Chronic ethanol and high glucose inducible CYP2E1 mediated oxidative stress leads to greater cellular injury in VL-17A cells: a potential mechanism for liver injury due to chronic alcohol consumption and hyperglycemia
Kavitha
Swaminathan
a,
S. Mathan
Kumar
a,
Dahn L.
Clemens
bc and
Aparajita
Dey
*a
aLife Science Division, AU-KBC Research Centre, MIT Campus of Anna University, Chromepet, Chennai-600044, India. E-mail: aparajitabhu@rediffmail.com; aparajitadey21@gmail.com; aparajita@au-kbc.org; Fax: +91 44 2223 2711; Tel: +91 44 2223 4885 Extension 149; +91 9790848299
bNebraska and Western Iowa Veterans Administration Medical Center, University of Nebraska Medical Center, Omaha, NE, USA
cDepartment of Internal Medicine, University of Nebraska Medical Center, Omaha, NE, USA
First published on 11th April 2013
Abstract
Diabetes, characterized by the presence of inherent oxidative stress, may be further complicated by the additional oxidative stress generated due to the metabolism of alcohol. This study focuses on the roles of alcohol and high glucose inducible ADH and CYP2E1, both of which function as ethanol metabolizing enzymes, in promoting oxidative stress and cellular damage under chronic alcohol plus hyperglycemic conditions in VL-17A cells over-expressing ADH and CYP2E1. A specific CYP2E1 inhibitor, diallyl sulfide, proved to be more effective in decreasing the toxicity in VL-17A cells exposed to chronic alcohol plus high glucose than the specific ADH inhibitor pyrazole and the dual ADH and CYP2E1 inhibitor 4-methyl pyrazole. Furthermore, the greatest decrease in several parameters for oxidative stress such as ROS level, lipid peroxidation, protein carbonyl and protein aldehyde adduct formation and apoptosis was observed in the diallyl sulfide plus chronic alcohol plus high glucose treated VL-17A cells. In addition, specific inhibition of CYP2E1 with phenethyl isothiocyanate or CYP2E1 siRNA led to significant restoration of viability, and decrease in oxidative stress and apoptosis. Thus, the inducibility of CYP2E1 with both ethanol and high glucose leads to greater oxidative stress and cellular dysfunction in liver.
Introduction
Both alcoholism and hyperglycemia are associated with liver injury as distinct causative agents.1,2 Alcohol or hyperglycemia mediated liver injury can occur through several mechanisms among which oxidative stress is a prominent mechanism.1,3–5 The oxidative metabolism of substrates through cytochrome P4502E1 (CYP2E1) catalyzed reactions leads to oxidative stress in the cellular environment due to the ability of CYP2E1 to generate reactive oxygen species (ROS).6,7 Further, both alcohol and high concentrations of glucose independently upregulate CYP2E1.8,9In a recent study, using the hepatoma cell line VL-17A whose cells over-express alcohol dehydrogenase (ADH) and CYP2E1, the enzymes involved in ethanol metabolism, we showed that the VL-17A cells exhibit greater oxidative stress and toxicity than HepG2 cells devoid of ADH and CYP2E1 expression when exposed to both chronic alcohol and hyperglycemia.10 The study, using an in vitro model, provided evidence that chronic alcohol aggravates liver injury due to hyperglycemia which is an underlying factor in diabetes.10 Further, oxidative stress may be critical in promoting the synergistic toxicity of both agents as both chronic alcohol and hyperglycemia lead to oxidative stress. Thus, overlapping domains occur between chronic ethanol plus high glucose mediated liver injury, CYP2E1 and oxidative stress. The present study, using specific inhibitors for CYP2E1 (diallyl sulfide) and ADH (pyrazole), and the dual ADH and CYP2E1 inhibitor 4-methyl pyrazole, investigates the relative contribution of ADH and CYP2E1 in mediating the greater oxidative stress and toxicity due to chronic ethanol plus high glucose in liver cells.
Materials and methods
HepG2 cells, which do not express ADH and CYP2E1, and VL-17A cells, which are HepG2 cells constitutively expressing ADH and CYP2E1, were used for the study.11 HepG2 cells were obtained from the National Centre for Cell Sciences, Pune, India. VL-17A cells were kindly provided by Dr DL Clemens, University of Nebraska Medical Center and Veterans Affairs Medical Center, Nebraska USA. Cultures of HepG2 and VL-17A cells were grown and experiments with 100 mM ethanol were performed as described previously.12Both HepG2 and VL-17A cells were each divided into ten experimental groups: (1) untreated cells in the first group served as the control; (2) cells treated with 50 mM glucose for 72 hours served as the hyperglycemic group; (3) cells treated with 100 mM ethanol for 72 hours served as the chronic alcohol group; (4) cells treated with 100 mM ethanol and 50 mM glucose for 72 hours served as the chronic alcohol plus high glucose group; (5) cells treated with 10 μM diallyl sulfide, a CYP2E1 inhibitor, for 72 hours; (6) cells treated with 2 mM pyrazole, an ADH inhibitor, for 72 hours; and (7) cells treated with 5 mM 4-methyl pyrazole, a dual CYP2E1 and ADH inhibitor, for 72 hours; (8) cells treated with 10 μM diallyl sulfide, a CYP2E1 inhibitor, plus 100 mM ethanol and 50 mM glucose for 72 hours; (9) cells treated with 2 mM pyrazole, an ADH inhibitor, plus 100 mM ethanol and 50 mM glucose for 72 hours; and (10) cells treated with 5 mM 4-methyl pyrazole, a dual CYP2E1 and ADH inhibitor , plus 100 mM ethanol and 50 mM glucose for 72 hours.
Experiments were performed with 10 μM diallyl sulfide (DAS), a specific CYP2E1 inhibitor;13,14 ADH-specific pyrazole (PYR, 2 mM)15 and the dual inhibitor of both ADH and CYP2E1 4-methyl pyrazole (4-MP, 5 mM).11,13
In some experiments, liver cells were treated with either another specific inhibitor of CYP2E1, phenethyl isothiocyanate (PIC, 0.1 mM)16,17 or siRNA CYP2E1.
The cell viability was assayed by MTT assay, using an MTT Cell Proliferation Assay Kit according to the manufacturer's instructions. Fluorescence spectrometry was used to measure the intracellular levels of ROS with 2′,7′-dichlorofluorescein diacetate (DCF-DA) as described previously.18 Lipid peroxidation was measured using a protocol of the Nanotechnology Characterization Laboratory using a thiobarbituric acid reactive (TBAR) method.19 Protein carbonyl content in cells was analyzed spectrophotometrically as described by Palmeira.20 Glutathione (GSH) content was determined in liver cells and the increase in absorbance at 412 nm was converted to GSH concentration by using a standard curve with known amounts of GSH.21
Caspase 3 activity was measured in the supernatant by detecting the hydrolysis of Ac-DEVD-p-nitroanilide (Ac-DEVD-pNA), resulting in the release of p-nitroaniline (pNA) as previously described.18 Apoptosis or necrosis was determined using an Annexin V-FITC apoptosis detection kit (Sigma Aldrich, India) as previously described.18 Apoptotic cells positively stained with Annexin V showed green fluorescence and necrotic cells appeared as red fluorescent cells.
Malondialdehyde (MDA)-, acetaldehyde (AA)- and malondialdehyde-acetaldehyde (MAA)-adduct formation were detected by modifying previously described methods and protein concentration was measured by the method of Lowry.22–24 CYP2E1 and ADH protein expression in liver cells were monitored by Western blotting.18 The antibody for ADH was procured from Abcam, USA and the antibody for CYP2E1 was kindly gifted by Dr AI Cederbaum, Mount Sinai School of Medicine, New York, USA.
Student's t-test was performed to analyze results between the different groups. Statistical significance between untreated and treated cells was calculated using ANOVA single factor analysis. A value of p < 0.05 was considered to be statistically significant.
Results
Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on cell viability of HepG2 and VL-17A cells
HepG2 and VL-17A cells treated with 10 μM DAS or 2 mM PYR or 5 mM 4-MP exhibited 95–102% viability (Fig. 1). While treatment of HepG2 cells with 100 mM ethanol did not significantly decrease the viability (88%), 50 mM glucose or 100 mM ethanol plus 50 mM glucose treatment resulted in 68–70% viability when compared with untreated HepG2 cells. In contrast, treatment of VL-17A cells with 50 mM glucose, 100 mM ethanol or 100 mM ethanol plus 50 mM glucose decreased the viability to 63%, 55% and 38% respectively when compared with untreated VL-17A cells. | ||
| Fig. 1 Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on viability of HepG2 and VL-17A cells. HepG2 and VL-17A cells were treated with 50 mM glucose or 100 mM ethanol or 100 mM ethanol plus 50 mM glucose or 10 μM diallyl sulfide (DAS); 2 mM pyrazole (PYR); and 5 mM 4-methyl pyrazole (4-MP); or 10 μM DAS plus 100 mM ethanol plus 50 mM glucose; or 2 mM PYR plus 100 mM ethanol plus 50 mM glucose; or 5 mM 4-MP plus 100 mM ethanol plus 50 mM glucose for 72 hours. Cell viability was measured through MTT assay. Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
Furthermore, pretreatment of 100 mM ethanol plus 50 mM glucose treated HepG2 cells with 10 μM DAS or 2 mM PYR or 5 mM 4-MP did not change the viability (67–70%) when compared with 100 mM ethanol plus 50 mM glucose treated HepG2 cells. However, VL-17A cells treated with 100 mM ethanol plus 50 mM glucose pretreated with 10 μM DAS or 2 mM PYR or 5 mM 4-MP exhibited distinct increases in viability i.e. 111%, 90% and 81% respectively when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on intracellular reactive oxygen species level in HepG2 and VL-17A cells
Untreated VL-17A cells or VL-17A cells treated with 10 μM DAS or 2 mM PYR or 5 mM 4-MP exhibited 1.4–1.7-fold higher ROS levels than the corresponding groups of HepG2 cells (Fig. 2(a)). HepG2 cells treated with 50 mM glucose or 100 mM ethanol plus 50 mM glucose exhibited 1.9–2.0-fold increases in their ROS levels when compared with untreated HepG2 cells. In contrast, HepG2 cells treated with 100 mM ethanol were characterized by unchanged ROS levels when compared with untreated HepG2 cells. Furthermore, treatment with 10 μM DAS or 2 mM PYR or 5 mM 4-MP did not cause significant changes in 100 mM ethanol plus 50 mM glucose treated HepG2 cells when compared with 100 mM ethanol plus 50 mM glucose treated HepG2 cells.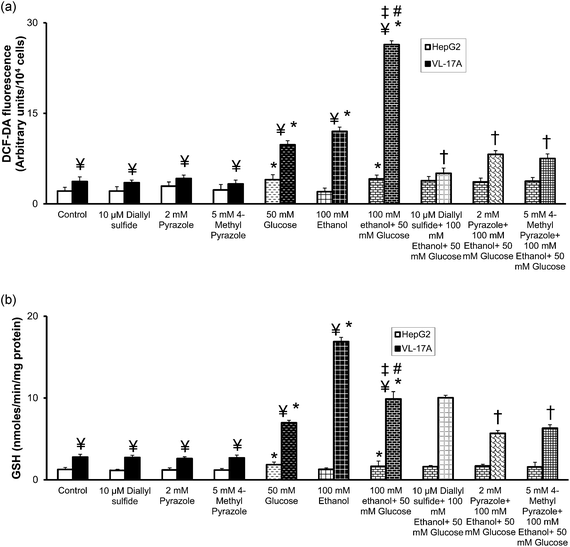 | ||
| Fig. 2 Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on (a) intracellular reactive oxygen species levels and (b) GSH levels in HepG2 and VL-17A cells. HepG2 and VL-17A cells were treated with 50 mM glucose or 100 mM ethanol or 100 mM ethanol plus 50 mM glucose or 10 μM diallyl sulfide (DAS); 2 mM pyrazole (PYR); and 5 mM 4-methyl pyrazole (4-MP); or 10 μM DAS plus 100 mM ethanol plus 50 mM glucose; or 2 mM PYR plus 100 mM ethanol plus 50 mM glucose; or 5 mM 4-MP plus 100 mM ethanol plus 50 mM glucose for 72 hours. (a) The levels of intracellular ROS were measured by fluorescence spectrometry using 2′,7′-DCF-DA as the probe. (b) The total GSH content of samples was assayed by measuring the rate of 2-nitro-5-thiobenzoic acid production. Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
However, VL-17A cells treated with 50 mM glucose, 100 mM ethanol or 100 mM ethanol plus 50 mM glucose exhibited 2.7-fold, 3.2-fold and 7.1-fold increases in their ROS levels when compared with untreated VL-17A cells. Furthermore, pretreatment of 100 mM ethanol plus 50 mM glucose treated VL-17A cells with 10 μM DAS or 2 mM PYR or 5 mM 4-MP caused 5.2-, 3.2- and 3.5-fold decreases in ROS levels when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
Treatment of HepG2 cells with 50 mM glucose or 100 mM ethanol plus 50 mM glucose caused 1.2-fold increases in lipid peroxidation when compared with untreated HepG2 cells, which was not observed in 100 mM ethanol treated HepG2 cells (Table 1). The inhibitors did not cause further changes in lipid peroxidation in HepG2 cells treated with the toxins. Further, VL-17A cells treated with 50 mM glucose, 100 mM ethanol or 100 mM ethanol plus 50 mM glucose exhibited 1.7-fold, 1.9-fold and 2.5-fold increases in lipid peroxidation when compared with untreated VL-17A cells. Pretreatment of 100 mM ethanol plus 50 mM glucose treated VL-17A cells with 10 μM DAS or 2 mM PYR or 5 mM 4-MP caused 2.8-, 2.3- and 1.9-fold decreases in lipid peroxidation when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
| Groups | Lipid peroxidation (arbitrary fluorescence units mg−1 protein) | |
|---|---|---|
| HepG2 | VL-17A | |
| Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
| Control | 0.48 ± 0.012 | 0.90 ± 0.007¥ |
| 10 μM diallyl sulfide | 0.47 ± 0.098 | 0.51 ± 0.015*¥ |
| 2 mM pyrazole | 0.49 ± 0.046 | 0.83 ± 0.12*¥ |
| 5 mM 4-methyl pyrazole | 0.48 ± 0.012 | 0.74 ± 0.010*¥ |
| 50 mM glucose | 0.58 ± 0.012* | 1.53 ± 0.14*¥ |
| 100 mM ethanol | 0.48 ± 0.013 | 1.36 ± 0. 12*¥ |
| 100 mM ethanol + 50 mM glucose | 0.56 ± 0.012* | 2.02 ± 0.013*¥‡# |
| 10 μM diallyl sulfide + 100 mM ethanol + 50 mM glucose | 0.60 ± 0.012 | 0.83 ± 0.012† |
| 2 mM pyrazole + 100 mM ethanol + 50 mM glucose | 0.55 ± 0.010 | 0.95 ± 0.015† |
| 5 mM 4-methyl pyrazole + 100 mM ethanol + 50 mM glucose | 0.57 ± 0.012 | 1.13 ± 0.12† |
Effects of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on protein carbonyl formation in HepG2 and VL-17A cells
Untreated VL-17A cells exhibited 1.7-fold higher protein carbonyl formation than the corresponding groups of HepG2 cells (Table 2). None of the inhibitors caused significant changes in protein carbonyl formation in HepG2 cells. VL-17A cells treated with 10 μM DAS or 5 mM 4-MP did not exhibit significant changes in protein carbonyl formation when compared with untreated VL-17A cells. Treatment of VL-17A cells with 2 mM PYR caused a 1.2-fold increase in protein carbonyl formation when compared with untreated VL-17A cells.| Groups | Protein carbonyl (nmol mg−1 protein) | |
|---|---|---|
| HepG2 | VL-17A | |
| Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
| Control | 7.19 ± 0.62 | 12.56 ± 0.98¥ |
| 10 μM diallyl sulfide | 8.71 ± 0.51 | 11.97 ± 0.51¥ |
| 2 mM pyrazole | 6.62 ± 0.98 | 15.52 ± 0.91*¥ |
| 5 mM 4-methyl pyrazole | 7.33 ± 0.51 | 12.65 ± 0.61¥ |
| 50 mM glucose | 17.88 ± 0.87* | 47.80 ± 0.22*¥ |
| 100 mM ethanol | 7.35 ± 0.45 | 52.40 ± 0.92*¥ |
| 100 mM ethanol + 50 mM glucose | 18.40 ± 0.86* | 70.71 ± 0.83*¥‡# |
| 10 μM Diallyl sulfide + 100 mM Ethanol + 50 mM Glucose | 17.80 ± 0.78 | 30.70 ± 0.16† |
| 2 mM pyrazole + 100 mM ethanol + 50 mM glucose | 17.65 ± 0.65 | 66.41 ± 0.19† |
| 5 mM 4-methyl pyrazole + 100 mM ethanol + 50 mM glucose | 17.36 ± 0.49 | 36.50 ± 0.78† |
Treatment of HepG2 cells with 50 mM glucose or 100 mM ethanol plus 50 mM glucose resulted in 2.5-fold increase in protein carbonyl formation, with 100 mM ethanol not exerting significant effects when compared with untreated HepG2 cells. Furthermore, treatment of VL-17A cells with 50 mM glucose or 100 mM ethanol or 100 mM ethanol plus 50 mM glucose resulted in 3.9-, 4.2- and 5.6-fold increases in protein carbonyl formation respectively when compared with untreated VL-17A cells.
While the inhibitors did not exert their inhibitory effects in 100 mM ethanol plus 50 mM glucose treated HepG2 cells, 10 μM DAS or 2 mM PYR or 5 mM 4-MP decreased protein carbonyl formation 2.3-, 1.1- and 1.7-fold respectively in 100 mM ethanol plus 50 mM glucose treated VL-17A cells when compared with VL-17A cells treated with both toxins.
Effects of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on GSH level in HepG2 and VL-17A cells
Untreated VL-17A cells or VL-17A cells treated with 10 μM DAS or 2 mM PYR or 5 mM 4-MP exhibited 2.1–2.4-fold higher GSH level than the corresponding groups of HepG2 cells (Fig. 2(b)). With 100 mM ethanol exerting no significant effect, 50 mM glucose or 100 mM ethanol plus 50 mM glucose increased GSH 1.3–1.5-fold in HepG2 cells when compared with HepG2 cells not subjected to treatment with either toxin. Treatment of VL-17A cells with 50 mM glucose or 100 mM ethanol resulted in 2.5- and 6.1-fold increases in GSH level respectively and 100 mM ethanol plus 50 mM glucose exerted an intermediate effect i.e. GSH level was increased 3.5-fold when compared with untreated VL-17A cells.While the inhibitors were ineffective in 100 mM ethanol plus 50 mM glucose treated HepG2 cells, 2 mM PYR or 5 mM 4-MP decreased GSH level 1.5–1.7-fold with 10 μM DAS causing less than 1.0-fold change in 100 mM ethanol plus 50 mM glucose treated VL-17A cells when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on MDA-, AA-, and MAA-adduct formation in VL-17A cells
The effects of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on MDA-, AA-, and MAA-adduct formation is shown in Tables 3–6. The basal or endogenous MDA-, AA-, and MAA-adduct formation in the absence of in vitro addition of MDA or acetaldehyde in untreated VL-17A cells was 1.8-fold higher than the corresponding HepG2 cells (Table 3). HepG2 or VL-17A cells treated with 10 μM DAS or 2 mM PYR or 5 mM 4-MP did not exhibit significant changes in endogenous MDA-, AA-, and MAA-adduct formation when compared with the corresponding groups of untreated liver cells.| Groups | Basal MDA, AA, MAA adduct formation (basal arbitrary fluorescence units mg−1 protein) | |
|---|---|---|
| HepG2 | VL-17A | |
| Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
| Control | 0.133 ± 0.044 | 0.242 ± 0.035¥ |
| 10 μM diallyl sulfide | 0.135 ± 0.023 | 0.238 ± 0.044¥ |
| 2 mM pyrazole | 0.130 ± 0.055 | 0.230 ± 0.032¥ |
| 5 mM 4-methyl Pyrazole | 0.137 ± 0.057 | 0.232 ± 0.043¥ |
| 50 mM glucose | 0.259 ± 0.058* | 0.601 ± 0.086*¥ |
| 100 mM ethanol | 0.139 ± 0.012 | 1.250 ± 0.047*¥ |
| 100 mM ethanol + 50 mM glucose | 0.248 ± 0.023* | 1.890 ± 0.076*¥‡# |
| 10 μM diallyl sulfide + 100 mM ethanol + 50 mM glucose | 0.262 ± 0.068 | 0.514 ± 0.048† |
| 2 mM pyrazole + 100 mM ethanol + 50 mM glucose | 0.235 ± 0.058 | 0.956 ± 0.023† |
| 5 mM 4-methyl pyrazole + 100 mM ethanol + 50 mM glucose | 0.241 ± 0.047 | 0.829 ± 0.052† |
Treatment of HepG2 and VL-17A cells with 50 mM glucose caused 1.9- and 2.5-fold increases in arbitrary fluorescence, which is indicative of endogenous protein adduct formation. Although basal MDA-, AA-, and MAA-adduct formation was not affected in HepG2 cells exposed to 100 mM ethanol, an increase of 5.0-fold was observed in the corresponding group of VL-17A cells when compared with the untreated liver cells. Furthermore, 100 mM ethanol plus 50 mM glucose treated HepG2 and VL-17A cells exhibited 1.8- and 7.8-fold increases in basal MDA-, AA- and MAA-adduct formation, respectively, when compared with untreated HepG2 and VL-17A cells respectively.
Although, the inhibitors did not decrease the basal protein adduct formation in 100 mM ethanol plus 50 mM glucose treated HepG2 cells, VL-17A cells pretreated with or 10 μM DAS or 2 mM PYR or 5 mM 4-MP and then treated with 100 mM ethanol plus 50 mM glucose showed 3.7-, 1.9- and 2.3-fold decreases in protein adduct formation, respectively.
The effect of in vitro incubation of cell lysates obtained from HepG2 and VL-17A cells with 1 mM MDA is shown in Table 4. Untreated VL-17A cells or VL-17A cells treated with 10 μM DAS or 2 mM PYR or 5 mM 4-MP exhibited 1.2–1.4-fold increased MDA-adduct formation when compared with the corresponding groups of HepG2 cells. HepG2 cells treated with 50 mM glucose or 100 mM ethanol plus 50 mM glucose exhibited 2.7–2.9-fold increases in MDA-adduct formation whereas 100 mM ethanol treated HepG2 cells were characterized by non-significant changes in MDA-adduct formation. VL-17A cells treated with 50 mM glucose or 100 mM ethanol or 100 mM ethanol plus 50 mM glucose were characterized by 4.3-, 7.8- and 9.1-fold increases in MDA-adduct formation, respectively.
| Groups | MDA adduct formation (arbitrary fluorescence units mg−1 protein) | |
|---|---|---|
| HepG2 | VL-17A | |
| Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
| Control | 5.19 ± 0.15 | 6.40 ± 0.52¥ |
| 10 μM diallyl sulfide | 5.10 ± 0.20 | 6.03 ± 0.76¥ |
| 2 mM pyrazole | 5.30 ± 0.22 | 6.20 ± 0.56¥ |
| 5 mM 4-methyl pyrazole | 5.12 ± 0.07 | 6.12 ± 0.45¥ |
| 50 mM glucose | 14.80 ± 0.58* | 25.98 ± 0.88*¥ |
| 100 mM ethanol | 5.64 ± 0.34 | 47.84 ± 0.40*¥ |
| 100 mM ethanol + 50 mM glucose | 14.95 ± 0.09* | 55.68 ± 0.82*¥‡# |
| 10 μM diallyl sulfide + 100 mM ethanol + 50 mM glucose | 14.50 ± 0.56 | 22.87 ± 0.36† |
| 2 mM pyrazole + 100 mM ethanol + 50 mM glucose | 14.90 ± 0.33 | 46.31 ± 0.54† |
| 5 mM 4-methyl pyrazole + 100 mM ethanol + 50 mM glucose | 14.40 ± 0.48 | 23.01 ± 0.47† |
While the inhibitors did not change the MDA-adduct formation in 100 mM ethanol plus 50 mM glucose treated HepG2 cells, 10 μM DAS or 5 mM 4-MP pretreated and 100 mM ethanol plus 50 mM glucose treated VL-17A cells showed 2.4-fold decrease and 2 mM PYR showed 1.2-fold decrease in MDA-adduct formation when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
AA-adduct formation due to in vitro incubation of cell lysates obtained from HepG2 and VL-17A cells with 1 mM acetaldehyde is shown in Table 5. Untreated VL-17A cells exhibited a 5.6-fold increase in AA-adduct formation when compared with untreated HepG2 cells. The CYP2E1, ADH or CYP2E1 and ADH inhibitors did not cause significant changes in AA-adduct formation in HepG2 cells. While VL-17A cells treated with 5 mM 4-MP did not exhibit significant changes in AA-adduct formation, treatment of VL-17A cells with 10 μM DAS or 2 mM PYR resulted in 1.1-fold decrease and 1.4-fold increase respectively in AA-adduct formation when compared with untreated VL-17A cells.
| Groups | AA adduct formation (arbitrary fluorescence units mg−1 protein) | |
|---|---|---|
| HepG2 | VL-17A | |
| Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
| Control | 1.216 ± 0.044 | 6.694 ± 0.044¥ |
| 10 μM diallyl sulfide | 1.301 ± 0.023 | 6.105 ± 0.035*¥ |
| 2 mM pyrazole | 1.029 ± 0.055 | 9.289 ± 0.042*¥ |
| 5 mM 4-methyl pyrazole | 1.110 ± 0.067 | 6.687 ± 0.086¥ |
| 50 mM glucose | 2.120 ± 0.058* | 14.090 ± 0.053*¥ |
| 100 mM ethanol | 1.560 ± 0.012 | 33.086 ± 0.035*¥ |
| 100 mM ethanol + 50 mM glucose | 2.166 ± 0.023* | 41.088 ± 0.044*¥‡# |
| 10 μM diallyl sulfide + 100 mM ethanol + 50 mM glucose | 2.185 ± 0.061 | 6.167 ± 0.076† |
| 2 mM pyrazole + 100 mM ethanol + 50 mM glucose | 2.172 ± 0.038 | 10.738 ± 0.047† |
| 5 mM 4-methyl pyrazole + 100 mM ethanol + 50 mM glucose | 2.123 ± 0.058 | 9.999 ± 0.023† |
HepG2 cells treated with 50 mM glucose or 100 mM ethanol plus 50 mM glucose exhibited a 1.8-fold increase in AA-adduct formation whereas 100 mM ethanol treated HepG2 cells were characterized by non-significant changes in AA-adduct formation. VL-17A cells treated with 50 mM glucose or 100 mM ethanol or 100 mM ethanol plus 50 mM glucose were characterized by 2.1-, 4.7- and 6.2-fold increases in AA-adduct formation, respectively. While the inhibitors did not change the AA-adduct formation in 100 mM ethanol plus 50 mM glucose treated HepG2 cells, 10 μM DAS or 5 mM 4-MP or 2 mM PYR pretreated 100 mM ethanol plus 50 mM glucose treated VL-17A cells showed 6.6-, 3.8- and 4.1-fold decreases in AA-adduct formation, respectively, when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
MAA-adduct formation due to in vitro incubation of cell lysates obtained from HepG2 and VL-17A cells with 1 mM MDA and 1 mM acetaldehyde is shown in Table 6. Untreated VL-17A cells exhibited 1.4-fold increased MAA-adduct formation when compared with untreated HepG2 cells. The inhibitors did not significantly affect MAA-adduct formation in HepG2 cells. Treatment of VL-17A cells with 10 μM DAS resulted in a 1.6-fold decrease and 2 mM PYR or 5 mM 4-MP did not cause significant changes in MAA-adduct formation when compared with untreated VL-17A cells.
| Groups | MAA adduct formation (arbitrary fluorescence units mg−1 protein) | |
|---|---|---|
| HepG2 | VL-17A | |
| Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
| Control | 6.70 ± 0.57 | 9.20 ± 0.45¥ |
| 10 μM diallyl sulfide | 6.70 ± 0.63 | 5.66 ± 0.17*¥ |
| 2 mM pyrazole | 6.84 ± 0.38 | 9.47 ± 0.84¥ |
| 5 mM 4-methyl pyrazole | 6.48 ± 0.77 | 9.70 ± 0.12¥ |
| 50 mM glucose | 16.40 ± 0.45* | 34.96 ± 0.27*¥ |
| 100 mM ethanol | 6.90 ± 0.82 | 60.95 ± 0.43¥ |
| 100 mM ethanol + 50 mM glucose | 16.90 ± 0.20* | 89.51 ± 0.93*¥‡# |
| 10 μM diallyl sulfide + 100 mM ethanol + 50 mM glucose | 16.30 ± 0.15 | 35.34 ± 0.32† |
| 2 mM pyrazole + 100 mM ethanol + 50 mM glucose | 16.200 ± 0.89 | 57.74 ± 0.52† |
| 5 mM 4-methyl pyrazole + 100 mM ethanol + 50 mM glucose | 16.800 ± 0.55 | 54.39 ± 0.46† |
HepG2 cells treated with 50 mM glucose or 100 mM ethanol plus 50 mM glucose exhibited 2.3–2.5-fold increase in MAA-adduct formation whereas 100 mM ethanol treated HepG2 cells were characterized by non-significant changes in MAA-adduct formation. VL-17A cells treated with 50 mM glucose or 100 mM ethanol or 100 mM ethanol plus 50 mM glucose were characterized by 2.4-, 6.6- and 9.7-fold increases respectively in MAA-adduct formation.
While the MAA-adduct formation was not affected by the inhibitors in 100 mM ethanol plus 50 mM glucose treated HepG2 cells, 10 μM DAS pretreated 100 mM ethanol plus 50 mM glucose treated VL-17A cells showed a 2.5-fold decrease and 2 mM PYR or 5 mM 4-MP pretreated 100 mM ethanol plus 50 mM glucose treated VL-17A cells showed a 1.6-fold decrease in MAA-adduct formation when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
Effects of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on caspase 3 activity in HepG2 and VL-17A cells
Untreated VL-17A cells exhibited 1.2-fold higher caspase 3 activity than untreated HepG2 cells (Fig. 3(a)). HepG2 and VL-17A cells treated with 10 μM DAS or 2 mM PYR or 5 mM 4-MP did not exhibit significant changes in caspase 3 activity when compared with untreated HepG2 and VL-17A cells, respectively. | ||
| Fig. 3 Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on (a) caspase-3 activity; and (b) Annexin V-propidium iodide staining in HepG2 and VL-17A cells. HepG2 and VL-17A cells were treated with 50 mM glucose or 100 mM ethanol or 100 mM ethanol plus 50 mM glucose or 10 μM diallyl sulfide (DAS); 2 mM pyrazole (PYR); and 5 mM 4-methyl pyrazole (4-MP); or 10 μM DAS plus 100 mM ethanol plus 50 mM glucose; or 2 mM PYR plus 100 mM ethanol plus 50 mM glucose; or 5 mM 4-MP plus 100 mM ethanol plus 50 mM glucose for 72 hours. (a) Caspase 3 activity was measured by the formation of pNA from Ac-DEVD-pNA. Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. (b) Photomicrographs of Annexin V-PI stained HepG2 and VL-17A cells visualized with fluorescence microscope are shown. One representative experiment of three is shown. | ||
Treatment of HepG2 and VL-17A cells with 50 mM glucose caused 1.0 and 1.2-fold increases in caspase 3 activity, respectively. While 100 mM ethanol increased the caspase 3 activity 1.5-fold in VL-17A cells, HepG2 cells were unaffected. Furthermore, 100 mM ethanol plus 50 mM glucose treated HepG2 and VL-17A cells were characterized by 1.1 and 2.0-fold increases in caspase 3 activity, respectively.
Although pretreatment of 100 mM ethanol plus 50 mM glucose treated HepG2 cells with 10 μM DAS or 2 mM PYR or 5 mM 4-MP did not affect caspase 3 activity, 10 μM DAS or 2 mM PYR or 5 mM 4-MP caused a 1.8-fold decrease in caspase 3 activity in 100 mM ethanol plus 50 mM glucose treated VL-17A cells when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on apoptosis in HepG2 and VL-17A cells
Untreated HepG2 and VL-17A cells or liver cells treated with 10 μM DAS or 2 mM PYR or 5 mM 4-MP were characterized by the presence of few AnnexinV-PI stained cells (+ and ++ respectively) (Fig. 3(b)). Although 100 mM ethanol treated HepG2 cells did not exhibit any change in the number of apoptotic cells (+), treatment of HepG2 cells with 50 mM glucose or 100 mM ethanol plus 50 mM glucose increased the number of apoptotic cells (++). VL-17A cells treated with 50 mM glucose or 100 mM ethanol or 100 mM ethanol plus 50 mM glucose showed increased numbers of Annexin V-PI stained cells: +++, ++++, and +++++ respectively.HepG2 cells treated with the inhibitors and different agents did not show any change in the number of apoptotic cells. While 10 μM DAS or 2 mM PYR were most effective in decreasing the number of Annexin V-PI stained 100 mM ethanol plus 50 mM glucose treated VL-17A (+++), 5 mM 4-MP decreased the number of apoptotic cells to a lesser extent (++++).
Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on ADH protein expression and catalytic activity in HepG2 and VL-17A cells
The effect of the different inhibitors of CYP2E1 and ADH on ADH protein expression and catalytic activity in HepG2 and VL-17A cells is shown in Fig. 4. VL-17A cells treated with 10 μM DAS or 2 mM PYR resulted in insignificant changes in ADH protein expression. However, 5 mM 4-MP caused a 1.0-fold decrease in ADH protein expression when compared with untreated VL-17A cells (Fig. 4(a)). | ||
| Fig. 4 Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on ADH protein expression and catalytic activity in HepG2 and VL-17A cells. HepG2 and VL-17A cells were treated with 100 mM ethanol plus 50 mM glucose or 10 μM diallyl sulfide (DAS); 2 mM pyrazole (PYR); and 5 mM 4-methyl pyrazole (4-MP); or 10 μM DAS plus 100 mM ethanol plus 50 mM glucose; or 2 mM PYR plus 100 mM ethanol plus 50 mM glucose; or 5 mM 4-MP plus 100 mM ethanol plus 50 mM glucose for 72 hours. (a) Top panel: the protein expression of ADH in VL-17A cells is shown. Pre-stained protein marker, medium range (16–98 kDa) obtained from Genei (Merck, USA) was used to identify the molecular weight of ADH. Lane 1: 5 mM 4-MP plus 100 mM ethanol plus 50 mM glucose treated VL-17A cells; lane 2: 10 μM DAS plus 100 mM ethanol plus 50 mM glucose treated VL-17A cells; lane 3: 2 mM PYR plus 100 mM ethanol plus 50 mM glucose treated VL-17A cells; lane 4: 100 mM ethanol plus 50 mM glucose treated VL-17A cells; lane 5: untreated VL-17A cells; lane 6: 5 mM 4-MP treated VL-17A cells; lane 7: 10 μM DAS treated VL-17A cells; and lane 8: 2 mM PYR treated VL-17A cells. One representative experiment of three is shown. Densitometric values for ADH under different treatments are shown in the panel underneath. Middle panel: Western blots showing β-actin protein bands for the corresponding groups represented in the top panel. Lower panel: densitometric values for ADH normalized to expression of β-actin under different treatments are shown. Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated VL-17A cells, † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. (b) ADH activity in HepG2 and VL-17A cells was determined. Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells, and † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
Treatment of VL-17A cells with 100 mM ethanol plus 50 mM glucose caused a 1.6-fold increase in ADH protein expression. Pretreatment of 100 mM ethanol plus 50 mM glucose treated VL-17A cells with 10 μM DAS did not decrease the ADH content but 2 mM PYR or 5 mM 4-MP treated VL-17A cells exhibited 1.0 and 1.2-fold decreases respectively in ADH expression when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
While 10 μM DAS did not affect ADH activity, 2 mM PYR or 5 mM 4-MP caused 1.9- and 1.6-fold decreases in ADH activity, respectively, when compared with untreated VL-17A cells (Fig. 4(b)). VL-17A cells treated with 50 mM glucose, 100 mM ethanol and 100 mM ethanol plus 50 mM glucose exhibited 1.1-, 1.3-, and 1.6-fold increases in ADH activity respectively when compared with untreated VL-17A cells. Pretreatment of 100 mM ethanol plus 50 mM glucose treated VL-17A cells with 10 μM DAS did not decrease ADH activity but 2 mM PYR or 5 mM 4-MP caused 1.5- and 1.2-fold decreases respectively when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on CYP2E1 protein expression and catalytic activity in HepG2 and VL-17A cells
The effect of the different inhibitors of CYP2E1 and ADH on CYP2E1 protein expression and catalytic activity in HepG2 and VL-17A cells is shown in Fig. 5. While 10 μM DAS or 5 mM 4-MP caused insignificant changes in CYP2E1 protein expression, 2 mM PYR caused a 1.3-fold increase in CYP2E1 content when compared with untreated VL-17A cells (Fig. 5(a)). | ||
| Fig. 5 Effect of CYP2E1 or ADH inhibition and CYP2E1 plus ADH inhibition on CYP2E1 protein expression and catalytic activity in HepG2 and VL-17A cells. HepG2 and VL-17A cells were treated with 100 mM ethanol plus 50 mM glucose or 10 μM diallyl sulfide (DAS); 2 mM pyrazole (PYR); and 5 mM 4-methyl pyrazole (4-MP); or 10 μM DAS plus 100 mM ethanol plus 50 mM glucose; or 2 mM PYR plus 100 mM ethanol plus 50 mM glucose; or 5 mM 4-MP plus 100 mM ethanol plus 50 mM glucose for 72 hours. (a) Top panel: the protein expression of CYP2E1 in VL-17A cells is shown. Pre-stained protein marker, medium range (16–98 kDa) obtained from Genei (Merck, USA) was used to identify the molecular weight of CYP2E1. Lane 1: untreated VL-17A cells; lane 2: 10 μM DAS treated VL-17A cells; lane 3: 5 mM 4-MP treated VL-17A cells; lane 4: 2 mM PYR treated VL-17A cells; lane 5: 10 μM DAS plus 100 mM ethanol plus 50 mM glucose treated VL-17A cells; lane 6: 5 mM 4-MP plus 100 mM ethanol plus 50 mM glucose treated VL-17A cells; lane 7: 2 mM PYR plus 100 mM ethanol plus 50 mM glucose treated VL-17A cells; and lane 8: 100 mM ethanol plus 50 mM glucose treated VL-17A cells. One representative experiment of three is shown. Middle panel: Western blots showing β-actin protein bands for the corresponding groups represented in the top panel. Lower panel: densitometric values for CYP2E1 normalized to expression of β-actin under different treatments are shown. Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated VL-17A cells, † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. (b) CYP2E1 activity in HepG2 and VL-17A cells was determined. Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated HepG2 or VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated HepG2 or VL-17A cells, ‡ p < 0.05 compared with 100 mM ethanol treated VL-17A cells, # p < 0.05 compared with 50 mM glucose treated VL-17A cells. | ||
Treatment of VL-17A cells with 100 mM ethanol plus 50 mM glucose caused a 4.8-fold increase in CYP2E1 protein expression. Pretreatment of 100 mM ethanol plus 50 mM glucose treated VL-17A cells with 10 μM DAS or 5 mM 4-MP caused 4.5 and 1.2-fold decreases respectively in CYP2E1 protein expression but 2 mM PYR was ineffective in inhibiting CYP2E1 protein expression when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
While 10 μM DAS and 5 mM 4-MP caused 1.1–1.2-fold decreases in CYP2E1 activity, 2 mM PYR caused a 1.2-fold increase in CYP2E1 activity when compared with untreated VL-17A cells (Fig. 5(b)). VL-17A cells treated with 50 mM glucose, 100 mM ethanol and 100 mM ethanol plus 50 mM glucose exhibited 1.5-, 1.8-, and 2.4-fold increases in CYP2E1 activity respectively when compared with untreated VL-17A cells. Pretreatment of 100 mM ethanol plus 50 mM glucose treated VL-17A cells with 10 μM DAS caused 1.4-fold decrease in CYP2E1 activity but surprisingly, 2 mM PYR or 5 mM 4-MP caused 1.0–1.2-fold increases in CYP2E1 activity when compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
Effect of CYP2E1 inhibition with 0.1 mM phenethyl isothiocyanate (PIC) on viability; ROS levels; and caspase-3 activity in HepG2 and VL-17A cells
PIC, another specific inhibitor of CYP2E1, was not toxic to HepG2 and VL-17A cells at the used concentration of 0.1 mM (92% and 102% viability respectively) (Fig. 6(a)). HepG2 cells pretreated with 0.1 mM PIC and treated with 100 mM ethanol plus 50 mM glucose did not exhibit any increase in viability (63% versus 62% viability). However, 0.1 mM PIC was effective in increasing the viability of 100 mM ethanol plus 50 mM glucose treated VL-17A cells from 38% to 106%.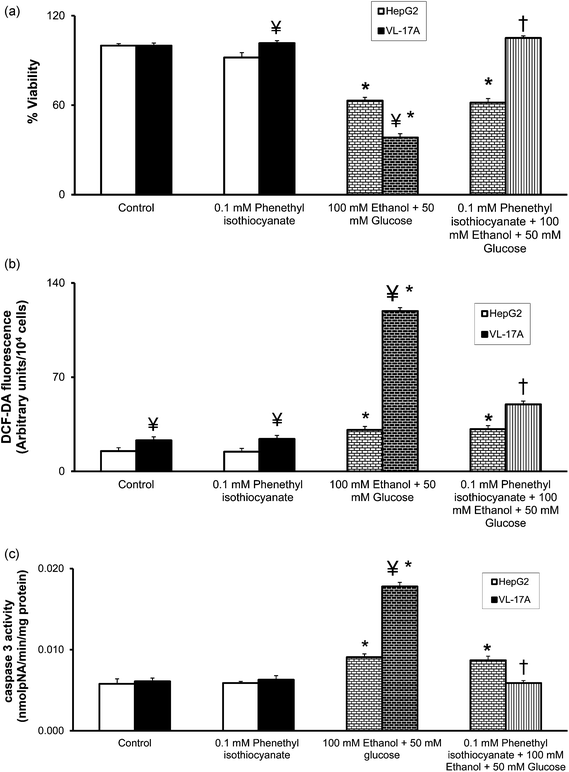 | ||
| Fig. 6 Effect of CYP2E1 inhibition with 0.1 mM phenethyl isothiocyanate (PIC) on (a) viability; (b) ROS levels; and (c) caspase-3 activity in HepG2 and VL-17A cells. HepG2 and VL-17A cells were treated with 100 mM ethanol plus 50 mM glucose or 0.1 mM PIC; or 0.1 mM PIC plus 100 mM ethanol plus 50 mM glucose for 72 hours. (a) Cell viability was measured through an MTT assay. (b) The levels of intracellular ROS were measured by fluorescence spectrometry using 2′,7′-DCF-DA as the probe. (c) The caspase 3 activity was measured by the formation of pNA from Ac-DEVD-pNA. Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated VL-17A cells, ¥ p < 0.05 compared with corresponding HepG2 cells, † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated HepG2 or VL-17A cells. | ||
Similarly, ROS levels in HepG2 and VL-17A cells treated with another specific CYP2E1 inhibitor, PIC, at a concentration of 0.1 mM were unaffected (Fig. 6(b)). While 0.1 mM PIC did not decrease the ROS level in 100 mM ethanol plus 50 mM glucose treated HepG2 cells, it caused a 2.4-fold decrease in the ROS level of 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
Treatment of HepG2 and VL-17A cells with 0.1 mM PIC did not change caspase 3 activity (Fig. 6(c)). While treatment of 100 mM ethanol plus 50 mM glucose treated HepG2 cells with 0.1 mM PIC did not decrease caspase 3 activity, 0.1 mM PIC caused a 3.0-fold decrease in caspase 3 activity of 100 mM ethanol plus 50 mM glucose treated VL-17A cells.
Effect of CYP2E1 inhibition with 10 μM CYP2E1 siRNA on viability; ROS levels; and protein expression in VL-17A cells
Fig. 7 shows the effects of the inhibition of CYP2E1 with CYP2E1 siRNA on cell viability, ROS levels and CYP2E1 protein expression in untreated VL-17A cells and VL-17A cells treated with 100 mM ethanol plus 50 mM glucose. VL-17A cells treated with CYP2E1 siRNA exhibited 108% viability and pretreatment of 100 mM ethanol plus 50 mM glucose treated VL-17A cells with CYP2E1 siRNA increased the viability from 48% to 81% (Fig. 7(a)). | ||
| Fig. 7 Effect of CYP2E1 inhibition with 10 μM CYP2E1 siRNA on (a) viability; (b) ROS levels; and (c) protein expression in VL-17A cells. VL-17A cells were treated with 100 mM ethanol plus 50 mM glucose or 10 μM CYP2E1 siRNA; or 10 μM CYP2E1 siRNA plus 100 mM ethanol plus 50 mM glucose for 72 hours. (a) Cell viability was measured through an MTT assay. (b) The levels of intracellular ROS were measured by fluorescence spectrometry using 2′,7′-DCF-DA as the probe. (c) The protein expression of CYP2E1 in VL-17A cells was determined. Top panel: lanes 1, 2, 3, 4 contain protein samples of 100 mM ethanol plus 50 mM glucose treated; CYP2E1 siRNA plus 100 mM ethanol plus 50 mM glucose treated; untreated; and CYP2E1 siRNA treated VL-17A cells, respectively. Middle panel: Western blots showing β-actin protein bands for the corresponding groups represented in the top panel. Lower panel: densitometric values for CYP2E1 normalized to expression of β-actin under different treatments are shown. Results are expressed as mean ± S.E. of three experiments. * p < 0.05 compared with untreated VL-17A cells, † p < 0.05 compared with 100 mM ethanol plus 50 mM glucose treated VL-17A cells. | ||
Similarly, CYP2E1 siRNA caused a 1.4-fold decrease in the ROS levels in VL-17A cells (Fig. 7(b)). ROS levels were 4.5-fold higher in 100 mM ethanol plus 50 mM glucose treated VL-17A cells when compared with untreated VL-17A cells and CYP2E1 siRNA caused a 2.8-fold decrease in the ROS levels.
The effect of CYP2E1 siRNA on inhibition of CYP2E1 in untreated VL-17A cells and 100 mM ethanol plus 50 mM glucose treated VL-17A cells is shown in Fig. 7(c). Inhibition of CYP2E1 with CYP2E1 siRNA caused a 1.2-fold decrease in CYP2E1 protein expression in untreated VL-17A cells. VL-17A cells treated with 100 mM ethanol plus 50 mM glucose exhibited 2.3-fold increase in CYP2E1 protein expression. CYP2E1 siRNA pretreated 100 mM ethanol plus 50 mM glucose treated VL-17A cells exhibited a 1.8-fold decreased CYP2E1 protein expression.
Discussion
We have previously shown in a study using VL-17A cells that chronic alcohol usage and high concentrations of glucose result in greater oxidative stress and injury in liver cells than either factor is responsible for alone.10 Furthermore, consistent with our observations, a study reported that prenatal ethanol exposure in rats leads to increased insulin resistance and gluconeogenesis in adult rat offsprings.25 Interestingly, in in vivo studies, chronic alcohol exposure inhibits the insulin signaling cascade at several levels in the liver.26 Chronic alcohol consumption leads to a fatty liver which promotes hepatic insulin resistance, oxidative stress and injury in animals,26 thus corroborating our in vitro findings.It may be noted that while ADH plays an important role at low alcohol concentrations, CYP2E1 is a high Km enzyme, assuming more relevance in the context of chronic alcohol exposure.27 Both ADH and CYP2E1 metabolize alcohol to acetaldehyde and CYP2E1 is a key component in the generation of free radicals or ROS species such as superoxide, hydroxyethyl radicals etc. from alcohol.28 Both ADH and CYP2E1 are inducible by alcohol or high glucose10,12,29 and hence their relative contributions to oxidative stress and toxicity due to both agents is not clear.
The present study investigates one of the mechanisms involved in cellular dysfunction due to chronic alcohol usage and high glucose and the first approach was to utilize different inhibitors for ADH and CYP2E1 to ascertain their relative contributions to the injury. Another interesting aspect of the study is the investigation of whether CYP2E1 by virtue of its inducibility by both agents, alcohol and hyperglycemia, and its ability to generate ROS is a key player in the whole phenomenon.
The specific inhibition of CYP2E1 with diallyl sulfide restored the viability of chronic alcohol plus high glucose treated VL-17A cells to the greatest extent as the inducibility of CYP2E1 due to ethanol and high glucose leads to greater ROS generation, an added factor being the ability of CYP2E1 to generate acetaldehyde. Both pyrazole and 4-methyl pyrazole were also effective in decreasing the toxicity in chronic alcohol plus high glucose treated VL-17A cells, though to a lesser extent than diallyl sulfide due to the ability of both ADH and CYP2E1 to yield the toxic metabolite of ethanol–acetaldehyde.
Further, diallyl sulfide was the most effective in decreasing ROS levels and lipid peroxidation in chronic alcohol plus high glucose treated VL-17A cells, than pyrazole and 4-methyl pyrazole which exerted similar effects confirming the role of CYP2E1 in ethanol and high glucose mediated oxidative stress. Diallyl sulfide inhibited protein carbonyl formation to the greatest extent followed by 4-methyl pyrazole and pyrazole treatment caused the least inhibition. Pyrazole, besides being a specific inhibitor of ADH, also acts as an inducer for CYP2E1.30–33 This may account for it having the lowest inhibitory effect among the three inhibitors studied on protein carbonyl formation, which reflects oxidative modification of cellular proteins due to persistent oxidative stress in the cell.
In contrast, diallyl sulfide caused the least decrease in upregulated GSH levels in chronic alcohol plus high glucose treated VL-17A cells, than pyrazole and 4-methyl pyrazole which exerted similar effects. GSH is upregulated as a protective mechanism against CYP2E1 mediated oxidative stress in chronic alcohol plus high glucose treated VL-17A cells and since elevated GSH levels implicate beneficial effects on the cell's redox system, therefore a further decrease was not observed on inhibition of CYP2E1.
However, both pyrazole and 4-methyl pyrazole caused similar decreases in GSH levels in chronic alcohol plus high glucose treated VL-17A cells to a similar level to that observed in 50 mM glucose treated VL-17A cells. We speculate that the level of oxidative stress in chronic alcohol plus high glucose treated VL-17A cells with the inhibition of ADH with pyrazole and ADH and CYP2E1 with 4-methyl pyrazole was decreased to the level observed with VL-17A cells treated with a high concentration of glucose alone, i.e. lack of severe oxidative stress in the presence of the inhibitors, and hence the GSH levels were not upregulated to the extent observed in chronic alcohol plus high glucose treated VL-17A cells.
The specific inhibition of CYP2E1 with diallyl sulfide led to the greatest decrease in endogenous aldehyde protein adduct formation and exogenous MDA-, AA-, or MAA-adduct formation, confirming the significant role of CYP2E1 in the metabolism of ethanol to acetaldehyde and ROS generation and its inducibility due to high glucose concentration in the cell leading to greater ROS formation.
All three inhibitors caused similar decreases in caspase 3 activity in chronic alcohol plus high glucose treated VL-17A cells suggesting that, besides caspase 3, a non caspase 3 mediated apoptotic pathway also mediates the toxicity observed which has been reported previously in an apoptotic model.34 Diallyl sulfide was the most effective in decreasing the number of Annexin V-PI stained cells suggesting the crucial role of CYP2E1 in promoting apoptosis in chronic alcohol plus high glucose treated VL-17A cells.
Marginal decreases in ADH protein expression and catalytic activity were observed with pyrazole and 4-methyl pyrazole in chronic alcohol plus high glucose treated VL-17A cells which were not observed with diallyl sulfide, consistent with the observations that both pyrazole and 4-methyl pyrazole act as potent inhibitors of ADH.11,14,15
However, although both diallyl sulfide and 4-methyl pyrazole were effective in inhibiting CYP2E1 protein expression in chronic alcohol plus high glucose treated VL-17A cells, with diallyl sulfide showing the greatest inhibitory effect, pyrazole did not decrease the CYP2E1 content, providing indirect evidence for its specificity as an inhibitor of ADH. Further, as expected, diallyl sulfide decreased the catalytic activity of CYP2E1. However, both pyrazole and 4-methyl pyrazole increased the CYP2E1 activity in chronic alcohol plus high glucose treated VL-17A cells. The observations with both pyrazole and 4-methyl pyrazole in chronic alcohol plus high glucose treated VL-17A cells are not surprising, taking into consideration that pyrazole is an inducer for CYP2E1 and 4-methyl pyrazole acts as a ligand for CYP2E1.17,30–33,35
Specific inhibition of CYP2E1 with phenethyl isothiocyanate was highly effective in restoring the viability, decreasing ROS and caspase 3 activity and CYP2E1 siRNA was highly effective in restoring the viability, decreasing ROS and decreasing the induced CYP2E1 protein content in chronic alcohol plus high glucose treated VL-17A cells, confirming the role of ethanol and high glucose inducible CYP2E1 in mediating oxidative stress and toxicity in chronic alcohol plus high glucose treated VL-17A cells.
In conclusion, specific inhibition of CYP2E1 with diallyl sulfide in VL-17A cells exposed to chronic alcohol plus high glucose leads to the restoration of the viability to the greatest extent than specific inhibition of ADH with pyrazole and the inhibition of ADH and CYP2E1 with 4-methyl pyrazole. Oxidative stress parameters such as ROS level, lipid peroxidation, protein carbonyl and protein aldehyde adduct formation are the lowest in the diallyl sulfide plus chronic alcohol plus high glucose treated VL-17A cells. Similar trends are observed with apoptotic parameters also. Further, the use of phenethyl isothiocyanate and CYP2E1 siRNA corroborate the earlier observations with the other chemical inhibitors. Thus, ethanol and high glucose inducible CYP2E1 is an integral component for chronic alcohol plus high glucose mediated oxidative stress which promotes cellular injury in liver cells.
Abbreviations
| Ac-DEVD-pNA | Ac-DEVD-p-nitroanilide |
| ADH | alcohol dehydrogenase |
| CYP2E1 | cytochrome P4502E1 |
| DCF-DA | 2′,7′-dichlorofluorescein diacetate |
| GSH | glutathione |
| MTT | thiazolyl blue tetrazolium bromide |
| MDA | malondialdehyde |
| AA | acetaldehyde adduct |
| MAA | malondialdehyde-acetaldehyde adduct |
| PI | propidium iodide |
| pNA | p-nitroaniline |
| ROS | reactive oxygen species |
| TBAR | thiobarbituric acid reactive |
Conflicts of interests
The authors declare that there are no conflicts of interests.Acknowledgements
The work was supported by funding from the Department of Science and Technology (FAST TRACK SCHEME FOR YOUNG SCIENTISTS) and Department of Biotechnology (RAPID GRANT FOR YOUNG INVESTIGATORS) awarded to AD. AD also acknowledges the financial support received from the KBC Research Foundation, Chennai.KS is grateful to the Council of Scientific and Industrial Research (CSIR), New Delhi, India for the awarding of the Junior and Senior Research Fellowships.
DLC was supported by a grant from the Veterans Administration.
We thank Dr Suvro Chatterjee, Life Sciences Division, AU-KBC Research Centre, Chennai, India for generously sharing his laboratory facilities for fluorescence microscopy.
References
- D. Wu and A. I. Cederbaum, Semin. Liver Dis., 2009, 29, 141–154 CrossRef CAS.
- B. W. Smith and L. A. Adams, Nat. Rev. Endocrinol., 2011, 7, 456–465 CrossRef CAS.
- J. I. Cohen, X. Chen and L. E. Nagy, Antioxid. Redox Signaling, 2011, 15, 523–534 CrossRef CAS.
- B. W. Smith and L. A. Adams, Crit. Rev. Clin. Lab. Sci., 2011, 48, 97–113 CrossRef CAS.
- A. Dey and K. Chandrasekaran, Curr. Diabetes Rev., 2009, 5, 67–78 CrossRef CAS.
- A. I. Cederbaum, Y. Lu and D. Wu, Arch. Toxicol., 2009, 83, 519–548 CrossRef CAS.
- G. Robertson, I. Leclercq and G. C. Farrell, Am. J. Physiol. Gastrointest. Liver Physiol., 2001, 281, G1135–G1139 CAS.
- Y. Wang, G. Millonig, J. Nair, E. Patsenker, F. Stickel, S. Mueller, H. Bartsch and H. K. Seitz, Hepatology, 2009, 50, 453–461 CrossRef CAS.
- A. Schäfer, P. Galuppo, D. Fraccarollo, C. Vogt, J. D. Widder, J. Pfrang, P. Tas, E. Barbosa-Sicard, H. Ruetten, G. Ertl, I. Fleming and J. Bauersachs, Diabetes, 2010, 59, 2001–2009 CrossRef.
- K. Chandrasekaran, K. Swaminathan, S. M. Kumar, D. L. Clemens and A. Dey, Alcohol.: Clin. Exp. Res., 2012, 36, 487–495 CrossRef.
- T. M. Donohue, N. A. Osna and D. L. Clemens, Int. J. Biochem. Cell Biol., 2006, 38, 92–101 CrossRef CAS.
- K. Chandrasekaran, K. Swaminathan, S. M. Kumar, S. Chatterjee, D. L. Clemens and A. Dey, Toxicol. in Vitro, 2011, 25, 969–978 CrossRef CAS.
- N. A. Osna, D. L. Clemens and T. M. Donohue, Hepatology, 2005, 42, 1109–1117 CrossRef CAS.
- D. Wu and A. I. Cederbaum, J. Biol. Chem., 1996, 271, 23914–23919 CrossRef CAS.
- D. L. Clemens, A. Forman, T. R. Jerrells, M. F. Sorrell and D. J. Tuma, Hepatology, 2002, 35, 1196–1204 CrossRef CAS.
- M. Morimoto, A. L. Hagbjörk, Y. J. Wan, P. C. Fu, P. Clot, E. Albano, M. Ingelman-Sundberg and S. W. French, Hepatology, 1995, 21, 1610–1617 CAS.
- S. M. Zimatkin, S. P. Pronko, V. Vasiliou, F. J. Gonzalez and R. A. Deitrich, Alcohol.: Clin. Exp. Res., 2006, 30, 1500–1505 CrossRef CAS.
- A. Dey and A. I. Cederbaum, J. Pharmacol. Exp. Ther., 2006, 317, 1391–1399 CrossRef CAS.
- S. T. Stern, T. M. Potter and B. W. Neun, Nanotechnology Characterization Laboratory-NCL Method GTA-4 Version 1.0, 2006 Search PubMed.
- C. M. Palmeira, A. P. Rolo, J. Berthiaume, J. A. Bjork and K. B. Wallace, Toxicol. Appl. Pharmacol., 2007, 225, 214–220 CrossRef CAS.
- F. Tietze, Anal. Biochem., 1969, 27, 502–522 CrossRef CAS.
- D. J. Tuma, G. M. Thiele and D. Xu, Hepatology, 1996, 23, 872–880 CrossRef CAS.
- V. B. Patel, S. Worrall and P. W. Emery, Alcohol Alcohol., 2005, 40, 485–493 CrossRef CAS.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- X. H. Yao, L. Chen and B. L. Nyomba, J. Appl. Physiol., 2006, 100, 642–648 CrossRef CAS.
- S. de la Monte, Z. Derdak and J. R. Wands, J. Gastroenterol. Hepatol., 2012,(Suppl 2), 33–41 CrossRef CAS.
- Y. Lu and A. I. Cederbaum, Free Radicals Biol. Med., 2008, 44, 723–738 CrossRef CAS.
- S. Zakhari, Alcohol Res. Health., 2006, 29, 245–254 Search PubMed.
- K. Chandrasekaran, K. Swaminathan, S. M. Kumar, D. L. Clemens and A. Dey, Integr. Biol., 2012, 4, 550–563 RSC.
- B. L. McVicker, P. L. Tuma, K. K. Kharbanda, S. M. L. Lee and D. J. Tuma, World J. Gastroenterol., 2009, 15, 2609–2616 CrossRef CAS.
- A. Dey and A. I. Cederbaum, Hepatology, 2007, 45, 1355–1365 CrossRef CAS.
- D. Kornbrust and D. Dietz, Chem. Biol. Interac., 1985, 56, 29–44 CrossRef CAS.
- M. X. Yang and A. I. Cederbaum, Arch. Biochem. Biophys., 1997, 341, 25–33 CrossRef CAS.
- M. M. Mc Gee, E. Hyland, G. Campiani, A. Ramunno, V. Nacci and D. M. Zisterer, FEBS Lett., 2002, 515, 66–70 CrossRef CAS.
- D. J. Tierney, A. L. Haas and D. R. Koop, Arch. Biochem. Biophys., 1992, 293, 9–16 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |