increased ROS can activate CaMKII42 and vice versa.43 Superoxide ion production and lipid peroxidation are established bio-markers for oxidative stress. We had previously reported arsenic-induced superoxide ion production and lipid peroxidation (malondialdehyde) in HKM.10 Here we studied the cross talk between CaM–CaMKII and ROS using superoxide ion production as the index. Our inhibitor studies with KN-93 and its structural analogue KN-92 suggested the role of CaMKII in superoxide ion levels and HKM apoptosis. NADPH oxidase is responsible for superoxide ion generation by arsenic-treated HKM and the effect can be blocked by the flavoprotein inhibitor DPI or the NADPH oxidase inhibitor Apo.10 The impact of these reagents on CaMKII activity was examined and we noted that pre-treatment with Apo or DPI had no effect on CaMKII levels but interfered with arsenic-induced HKM apoptosis.10 Our results suggest that CaMKII activation is critical for superoxide ion generation in arsenic-treated HKM and support our earlier findings10 that modulating the intracellular levels of superoxide ions can affect arsenic-toxicity.
We observed elevated cAMP levels in arsenic-treated HKM. There are reports on the interplay between ROS and cAMP/PKA activation16,17 and we were keen to investigate the same in our model. We failed to observe the effect of PKA inhibitor H-89 on superoxide ion generation. On the other hand, pre-incubation with both Apo and DPI led to reduction in cAMP levels and cell death which implies the role of superoxide ions in cAMP production in arsenic-treated HKM. Although it is not possible from this study to conclude on how superoxide ions affect cAMP production we speculate that they could be phosphorylating the ACs or inhibiting the activation of PDEs leading to increased accumulation of the cyclic nucleotide inside the HKM. Studies indicate that cAMP via different mediators regulates a broad range of cellular responses that includes its central role as both a pro- and anti-apoptotic agent.18
In eukaryotic cells, PKA is the principal effector of cAMP action and we hypothesized that the cAMP-induced effects observed were mediated via PKA. To prove this, we used H-89 and, indeed, our results clearly indicate the importance of cAMP/PKA activation in arsenic-induced HKM apoptosis. Although the role of cAMP/PKA in arsenic-induced apoptosis has been reported,43 the molecular mechanisms are not well understood. In APL cells, it has been suggested that arsenic-induced cAMP/PKA activation releases SMRT co-repressor and induces RARA transcriptional activation, eventually leading to cell death and remission.44 Further study on arsenic and cAMP modulated gene expression profiles may reveal how they cross-talk to promote HKM apoptosis.
Though the role of NF-κB in apoptosis has been reported during stress the cellular mechanisms are not well understood. Existing reports suggest that arsenic can inhibit as well as activate NF-κB under different conditions.28 Our results suggested that arsenic-induced up-regulation in NF-κB activity plays a pro-active role in initiating HKM apoptosis. We opine that this contradiction is due to the use of different cell models and different times and doses of exposure. Given that superoxide ions are involved in the signal transduction mechanisms for NF-κB activation, we thought it logical to investigate this possibility in our model. We observed that pre-treatment with both Apo and DPI led to marked decreases in NF-κB activity in arsenic-treated HKM. Thus, we propose that oxidative stress-induced NF-κB activation is a major factor in the development and pathogenesis of arsenic-toxicity in HKM. Although it is difficult to explain how NF-κB gets activated in HKM, superoxide ion-induced protein phosphorylation and proteolysis could be important. It has been suggested that PKA can enhance p65 transcriptional activity.28 However, in contrast to our expectations H-89 pre-treatment had no effect on p65 phosphorylation. Similarly, NF-κB inhibition exerted no significant effect on cAMP release. Thus, it would be important to identify the other kinase or protease activity on p65 phosphorylation during arsenic exposure in HKM.
There are several reports on arsenic-mediated activation of MAPKs in different cell types.45 We earlier observed the role of arsenic in the ERK 1/2 pathway in HKM and suggested the role of Ca2+-dependent calpain-2 and ROS in initiating the process. In the present study, we extend our findings and show that cAMP/PKA generated due to activation of the CaM–CaMKII–superoxide ion axis was also important for ERK 1/2 activation in the exposed HKM. Thus, it would be interesting to check whether calpain-2 also has any role in cAMP/PKA activation in arsenic-treated HKM.
Arsenic can inhibit or stimulate NO production46,47 in different cell types. The involvement of arsenic-induced NO production is not evident in fish. Here, we report iNOS induced NO production in the exposed HKM. Our inhibitor assays and immunofluorescence studies further indicated NO as pro-apoptotic in exposed HKM. How NO exerts its pro-apoptotic effect in our system is difficult to explain. It might react with superoxide ions triggering further oxidative stress; alternatively it might directly modulate the inherent intracellular ROS quenching machinery, thereby rendering the cells prone to apoptotic death.14 NF-κB and MAPK are critical for iNOS expression.23,24 From our observations it is apparent that though activation of both ERK 1/2 and NF-κB were independent of each other, the two pathways converge at iNOS to instigate HKM pathology.
The involvement of the mitochondrial pathway of apoptosis in arsenic-toxicity was not well reported in fish. Recently, it was reported that the mitochondrial pathway is important for arsenic-induced apoptosis of the fish hepatocellular cell line PLHC-1.48 Here, by using specific inhibitors we have conclusively demonstrated that arsenic-induced alteration in ψm of HKM is time dependent and under the influence of superoxide ions. We observed that the alteration in ψm resulted in release of pro-apoptotic cyt C to the cytosol. Our next goal was to establish the involvement of the mitochondrial pathway of apoptosis in our model and for that caspase-9 was the prime target. We observed increased caspase-9 activity in the HKM and pre-treatment with L-Nil significantly reduced the enzyme activity in exposed cells. How NO activates caspase-9 in our system is not clear. We presume that the nitrosative stress induced by arsenic affects mitochondrial membrane permeability and releases cyt C triggering caspase-9 activity in the HKM. Our observations that (i) CsA effectively prevents the loss of ψm, inhibits cyt C release into the cytosol and caspase-9 activity and (ii) Z-LEHD-FMK significantly blocks caspase-3 activity and apoptosis together suggest the role of MPTP formation in activating caspase-9 and caspase-3 to be a substrate for caspase-9 in our model.
Materials and methods
Animal care and maintenance
Catfish (Clarias batrachus, Siluriformes, 100 ± 20 g) were maintained in 50 L glass tanks (2–3 fish per tank) as per the guidelines set forth by the Animal Ethics Committee of Government of India and University of Delhi. The water quality, dissolved oxygen content and pH in each tank were monitored regularly. The fish were fed boiled chicken liver ad libitum and acclimatized to laboratory conditions for 15 days prior to setting up the experiments. Fish health was routinely monitored by appearance and pathological examinations. Diseased fish or fish showing any abnormal behaviour were removed immediately from the tanks.Chemicals, reagents and antibodies
The different chemicals, inhibitors and antibodies alongside their concentrations used for the study are as follows: arsenic trioxide (As2O3, 0.50 μM), Hoechst 33342 (3.25 μM), cAMP-dependent PKA inhibitor (H-89, 20 μM), CaMKII inhibitor (KN-93, 20 μM), inactive analogue of KN-93 (KN-92, 20 μM), nitroblue tetrazolium (NBT, 1.374 mM), ethyl 3-aminobenzoate methanesulfonate (MS 222), Percoll, ROS inhibitors – apocynin (Apo, 100 μM) and diphenyleneiodonium chloride (DPI, 10 μM) were purchased from Sigma-Aldrich. NF-κB activation inhibitor 6-amino-4-(4-phenoxyphenylethylamino) quinazoline (NF-κBi, 10 nM) and caspase-9 inhibitor (Z-LEHD-FMK, 10 μM) were from Calbiochem. Caspase-3 inhibitor (Ac-DEVD-CHO, 10 μM), ERK 1/2 inhibitor (U0126, 20 μM) and caspase-3 activity assay kits were from Promega. Cyclosporin A [CsA, mitochondrial permeability transition pore (MPTP) inhibitor, 5 μM] was a gift from D. Bhowmik, AIIMS, India. JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethybenzimidazol carbocyanine iodide, mitochondrial membrane potential dye, 20 μM), RPMI-1640 and foetal bovine serum (FBS) were from Invitrogen-Molecular Probes. Inducible NOS specific inhibitor [dihydrochloride L-N6-(1-iminoethyl) lysine, L-Nil, 50 μM] was from Cayman Chemicals. The ERK 1/2 EIA kit for measuring total ERK 1/2 and the pThr202/Tyr204 ERK 1/2 EIA kit for measuring phosphorylated-ERK (pERK) were purchased from Enzo Life Sciences. Detection of CaM and CaMKII activity was done using EIA kits from USCN Life Sciences. Total and phosphorylated NF-κB levels were measured using Pathscan total NF-κB p65 and phospho NF-κB p65 EIA kits respectively from Cell Signaling Technology. The cytochrome C (cyt C) detection kit was purchased from Biovision. The caspase-9 activity kit was purchased from Gene Script. The apoptosis detection kit AV-FITC-PI (Annexin V-FITC-propidium iodide) was purchased from BD Pharmingen. Mouse polyclonal antibody against inducible NOS (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :250) from Abcam and FITC-conjugated secondary antibody against mouse raised in goat (1:200) from Cell Signalling Technology were kind gifts from B. Ghosh, IGIB, India. Mouse polyclonal antibody against β-actin (1:10000) and alkaline phosphatase (AP)-conjugated secondary antibody raised in goat (1:1000) were purchased from Santacruz.
:250) from Abcam and FITC-conjugated secondary antibody against mouse raised in goat (1:200) from Cell Signalling Technology were kind gifts from B. Ghosh, IGIB, India. Mouse polyclonal antibody against β-actin (1:10000) and alkaline phosphatase (AP)-conjugated secondary antibody raised in goat (1:1000) were purchased from Santacruz.Preparation of head kidney macrophages
Fish were euthanized using an overdose of MS 222; the HKs were removed aseptically and placed in RPMI-1640 with phenol-red indicator supplemented with 25 mM HEPES containing 1% penicillin–streptomycin. The HK was passed through a 100 μm wire mesh; the single cell suspension was layered over a 34/51% Percoll gradient and centrifuged at 400g at 4 °C for 20 min. The phagocyte-rich fraction appearing above the 34/51 interface was collected and enriched for HKM as described earlier.11 The HKM viability and purity was checked by the trypan blue (0.4%) dye exclusion method and Wright-Giemsa stain respectively and the cells maintained in RPMI with phenol-red indicator supplemented with 25 mM HEPES, 10% FBS and 1% penicillin–streptomycin (complete RPMI).Detection of apoptosis
Apoptosis was measured using AV-FITC-PI and Hoechst 33342 staining. Briefly, the HKM (1 × 106) were pre-treated with indicated concentrations of different inhibitors for indicated time periods prior to addition of 0.50 μM arsenic. The process for preparing an arsenic solution has been described earlier.8 The HKM were incubated with arsenic for 24 h in complete RPMI at 30 °C and 5% CO2, washed and stained separately with Hoechst 33342 (in 1 × PBS) and AV-FITC-PI and visualized under a fluorescence microscope (×40, Nikon Eclipse 400) within 30 min of adding the stains.11 A total of 100 cells were studied in each field and three such fields were included to determine the percentage of apoptotic HKM. The doses of different inhibitors were selected on the basis of inhibitor specificity and cytotoxicity. The HKM treated with the indicated concentrations of the inhibitors remained as viable as control HKM at all time points as determined by the trypan blue (0.4%) dye exclusion method and were maintained during the entire course of the experiment.CaM and CaMKII assay
The quantitative estimation of CaM and CaMKII was done from the cell lysates using specific EIA kits as per the manufacturer's instructions using chemicals supplied with each kit. Briefly, the HKM (1×106) were pre-treated with or without the indicated concentrations of different inhibitors for indicated time periods and then incubated with 0.50 μM arsenic in complete RPMI at 30 °C and 5% CO2. Following incubation, the HKM were collected at indicated time periods, re-suspended in chilled lysis buffer provided with each kit and incubated on ice for 30 min. The cell lysates were collected by centrifugation at 16000g for 20 min at 4 °C. One hundred microlitres each of standards and cell lysates were added into the wells of specific plates and incubated for 5 h at 30 °C. Following incubation the liquid from each well was removed and 100 μL of Detection Reagent A specific for each assay added to the wells and further incubated for 1 h at 30 °C. The Detection Reagent A was removed and the wells washed several times with 200 μL of 1 × wash solution followed by addition of 100 μL of specific Detection Reagent B to the corresponding wells and incubated for 30 min at 30 °C. The wells were washed, and 90 μL of the substrate added to each well and incubated for 30 min at room temperature. The reaction was stopped by adding 50 μL of stop solution and the readings taken at 450 nm in a microplate reader (BMG Labtech). The amounts of CaM and CaMKII were interpolated from their respective standard curves obtained by plotting the O.D. of the standards.Measurement of superoxide ions
The HKM (1 × 105) were pre-treated with or without 0.50 μM arsenic in complete RPMI at 30 °C and 5% CO2 and superoxide ion generation checked by an NBT assay. Briefly, the culture supernatants were carefully removed at 2 h post incubation without disturbing the cells and 100 μL of NBT was added to each well and further incubated for another 5 h at 30 °C.7 The supernatants were removed and the formazan crystals solubilized by adding 60 μL of KOH (2 M) and 70 μL of DMSO, respectively, and the absorbance read at 620 nm using a microplate reader (BMG Labtech). In a parallel study, the HKM (1 × 105) were pre-treated with indicated concentrations of different inhibitors for indicated time periods prior to arsenic exposure and superoxide ion generation checked as described above.Measurement of intracellular cAMP levels
Arsenic-induced alterations in the intracellular cAMP levels were checked using an EIA kit as per the manufacturer's instructions using chemicals supplied with the kit. Briefly, the HKM (1 × 106) were pre-treated with or without 0.50 μM arsenic in complete RPMI at 30 °C and 5% CO2 for indicated time periods. Following incubation, the HKM were collected and re-suspended in 100 μL of 0.1 N HCl for 10 min at room temperature. The unwanted debris were removed by centrifugation at 800g and 100 μL each of the standards (supplied with the kit) and supernatants obtained after centrifugation were added separately to the bottom of appropriate wells of the cAMP assay plate followed by addition of 50 μL each of the conjugate and the antibody to each well. The plates were maintained on a plate shaker at room temperature for 4 h. The plates were washed and 200 μL of substrate solution added to each well and incubated for 1 h at room temperature. Finally, 50 μL of stop solution was added to the wells and the absorbance read at 405 nm using a microplate reader (BMG Labtech). The HKM (1 × 106) were also pre-treated with indicated concentrations of different inhibitors for indicated time periods prior to arsenic exposure and assayed for cAMP production. The cAMP concentrations were interpolated from the standard curves obtained by plotting the O.D. of the standards.ERK-1/2 assay
Total ERK 1/2 and pERK 1/2 was measured using specific EIA kits as per the manufacturer's instructions using chemicals supplied with the kit.11 Briefly, the HKM (1 × 106) were pre-treated with indicated concentrations of different inhibitors for indicated time periods in complete RPMI at 30 °C and 5% CO2 prior to addition of 0.50 μM arsenic. Following 24 h of incubation, the HKM were washed, re-suspended in chilled lysis buffer and incubated on ice for 30 min. The cell lysates were collected by centrifugation at 16000g for 20 min at 4 °C and the assays performed in a total volume of 100 μL in ERK 1/2 clear microtiter plates coated with mouse monoclonal antibody specific to total ERK 1/2 and pERK 1/2. One hundred microlitres of the samples along with standards were added to the wells and incubated overnight at 4 °C. The wells were washed and 100 μL of polyclonal ERK 1/2 and pERK 1/2 antibody added separately into the respective wells and incubated for 2 h at 30 °C with shaking. After extensive washing, 100 μL of HRP-conjugated secondary antibody was added to the wells, incubated for 45 min at room temperature following which 100 μL of TMB substrate containing hydrogen peroxide was added to each well and incubated for 30 min. Finally, 100 μL of stop solution was added and the absorbance read at 450 nm. Triplicate sets were prepared containing serially diluted standards, blank (no cell extract), negative control (extract from untreated cells) and arsenic-treated HKM. The amounts of ERK 1/2 and pERK 1/2 in the cell lysates were interpolated from the standard curves obtained by plotting the O.D. of the standards.Nitric oxide assay and immunocytochemistry of iNOS
The HKM (1 × 106) were incubated with or without 0.50 μM arsenic in complete RPMI at 30 °C and 5% CO2 for different time periods. Following incubation, 50 μL of the cell-free culture supernatant was collected and mixed with an equal volume of Griess’ reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride, 2.5% H3PO4) and incubated at 30 °C for 10 min. The absorbance was read at 540 nm in a microplate reader (μQuant, Biotek) and the amount of nitrite generated was calculated from the NaNO2 standard curve. The HKM were then pre-incubated with the iNOS-specific inhibitor L-Nil prior to arsenic exposure and checked for NO release at 24 h of incubation as mentioned earlier.To detect iNOS activity, the HKM were pre-treated with or without different inhibitors and then incubated with 0.50 μM arsenic in complete RPMI at 30 °C and 5% CO2 for 24 h. Following incubation the HKM were washed with PBS and fixed in 3% paraformaldehyde in PBS for 30 min at room temperature. The fixed cells were subsequently incubated with blocking and permeabilizing solution (PBS, 2 mg mL−1 BSA, 0.2 mg mL−1 saponin) for 1 h at room temperature and then washed and incubated with mouse anti-iNOS primary antibody (1:200) in blocking permeabilizing solution overnight at 4 °C. Next day, the HKM were washed in PBST (PBS containing 0.1% Tween-20) and stained with FITC-conjugated anti-mouse secondary antibody (1:250) for 3 h at 30 °C and visualized under a confocal microscope (×40 oil immersion, 1.30 NA, Nikon Eclipse A1Rsi-TiE-300).
NF-κB assay
Total and phosphorylated NF-κB p65 was measured using EIA kits as per the manufacturer's instructions using chemicals supplied with the kit. Briefly, the HKM (1 × 106) were pre-treated with indicated concentrations of different inhibitors for indicated time periods in complete RPMI at 30 °C and 5% CO2 prior to addition of 0.50 μM arsenic. Following 24 h of incubation, the HKM were washed and re-suspended in chilled lysis buffer containing 1 mM PMSF. The HKM were briefly sonicated at 4 °C, the samples centrifuged at 10000g for 5 min and the supernatant collected in another microcentrifuge tube. One hundred microlitres of sample diluents was added to the microcentrifuge tube and vortexed. Following this, 100 μL of the diluted cell lysate was added to the appropriate well and left overnight at 4 °C. The next day the supernatant was discarded, the wells were washed and 100 μL of detection antibody added to each well and incubated for 4 h at 30 °C. The wells were washed extensively and 100 μL of HRP-linked secondary antibody added to each well and further incubated for 30 min at 30 °C. After extensive washing, 100 μL of TMB substrate was added and incubated for 30 min at 30 °C. Finally, the reaction was terminated by addition of 100 μL of stop solution, the absorbance read at 450 nm using a microplate reader (BMG Labtech) and the results plotted as their respective O.D. values.Detection of mitochondrial transmembrane potential and release of cytochrome C
Arsenic-induced alteration in mitochondrial transmembrane potential of the exposed HKM was studied using the cationic dye JC-1. Briefly, the HKM (1 × 106) were pre-treated with indicated concentrations of different inhibitors for indicated time periods in complete RPMI at 30 °C and 5% CO2 prior to addition of 0.50 μM arsenic. The HKM were collected at different time intervals, washed, the dye added and incubated at 30 °C for 15 min. The cells were washed and examined under an inverted confocal microscope (A1Rsi Nikon Eclipse TiE-300). JC-1 loaded cells were excited at 488 nm and the green and red emissions were recorded simultaneously at FITC (green, 530 nm) and TRITC (red, 590 nm). The red/green fluorescence was digitized at ×40, 1.3 NA oil immersion objectives.Cytochrome C release was checked using an assay kit (Biovision) as per the manufacturer's instructions with chemicals supplied with the kit. Briefly, HKM (4×107) were pre-treated with or without inhibitor prior to 0.50 μM arsenic exposure and incubated in complete RPMI at 30 °C and 5% CO2 for 24 h. Following incubation, the HKM were centrifuged at 700g for 10 min at 4 °C, the pellet resuspended in 20 μL cytosolic extraction buffer containing protease inhibitor cocktail and incubated for 10 min at 4 °C. The pellet was homogenized and centrifuged at 700g for 10 min at 4 °C. The supernatant collected was centrifuged at 10000g for 30 min at 4 °C. The supernatant thus obtained was collected as a cytosolic fraction. The presence of cyt C released in the cytosolic fraction was checked by immunoblotting using mouse cyt c antibody (1 μg mL−1) provided with the kit.
Caspase-9 and caspase-3 assay
Caspase-9 and caspase-3 activities were measured by activity assay kits as per the manufacturer's instructions using chemicals supplied with the kit. Briefly, the HKM (1 × 106) were pre-treated with indicated concentrations of different inhibitors for indicated time periods in complete RPMI at 30 °C and 5% CO2 prior to addition of 0.50 μM arsenic. Following 24 h of incubation, the HKM were washed, re-suspended in 50 μL of chilled lysis and incubated on ice for 10 min. The cell lysates were collected by centrifugation at 10000g for 5 min at 4 °C and the assays performed. The caspase-9 assay was performed in a total volume of 100 μL; triplicate wells were prepared containing blank (no cell extract), negative control (extract from untreated cells) and arsenic treated cell extract. To 50 μL of cell extract, 50 μL of 2× reaction buffer containing 0.50 μL of DTT (10 mM), 0.25 μL of PMSF (5 mM) and 5 μL of LEHD-pNA substrate was added. The plates were incubated at 37 °C for 5 h and the absorbance read at 405 nm (BMG Labtech). The result was plotted as relative increase in O.D. values. For the caspase-3 assay, triplicate wells were prepared containing blank (no cell extract), negative control (extract from untreated cells) and arsenic-treated cell extract. In 10 μL of cell extract 32 μL of caspase buffer, 2 μL of DMSO, 10 μL of DTT (100 mM) and 2 μL of the DEVD-pNA substrate were added. The result was plotted in terms of cleaved caspase-3 activity detected.Statistical analysis
Data are presented as mean ± SE of the number of experiments performed, as indicated in the corresponding figure. Pair-wise comparison was made between groups employing a paired t-test with P < 0.05 as the minimum significant level.Conclusion
Arsenic exposure activates the CaM-CaMKII pathway to induce superoxide ion production in the HKM. The superoxide ion activates the pro-apoptotic cAMP/PKA pathway leading to phosphorylation of ERK 1/2 and NF-κB; the two pathways independently converge at iNOS to produce NO. Oxidative stress also alters the ψm releasing cyt C in the cytosol to activate caspase-9. NO was also observed to have a positive effect on caspase-9 activity in arsenic-exposed HKM. Eventually, activated caspase-9 activated caspase-3 to execute HKM apoptosis (Fig. 10). We conclude that multiple oxidative effects of arsenic converge to promote HKM apoptosis, and modulating the intracellular superoxide ion levels can attenuate arsenic-toxicity. | ||
| Fig. 10 Overview of the work. Arsenic-induced alterations in intracellular Ca2+ levels activate CaM-CaMKII, inducing downstream production of superoxide ions and activation of the cAMP/PKA pathway in HKM. The cAMP/PKA pathway is critical for the activation of ERK 1/2 and iNOS expression. Superoxide ions, besides inducing NF-κB activation, alter mitochondrial membrane permeability, releasing cyt C into the cytosol of exposed HKM. Cytochrome C and NO activate caspase-9 to execute caspase-3 mediated HKM apoptosis. | ||
Acknowledgements
We would like to acknowledge D. Ghosh, IITR India, for critically analysing the manuscript and A. Pal for technical assistance. The authors have declared no conflict of interest. This work was supported by a Department of Science and Technology (DST), Government of India grant (SR/SO/AS-51/2007) and a Delhi University Post-Doctoral Research and Development grant. C.B. and A.S. were supported by fellowships from DST-INSPIRE (Govt. of India) and UGC (Govt. of India) respectively.References
- M. F. Hughes, B. D. Beck, Y. Chen, A. S. Lewis and D. J. Thomas, Toxicol. Sci., 2011, 123, 305–332 CrossRef CAS.
- P. B. Tchounwou, A. K. Patlolla and J. A. Centeno, Toxicol. Pathol., 2003, 31, 575–588 CAS.
- A. Lemarie, C. Morzadec, E. Bourdonnay, O. Fardel and L. Vernhet, J. Immunol., 2006, 177, 3019–3027 CAS.
- G. Galicia, R. Leyva, E. P. Tenorio, P. Ostrosky-Wegman and R. Saavedra, Int. Immunopharmacol., 2003, 3, 671–682 CrossRef CAS.
- C. Yedjou, P. Tchounwou, J. Jenkins and R. J. McMurray, Hematol. Oncol., 2010, 3, 28 CrossRef.
- K. Ahmed, A. A. Akhand, M. Hasan, M. Islam and A. Hasan, Am. Eurasian J. Agric. Environ. Sci., 2008, 4, 18–22 Search PubMed.
- S. Datta, D. Ghosh, D. R. Saha, S. Bhattacharya and S. Mazumder, Aquat. Toxicol., 2009, 92, 86–94 CrossRef CAS.
- D. Ghosh, S. Datta, S. Bhattacharya and S. Mazumder, Aquat. Toxicol., 2007, 81, 79–89 CrossRef CAS.
- A. S. Nayak, C. R. Lage and C. H. Kim, Toxicol. Sci., 2007, 98, 118–124 CrossRef CAS.
- S. Datta, S. Mazumder, D. Ghosh, S. Dey and S. Bhattacharya, Toxicol. Appl. Pharmacol., 2009, 241, 329–338 CrossRef CAS.
- J. M. Schmitt, E. S. Guire, T. Saneyoshi and T. R. Soderling, J. Neurosci., 2005, 25, 1281–1290 CrossRef CAS.
- J. M. Timmins, L. Oczan, T. A. Seimon, G. Li, C. Malagelada, J. Backs, R. Bassel-Duby, E. N. Olson, M. E. Anderson and I. Taba, J. Clin. Invest., 2009, 119, 2925–2941 CrossRef CAS.
- C. Banerjee, R. Goswami, S. Datta, R. Rajagopal and S. Mazumder, Toxicol. Appl. Pharmacol., 2011, 256, 44–51 CrossRef CAS.
- W. Droge, Physiol. Rev., 2002, 82, 47–95 CAS.
- H. U. Simon, A. Haj-Yehia and F. Levi-Schaffer, Apoptosis, 2000, 5, 415–418 CrossRef CAS.
- C. A. Isoni, E. A. Borges, C. A. Veloso, R. T. Mattos, M. M. Chaves and J. A. Nogueira-Machado, Oxid. Med. Cell. Longevity, 2009, 2, 317–321 CrossRef.
- C. I. Ezeamuzie and N. Taslim, Med. Princ. Pract., 2008, 17, 468–474 CrossRef.
- P. A. Insel, L. Zhang, F. Murray, H. Yokouchi and A. C. Zambon, Acta Physiol., 2012, 204, 277–287 CrossRef CAS.
- L. Li, J. Wang, R. D. Ye, G. Shi, H. Jin, X. Tang and J. Li, J. Cell. Physiol., 2008, 217, 486–493 CrossRef CAS.
- J. L. Luo, H. Kamata and M. Karin, J. Clin. Invest., 2005, 115, 2625–2632 CrossRef CAS.
- T. Gotoh, S. Oyadomari, K. Mori and M. Mori, J. Biol. Chem., 2002, 277, 12343–12350 CrossRef CAS.
- C. R. Majhi, S. Khan, M. D. M. Leo, A. Manimaran and P. Sankar, Food Chem. Toxicol., 2011, 49, 974–982 CrossRef CAS.
- A. F. Mendes, A. P. Carvalho, M. M. Caramona and M. C. Lopes, Mediators Inflammation, 2001, 10, 209–215 CrossRef CAS.
- A. F. Mendes, M. M. Caramona, Y. P. Carvalho and M. C. Lopes, Nitric Oxide, 2002, 6, 35–44 CrossRef CAS.
- Y. P. Yen, K. S. Tsai, Y. W. Chen, C. F. Huang, R. S. Yang and S. H. Liu, Arch. Toxicol., 2012, 86, 923–933 CrossRef CAS.
- S. J. Riedl and G. S. Salvesen, Nat. Rev. Mol. Cell Biol., 2007, 8, 405–413 CrossRef CAS.
- L. Racioppi, P. K. Noeldner, F. Lin, S. Arvai and A. R. Means, J. Biol. Chem., 2012, 287, 11579–11591 CrossRef CAS.
- Y. Yamamoto and R. B. Gaynor, Curr. Mol. Med., 2001, 1, 287–296 CrossRef CAS.
- S. Choi, J. H. Kim, E. J. Roh, M. J. Ko, J. E. Jung and H. J. Kim, J. Biol. Chem., 2006, 281, 12722–12728 CrossRef CAS.
- W. H. Kim, W. B. Park, B. Gao and M. H. Jung, Mol. Pharmacol., 2004, 66, 1383–1396 CrossRef CAS.
- D. H. Walter, J. Haendeler, J. Galle, A. M. Zeiher and S. Dimmeler, Circulation, 1998, 98, 1153–1157 CrossRef CAS.
- J. G. Kiang, P. D. Bowman, X. Lu, Y. Li, B. W. Wu, H. H. Loh, K. T. Tsen and G. C. Tsokos, J. Appl. Physiol., 2007, 103, 1045–1055 CrossRef CAS.
- R. L. Cox, T. Mariano, D. E. Heck, J. D. Laskin and J. J. Stegeman, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2001, 130, 479–491 CrossRef CAS.
- C. Y. Chai, Y. C. Huang, W. C. Hung, W. Y. Kang and W. T. Chen, Toxicol. Lett., 2007, 173, 48–56 CrossRef CAS.
- M. Yadav, S. K. Roach and J. S. Schorey, J. Immunol., 2004, 172, 5588–5597 CAS.
- J. Si and S. J. Collins, Cancer Res., 2008, 68, 3733–3742 CrossRef CAS.
- L. M. Livshitz and Y. Rudy, Am. J. Physiol.: Heart Circ. Physiol., 2007, 292, 2854–2866 CrossRef.
- G. Mellgren, T. Bruland, A. P. Doskeland, T. Flatmark, O. K. Vintermyr and S. O. Doskeland, Endocrinology, 1997, 138, 4373–4383 CrossRef CAS.
- S. Chen, Y. Xu, B. Xu, M. Guo, Z. Zhang and L. Liu, J. Neurochem., 2011, 119, 1108–1118 CrossRef CAS.
- S. L. Bissonnette, A. Haas, K. K. Mann and J. J. Schlezinger, Toxicol. Sci., 2010, 118, 108–118 CrossRef CAS.
- S. C. Wright, U. Schellenberger, L. Ji, H. Wang and J. W. Larrick, FASEB J., 1997, 11, 843–849 CAS.
- J. F. Erickson, M. I. A. Joiner, X. Guan, W. Kutschke, J. Yang and C. V. Oddis, Cell, 2008, 133, 462–474 CrossRef CAS.
- S. Nishio, Y. Teshima, N. Takahashi, L. C. Thuc, S. Saito, A. Fukui, O. Kume, N. Fukunaga, M. Hara, M. Nakagawa and T. Saikawa, J. Mol. Cell. Cardiol., 2012, 52, 1103–1111 CrossRef CAS.
- R. Nasr and H. de The, Int. J. Hematol., 2010, 91, 742–747 CrossRef.
- R. Eguchi, Y. Fujimori, H. Takeda, C. Tabata, T. Ohta, K. Kuribayashi, K. Fukuoka and T. Nakano, J. Cell. Physiol., 2011, 226, 762–768 CrossRef CAS.
- M. Aggarwal, S. B. Naraharisetti, S. Dandapat, J. H. Degen and J. K. Malik, Toxicology, 2008, 251, 51–60 CrossRef CAS.
- S. Lynn, J. N. Shiung, J. R. Gurr and K. Y. Jan, Free Radical Biol. Med., 1998, 24, 442–449 CrossRef CAS.
- V. Selvaraj, M. A. Armistead, M. Cohenford and E. Murray, Chemosphere, 2013, 90, 1201–1209 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |