 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bismuth sulphide–polymer nanocomposites from a highly soluble bismuth xanthate precursor†
Verena
Kaltenhauser
ab,
Thomas
Rath
*ab,
Wernfried
Haas
bc,
Ana
Torvisco
d,
Stefan K.
Müller
d,
Bettina
Friedel
e,
Birgit
Kunert
e,
Robert
Saf
a,
Ferdinand
Hofer
c and
Gregor
Trimmel
ab
aInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: thomas.rath@tugraz.at; Fax: +43 316-8731032331; Tel: +43 316-87332281
bChristian Doppler Laboratory for Nanocomposite Solar Cells, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
cInstitute for Electron Microscopy and Nanoanalysis, Graz University of Technology, Steyrergasse 17, 8010 Graz, Austria
dInstitute of Inorganic Chemistry, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
eInstitute of Solid State Physics, Graz University of Technology, Petersgasse 16, 8010 Graz, Austria
First published on 3rd October 2013
Abstract
Bismuth sulphide nanocrystal–polymer hybrid layers are of interest for various optoelectronic, thermoelectric or sensing applications. In this work, we present a ligand-free in situ route for the formation of Bi2S3 nanorods directly within a polymer matrix. For this purpose, we introduce a novel bismuth xanthate (bismuth(III) O-3,3-dimethylbutan-2-yl dithiocarbonate), which is highly soluble in non-polar organic solvents. The analysis of the crystal structure revealed that the prepared bismuth xanthate crystallises in the monoclinic space group C2/c and forms dimers. The bismuth xanthate can be converted into nanocrystalline Bi2S3 with an orthorhombic crystal structure via a thermally induced solid state reaction at moderate temperatures below 200 °C. In combination with the high solubility in non-polar solvents this synthetic route for Bi2S3 is of particular interest for the preparation of Bi2S3–polymer nanocomposites as exemplarily investigated on Bi2S3–poly(methyl methacrylate) and Bi2S3–poly(3-hexylthiophene-2,5-diyl) (P3HT) nanocomposite layers. Atomic force and transmission electron microscopy revealed that Bi2S3 nanorods are dispersed in the polymer matrix. Photoluminescence experiments showed a quenching of the P3HT fluorescence with increasing Bi2S3 content in the hybrid layer.
1. Introduction
Organic–inorganic hybrid materials are a highly interesting and versatile class of materials that offer a plethora of applications due to their variety of physical and chemical properties. These can be tailored in a wide range by using different material combinations, morphologies and nanostructures. Of particular interest are nanocrystal–polymer hybrid materials, often called nanocomposites, in which inorganic nanocrystals with specific properties e.g. concerning light emission or light absorption are integrated into a polymer matrix. This allows combining the advantages of easy processing and structural flexibility of polymers, with the optical properties and stability provided by the inorganic material.1 Semiconducting quantum dots offer the unique possibility of tuning their properties by controlling their size and shape.2 Organic–inorganic nanocomposites find applications, for example, in photochromic devices,3 non linear optics (NLO),4 electroluminescent devices,5 hybrid optical sensors6 and also in polymer–nanoparticle hybrid solar cells.7–9Bismuth sulphide (Bi2S3) is a non-toxic semiconductor, consisting of abundant elements, with a direct band gap of 1.3 to 1.7 eV,10 and a high absorption coefficient in the order of 105 cm−1.11 This makes Bi2S3 also an interesting candidate for a variety of applications, e.g. for polymer–nanoparticle hybrid solar cells. There are already some reports discussing Bi2S3 as an acceptor material in bulk-heterojunction hybrid solar cells;12,13 however, the efficiencies are still relatively low. Furthermore, Bi2S3 nanoparticles and nanocomposites can find numerous applications in thermoelectric devices,14 memory devices,10 photodetectors,15 as electrode materials in lithium-ion batteries,16 as well as in electronic, optoelectronic and gas sensor devices.17–20
Several synthetic methods to prepare Bi2S3 nanostructures exist starting from metal salts and a sulphur source21,22 or single source precursors.23–25 Thus, an obvious and common way to prepare bismuth sulphide–polymer nanocomposites is to synthesise Bi2S3 nanostructures first and then mix them in a second step with a polymer to obtain the organic–inorganic hybrid nanocomposite. Following this strategy several types of nanoparticle syntheses have been applied so far. A quite common approach is the synthesis of Bi2S3 nanoparticles via a colloidal synthesis using metal salts, a sulphur source and ligands stabilizing the nanoparticles. Wang et al.,5 for example, used Bi(NO3)3 with sodium octanoate and elemental sulphur to form Bi2S3 nanoparticles, which were subsequently mixed with a conjugated polymer.12 In further approaches, the polymer matrix was polymerised in the presence of Bi2S3 nanoparticles,26 or pre-formed nanostructured Bi2S3 layers were coated with a polymer.27
All these approaches are rather tedious and require several steps. In contrast, in this work we discuss a convenient and fast route to prepare metal sulphide–polymer nanocomposites by synthesising Bi2S3 nanocrystals directly within a polymer matrix in the solid state without the necessity of a separate nanoparticle synthesis step. This method is based on metal xanthates, which are highly soluble in a variety of organic solvents depending on the alkyl moiety of the xanthate group. The good solubility makes this route very versatile in terms of selecting the matrix polymers, which are also inherently different in solubility and, even more crucial, it provides the prerequisite for a homogeneous distribution of both materials in the nanocomposite. A second beneficial property of metal xanthates in this regard is that they decompose at low temperatures (100–200 °C) and the side products of the decomposition are volatile, which leads to tidy layers without impurities.28 Thus, this method is well suited for the preparation of metal sulphide nanostructures in a polymer matrix at low temperatures, which also allows using thermally less stable conjugated polymers as matrix without harming their optoelectronic properties. Metal sulphide–conjugated polymer nanocomposites, prepared via metal xanthates, can be applied as absorber layers in polymer–nanoparticle hybrid solar cells. This has recently been demonstrated for several nanocomposite materials29 containing CdS,30,31 CuInS2 (ref. 28 and 32) or Sb2S3 (ref. 33) nanoparticles and conjugated polymers or small molecules.34
The synthesis of bismuth xanthates with methyl, ethyl, i-propyl, n-butyl, cyclohexyl and benzyl-moieties has been reported in the literature several decades ago.35–37 Quite recently, some of these compounds have been applied for the synthesis of Bi2S3 nanowires and nanoparticles.23,25 In this contribution, we introduce a new bismuth xanthate (bismuth(III) O-3,3-dimethylbutan-2-yl dithiocarbonate, BiHexXan), which is highly soluble in non-polar organic solvents due to its asymmetric, branched alkyl side chain. Therefore, this compound is well suited for the preparation of Bi2S3–polymer nanocomposite materials. The formation of Bi2S3 from this precursor is investigated in detail and first Bi2S3–polymer nanocomposites are prepared and studied.
2. Experimental
2.1. Synthesis
All chemicals were purchased from Sigma-Aldrich and used without further purification. Potassium xanthate (potassium O-3,3-dimethylbutan-2-yl dithiocarbonate) was prepared according to the literature,28 for further information see the ESI.†Synthesis of bismuth( III ) O-3,3-dimethylbutan-2-yl dithiocarbonate: bismuth(III) chloride (3.64 g, 11.6 mmol, 1.0 eq.) was dissolved in 125 mL of deionised water. A solution of potassium xanthate (8.0 g, 37.0 mmol, 3.2 eq.) in 40 mL deionised water was added dropwise under stirring and a sticky brown solid was formed immediately. After the reaction solution was stirred for 3 hours, the brownish-orange precipitate was filtered and dried under vacuum. The residue was dissolved in 30 mL chloroform and the insoluble side products were removed by filtration. After recrystallisation (chloroform) orange crystals were obtained which were dried in vacuum (yield: 7.1 g, 83%).
1 H NMR (300 MHz, 20 °C, CDCl 3 , δ): 5.46–5.39 (q, 1H, CH), 1.37–1.35 (d, 3H, CH3), 0.98 (s, 9H, 3× CH3, tert-butyl) ppm. 13C NMR (75 MHz, 20 °C, CDCl3, δ): 225.5 (CS2O), 91.1 (CH), 35.3 (C(CH3)), 25.9 (3C, C(CH3)3), 14.5 (CH–CH3) ppm. IR (on Si-wafer): 2965, 2873, 1479, 1397, 1378, 1366, 1340, 1235, 1211, 1113, 1074, 1049, 1018, 879 cm−1. Elem. anal. calc. for Bi2S12O6C42H78: C 34.08, H 5.31, S 26.00; found: C 32.45, H 5.04, S 25.09%.
Preparation of Bi
2
S
3
–polymer nanocomposite layers: nanocomposite layers were prepared on different substrates (NaCl substrates for transmission electron microscopy images (TEM), CaF2 substrates for optical measurements and silicon wafers for IR-measurements) by spin coating a polymer–bismuth-xanthate chloroform solution, followed by a thermal heating step (heating rate 11 °C min−1 to 195 °C, holding time 15 min). The concentration of the polymer was 5 mg mL−1 and the weight ratio of polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bi2S3 was 1:5. For the preparation of the TEM samples, poly(3-hexylthiophene-2,5-diyl) (P3HT) was used as a polymer and the thermal heating step was performed in a tube furnace (Hereaus RO 4/25). For optical measurements and IR measurements, PMMA was used as a transparent matrix polymer, and the conversion was accomplished on a programmable heating plate (CAT Ingenieurbüro M. Zipperer GmbH, Germany).
:Bi2S3 was 1:5. For the preparation of the TEM samples, poly(3-hexylthiophene-2,5-diyl) (P3HT) was used as a polymer and the thermal heating step was performed in a tube furnace (Hereaus RO 4/25). For optical measurements and IR measurements, PMMA was used as a transparent matrix polymer, and the conversion was accomplished on a programmable heating plate (CAT Ingenieurbüro M. Zipperer GmbH, Germany).
2.2. Characterisation
1H NMR and 13C NMR spectra were recorded on a Bruker Ultrashield 300 MHz spectrometer. Referencing the NMR spectra to the corresponding values given in the literature was done by using solvent residual peaks (CDCl3: 1H NMR: 7.26 ppm; 13C NMR: 77.16 ppm). Elemental analyses were carried out on a Universal CHNS Elemental Analyzer Vario El III.FT-IR spectra were acquired using a Perkin-Elmer Spectrum One instrument. All FT-IR spectra of the samples were recorded in transmission mode (films on silicon wafers, spectral range between 4000 and 800 cm−1).
X-ray crystallography: a suitable crystal for single crystal X-ray diffractometry was removed from a Schlenk flask under a stream of N2 and immediately covered with a layer of silicone oil. A single crystal was selected, mounted on a glass rod on a copper pin, and placed in the cold N2 stream provided by an Oxford Cryosystems cryometer. Single crystal data collection was performed on a BRUKER APEX II diffractometer with the use of Mo Kα radiation (λ = 0.71073 Å) and a CCD area detector. Empirical absorption corrections were applied using SADABS.38,39 The structure was solved with the use of direct methods in SHELXS and refined by the full-matrix least-squares procedures in SHELXL.40 The space group assignments and structural solutions were evaluated using PLATON.41 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were located in calculated positions corresponding to standard bond lengths and angles. Attempts to model disorder around one of the xanthate ligands and the solvent of crystallisation CHCl3 resulted in unstable refinement.
Crystal data: C43H78Bi2Cl4O6S12, M = 1635.53 g mol−1, orange block, crystal size 0.20 × 0.10 × 0.10 mm, monoclinic, space group C2/c: a = 24.5050(8) Å, b = 9.0961(3) Å, c = 29.5784(10) Å, α = 90°, β = 105.123(2)°, γ = 90°, V = 6364.7(4) Å3, Z = 4, Dc = 1.707 Mg m−3, F(000) 3240, μ = 6.123 mm−1, T = 100(2) K, 43646 reflections (θmax = 25°), 5602 unique (Rint = 0.0261), 329 parameters, 0 restraints, R1 (all data) = 0.0396, wR2 (all data), = 0.0867 largest difference peak and hole: 1.716 and −1.927 e Å−3.
The X-ray powder diffraction profiles were obtained on a Siemens D 501 diffractometer in Bragg–Brentano geometry operated at 40 kV and 30 mA, using Cu Kα radiation (λ = 1.54178 Å) and a graphite monochromator at the secondary side.
Thermogravimetric measurements were performed on a Netzsch Jupiter STA 449C. All measurements were carried out in a helium atmosphere and at a heating rate of 10 °C min−1. High resolution mass spectra (EI, 70 eV; pion source = 1.05 × 10−6 mbar; mass range: 50–800 Da; 1 spectrum per s) were recorded on a Waters GCT Premier equipped with a direct insertion probe (temperature program of the DI: start at 40 °C, 10 °C min−1 to 260 °C). Raman spectra were recorded on a Perkin-Elmer Raman Station 400F with a 350 mW 785 nm near infrared laser.
Transmittance and reflectance spectra for the determination of the optical absorption coefficient as well as absorption spectra were recorded on a Perkin-Elmer Lambda 35 equipped with an integrating sphere. The optical band gap was estimated from the (αhν)2versus hν plots by extrapolating the linear part of the function. PL-quenching studies were performed on a Fluorescence Spectrophotometer F-7000 from Hitachi.
Layer thicknesses were specified on a Bruker DekTak XT surface profiler. Atomic force microscopy (AFM) of the prepared films was performed under ambient conditions using a Nanosurf Easyscan 2 scanning probe microscope in tapping mode. Bi2S3–PMMA layers for AFM measurements were prepared via doctor-blading on glass substrates and Bi2S3–P3HT layers were prepared on PEDOT:PSS coated glass/ITO substrates.
Transmission electron microscopy images were acquired on a Tecnai F 20 microscope (FEI Company) equipped with a Schottky emitter, an energy dispersive X-ray spectrometer and a high resolution Gatan imaging filter (HR-GIF) with an UltraScanCCD camera. TEM specimens were prepared by spin coating a solution of polymer and precursor on NaCl single crystals, annealing at 200 °C, subsequent dissolution of the NaCl crystal in deionised water and transfer of the nanocomposite layer, floating on the water surface, to a TEM grid.
3. Results and discussion
The synthesis of new bismuth(III) O-3,3-dimethylbutan-2-yl dithiocarbonate (BiHexXan) was performed in analogy to previously reported Cu and In xanthates,28 see Scheme 1. The prepared Bi-xanthate complex was analysed with 1H, 13C-NMR and IR spectroscopy and by elementary analysis (see Section 2.1.). The NMR spectra are in agreement with the proposed structure with a chemical shift of 225.5 ppm for the quaternary carbon of the xanthate group in the 13C NMR spectrum. | ||
| Scheme 1 Synthesis of BiHexXan and Bi2S3–polymer hybrid materials. | ||
Single crystals of BiHexXan have been obtained from solutions in chloroform and were used for determination of the crystal structure, depicted in Fig. 1.
 | ||
| Fig. 1 Crystal structure of BiHexXan (Bi2S12O6C42H78). | ||
The BiHexXan compound crystallises in the monoclinic space group C2/c. The crystal structure contains residual amounts of chloroform as the solvent of crystallisation. The Bi central atom is surrounded by six sulphur atoms derived from the three asymmetrically bound xanthate ligands. An additional interaction with a bridging sulphur atom from the neighbouring Bi(xanthate)3 unit results in the formation of a dimer complex with a Bi–S interatomic distance of 3.205(2) Å which is significantly longer than the direct chelating Bi–S bonds (Table 1). Thus, the environment of the bismuth atom is seven-coordinate, and a four membered ring is formed between the bismuth metal and the bridging xanthate ligand, with all angles close to 90°. Similar dimeric structures with a bridging sulphur bond have been found for Bi(S2COCH3)3 (ref. 36) as well as for the dithiocarbonate Bi[S2CN(C2H5)2]3.42 In contrast, a polymeric structure caused by a similar bridging Bi–S-interaction has been found in tris(O-isopropylxanthato)-bismuth(III) presented by B. F. Hoskins et al.37
| Bi(1)–S(1) | 2.7782(15) | Bi(1)–S(2) | 2.9772(14) |
| Bi(1)–S(3) | 2.7134(15) | Bi(1)–S(4) | 2.8350(16) |
| Bi(1)–S(5) | 2.6032(15) | Bi(1)–S(6) | 2.9614(16) |
| Bi(1)–S(2a) | 3.205(2) | S(1)–C(1) | 1.692(6) |
| S(2)–C(1) | 1.709(6) | C(1)–O(1) | 1.308(7) |
| O(1)–C(2) | 1.503(9) | S(3)–C(8) | 1.702(7) |
| S(1)–C(1)–S(2) | 123.2(4) | S(1)–Bi(1)–S(2) | 62.51(4) |
| S(3)–C(8)–S(4) | 122.7(4) | S(3)–Bi(1)–S(4) | 64.79(5) |
| S(5)–C(15)–S(6) | 124.0(4) | S(5)–Bi(1)–S(6) | 64.65(4) |
| S(2)–Bi(1)–S(2a) | 90.53(2) | Bi(1)–S(2)–Bi(1a) | 89.22(2) |
In Table 1, the selected interatomic distances for BiHexXan are listed. The Bi–S distances vary between 2.6032(15)–2.9772(14) Å. All measured interatomic distances correlate very well with the distances found for other reported Bi-xanthates. It is noticeable that there are two groups of Bi–S distances; one group exhibits short M–S bonds and the other long M–S ones, due to the intermediate mode of coordination of the xanthate ligand.36 The C–O bonds of the S2CO-group (e.g. C(1)–O(1)) show interatomic distances of approx. 1.31 Å typical of carbonate type bonding, whereas the C–O bonds to the alkyl moiety exhibit significantly larger values of 1.50 Å (e.g. O(1)–C(2)).
In a next step, the thermal decomposition of the BiHexXan complex was investigated. Thermogravimetric analysis revealed that the onset of decomposition is at approximately 150 °C (see Fig. 2A). The observed weight loss of 65.6% is in good agreement with the calculated one (67.2%) for the formation of Bi2S3. An additional minor mass loss can be detected at a temperature of 120 °C, which does not stem from the decomposition of the xanthate but from the evaporation of the remaining solvent, as indicated from the crystal structure. In addition, this was confirmed by high resolution mass spectrometric (HRMS) investigations using a direct insertion probe (DI) and electron impact ionisation (EI). For this, volatile products were continuously monitored, while a sample of BiHexXan was heated from 40 °C to 250 °C directly in the mass spectrometer. Starting approximately at 150 °C COS, CS2 and 3,3-dimethylbut-1-ene were detected which are the typical species of the decomposition of xanthates via the Chugaev rearrangement.43 Additionally, fragment ions containing Bi e.g. C14H26O2S4Bi+ and C2H2O2S4Bi+ were identified. This means that BiHexXan partly evaporates under the high vacuum conditions in the mass spectrometer (for more details see the ESI†).
 | ||
| Fig. 2 (A) Thermogravimetric curve of the bismuth xanthate BiHexXan, decomposition temperature: 150 °C, weight loss: 65.6%; (B) XRD pattern (reference data: PDF 17-320) and (C) Raman spectrum of the in situ prepared Bi2S3 nanoparticles. | ||
To confirm that Bi2S3 was successfully formed by the solid-state reaction induced by the thermal decomposition of BiHexXan, a sample annealed at 195 °C for 30 min was subjected to X-ray powder diffractometry. Fig. 2B shows the XRD pattern of the obtained nanocrystalline Bi2S3. The diffraction peaks (main peaks: 25°, 28.5°, 31.8°, 46.6° and 52.7° 2θ) correlate very well with the reference diffractions of a pure orthorhombic Bi2S3 phase (a = 1.114 nm, b = 1.130 nm and c = 0.3981 nm; PDF 17-320). No additional peaks which would indicate impurities or secondary phases could be detected. The broadening of the peaks reveals the nanocrystalline character in the order of 25–29 nm (estimated from the 211, 220 and 120 reflections using the Scherrer formula). The Raman spectrum (Fig. 2C) of the Bi2S3 nanoparticles exhibits three broad Raman peaks which correspond to the Ag (187, 237 cm−1) and B1g (259 cm−1) Raman modes and are consistent with literature data.44–46
In Fig. 3A the absorption coefficient of a 30 nm thick Bi2S3 film formed at 195 °C is plotted, exhibiting values higher than 1 × 105 cm−1. The optical band gap (Eg) was determined by plotting the straight line of (αhν)2 against hν, revealing a band gap of 1.65 eV which is in the range of the values (1.3–1.7 eV) commonly reported in the literature.23,24
 | ||
| Fig. 3 (A) Optical absorption coefficient and band gap determination of a pure Bi2S3 layer; (B) UV-Vis spectra of BiHexXan before (BiHexXan–PMMA) and after heating to 195 °C (Bi2S3–PMMA) in the PMMA matrix. | ||
The low conversion temperature of BiHexXan and the formation of a pure Bi2S3 layer were encouraging to explore also the formation of polymer–nanoparticle hybrid materials following this solid state in situ formation route (see Scheme 1). For the optical characterisation of the hybrid nanocomposite formation poly(methyl methacrylate) (PMMA) was used as the matrix polymer because of its transparency in the near UV and visible wavelength range.
In Fig. 3B, the UV-Vis spectra of BiHexXan before (BiHexXan–PMMA) and after heating to 195 °C (Bi2S3–PMMA) in the PMMA matrix are depicted, which clearly illustrate the differences in optical properties of the material before and after thermal conversion. In the BiHexXan sample, an intensive absorption in the UV-range between 250 and 450 nm is observed. This distinct UV absorption vanishes after the thermal treatment and a typical absorption profile of Bi2S3 nanoparticles with an absorption onset between 800 and 900 nm is visible in the spectrum. PMMA, which was used as the matrix polymer for the nanocomposite formation, does not exhibit significant absorption in this wavelength range.
A comparison of the FTIR spectra of the pristine BiHexXan–PMMA sample, the Bi2S3–PMMA sample (annealed at 195 °C) and pure PMMA is shown in Fig. 4. In BiHexXan–PMMA typical vibrations of xanthates are detected. The most intense peaks at 1238 and 1203 cm−1 can be attributed to the asymmetric C–O–C stretching vibrations, and the bands at 1047 and 1017 cm−1 to the C–S stretching vibrations (for more details see the ESI, Table S1†).47 After the annealing step, all vibrational bands of BiHexXan fully vanish and the spectrum of Bi2S3–PMMA resembles the pure PMMA spectrum with its most prominent peak at 1732 cm−1 attributable to the carbonyl stretching vibration.
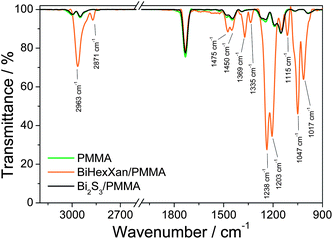 | ||
| Fig. 4 IR spectra of BiHexXan before (BiHexXan–PMMA) and after heating to 195 °C (Bi2S3–PMMA) in the PMMA matrix. | ||
Considering possible applications, nanocomposite layers containing the conjugated polymer P3HT as a matrix material were also prepared similar to the Bi2S3–PMMA samples. The absorption spectrum of the Bi2S3–P3HT hybrid layer is characterised by the absorption of both components, a broad absorption peak of P3HT around 560 nm and the absorption profile of Bi2S3 nanoparticles with an onset wavelength between 800 and 900 nm (see the ESI†).
The morphology of the prepared Bi2S3–polymer nanocomposite layers was characterised by atomic force microscopy (AFM). Fig. 5 shows surface images of a Bi2S3–PMMA layer as well as of a Bi2S3–P3HT layer. The images of the prepared layers reveal that the polymer forms a few 100 nm large domains, which can be clearly seen in the topographic image as well as in the phase contrast image. In the Bi2S3–PMMA layers structures with smaller sizes of about 50 nm are also visible, which can be ascribed to the Bi2S3 nanostructures present in the nanocomposite. These smaller structures are not observed in the AFM image of the Bi2S3–P3HT hybrid layer, which might be due to surface-energetically preferential diffusion of P3HT-rich phases towards the air–film interface and thus formation of a P3HT-rich cover layer at the surface of the nanocomposite film. For further investigation of this issue, transmission electron microscopy (TEM) measurements of the in situ prepared Bi2S3–P3HT layers were performed.
 | ||
| Fig. 5 AFM images ((A and B) topography; (C) phase contrast) of a Bi2S3–PMMA hybrid layer and a Bi2S3–P3HT layer ((D and E) topography; (F) phase contrast). | ||
Bright field TEM images of the Bi2S3–P3HT sample at different magnifications are shown in Fig. 6, which exhibit that nanorods are formed in the conjugated polymer matrix. The Bi2S3 nanorods reveal regular shapes with a diameter of 7–22 nm and a length of 40–70 nm. The formation of nanorods in the matrix of a conjugated polymer via a ligand-free in situ process in the solid state is a highly interesting result, as it was not possible before to realise nanorod structures via ligand-free in situ processes.29,48 Until now, only spherical particles could be obtained. In particular, nanorods are very desirable, thinking of a possible application of the nanocomposites as absorber layers in hybrid solar cells, because they inherently facilitate charge transport through the layer, while retaining a high polymer–nanoparticle interfacial area.49 Current record power conversion efficiencies of hybrid solar cells of 4.7% are also reached using nanorods of CdSe instead of spherical nanoparticles in combination with the low band gap polymer PCPDTBT.50
 | ||
| Fig. 6 TEM images of Bi2S3 nanorods in a P3HT matrix at different magnifications. | ||
The reason for the formation of Bi2S3 nanorods in this solid state reaction is most presumably the predominant growth of the orthorhombic Bi2S3 nanocrystals along the [001] direction.10,51 Therefore, these elongated nanostructures are quite common in solution grown Bi2S3 nanocrystals and there exist several publications reporting on Bi2S3 nanorods or nanowires yielded by different synthetic approaches.11,12,23,24,52
A further interesting aspect is that no aggregation of the nanorods and thus phase separation between the organic and inorganic phase occurs. The Bi2S3 nanorods are homogeneously distributed over the whole layer. A possible explanation for that could be that the Bi2S3 nanorods are formed directly in the polymer matrix from a homogeneous mixture of bismuth xanthate and polymer. Nevertheless, the nanorods are in close proximity to each other; therefore, continuous pathways should be present in both phases.
To investigate the optical properties of the prepared P3HT–Bi2S3 nanocomposites in more detail, we performed photoluminescence (PL) quenching measurements on P3HT–Bi2S3 nanocomposites with different contents of the inorganic component. The PL of the P3HT is found to be strongly quenched when Bi2S3 is present in the composites. The detailed results of the PL-quenching study are summarised in Fig. 7, which shows the PL spectra of pure P3HT and P3HT–Bi2S3 nanocomposite layers with different polymer:Bi2S3 weight ratios of 1:3, 1:6, 1:9 and 1:12. To obtain reliable results, we increased the thickness of the layers with increasing Bi2S3 content to ensure that the polymer content was equal in each sample. Optical excitation of the pristine P3HT film at 560 nm resulted in the appearance of an emission band at 670 nm. This emission is quenched after introduction of the Bi2S3 nanorods into the polymer film (see Fig. 7A). The PL emission was quenched to 22% of the initial emission intensity at a P3HT:Bi2S3 weight ratio of 1:3 and is further reduced to 6% (P3HT:Bi2S3 = 1:6), to 3% (P3HT:Bi2S3 = 1:9) and to 0.4% (P3HT:Bi2S3 = 1:12) of the original level (see Fig. 7B). It has to be considered that part of the reduction of the PL intensity may stem from absorption of the emitted light by the Bi2S3 nanorods. However, the qualitative quenching of the PL of P3HT by the Bi2S3 nanoparticles in the nanocomposite seems to be quite efficient and is comparable to or even higher than PL quenching efficiencies which were obtained by PL quenching studies on other polymer–nanoparticle hybrid materials used in hybrid photovoltaics.28,53,54
 | ||
| Fig. 7 (A) Photoluminescence quenching in P3HT–Bi2S3 nanocomposites; (B) normalised integrated PL vs. Bi2S3 volume ratio in the nanocomposite. | ||
The photoluminescence quenching study suggests good suitability of the prepared Bi2S3–P3HT nanocomposite layers as an absorber material in polymer–nanoparticle hybrid solar cells. Thus, we prepared polymer–nanoparticle hybrid solar cells with Bi2S3–P3HT nanocomposite layers as an absorber material in the configuration glass/ITO/PEDOT:PSS/P3HT:Bi2S3/Al with different P3HT:Bi2S3 ratios. However, all prepared solar cells were short-circuited and no diode characteristics could be observed. To prevent short circuits in the solar cells we introduced additional interlayers between electrodes and the absorber layer. Also with this improved geometry, we could not realise working solar cells with the in situ prepared Bi2S3–P3HT hybrid layers so far, even though Bi2S3–P3HT hybrid solar cells with a power conversion efficiency of 1% were already reported with a similar device architecture using pre-synthesised Bi2S3 nanoparticles.13
4. Conclusions
The novel bismuth xanthate, bismuth(III) O-3,3-dimethylbutan-2-yl dithiocarbonate, is a valuable precursor for the preparation of Bi2S3 at temperatures below 200 °C. In combination with the high solubility in non-polar organic solvents caused by the branched alkyl side chains, this synthetic route for Bi2S3 is of particular interest for the preparation of Bi2S3–polymer nanocomposites. By mixing the xanthate with PMMA as well as with the conjugated polymer P3HT in a solvent, coating of precursor layers and heating them to 200 °C in an inert atmosphere, Bi2S3 nanorods are formed within the polymer matrix by a ligand-free solid state in situ reaction pathway. Such metal sulphide nanorods may be of special interest for application in hybrid solar cells as nanorods facilitate charge transport and also provide a high interfacial area towards the conjugated polymer. Studying Bi2S3–P3HT nanocomposites concerning their applicability in hybrid solar cells, it was not possible to realise working solar cells using the in situ prepared Bi2S3–P3HT hybrid layers, even though we observed that the fluorescence of the conjugated polymer is efficiently quenched by the Bi2S3 nanorods, which points to an efficient charge transfer at the hybrid interface.Acknowledgements
Financial support by the Christian Doppler Research Association, the Austrian Federal Ministry of Economy, Family and Youth (BMWFJ), and ISOVOLTAIC AG is gratefully acknowledged. The authors thank Kathrin Bohnemann, Josefine Hobisch and Andrea Anusic for technical help.References
- L. Persano, A. Camposeo, F. Di Benedetto, R. Stabile, A. M. Laera, E. Piscopiello, L. Tapfer and D. Pisignano, Adv. Mater., 2012, 24, 5320–5326 CrossRef CAS PubMed.
- C. De Mello Donegá, Chem. Soc. Rev., 2011, 40, 1512–1546 RSC.
- R. Pardo, M. Zayat and D. Levy, Chem. Soc. Rev., 2011, 40, 672–687 RSC.
- B. Lebeau, S. Brasselet, J. Zyss and C. Sanchez, Chem. Mater., 1997, 9, 1012–1020 CrossRef CAS.
- T.-P. Nguyen, Surf. Coat. Technol., 2011, 206, 742–752 CrossRef CAS PubMed.
- C. Sanchez, B. Lebeau, F. Chaput and J.-P. Boilot, Adv. Mater., 2003, 15, 1969–1994 CrossRef CAS.
- T. Xu and Q. Qiao, Energy Environ. Sci., 2011, 4, 2700–2720 CAS.
- Y. Zhou, M. Eck and M. Krüger, Energy Environ. Sci., 2010, 3, 1851–1864 CAS.
- X. Fan, M. Zhang, X. Wang, F. Yang and X. Meng, J. Mater. Chem. A, 2013, 1, 8694–8709 CAS.
- G.-Y. Liu, L.-Y. Xu, F. Zhou, Y. Zhang, H. Li, Y. F. Xu and J. M. Lu, Phys. Chem. Chem. Phys., 2013, 15, 11554–11558 RSC.
- A. K. Rath, M. Bernechea, L. Martinez and G. Konstantatos, Adv. Mater., 2011, 23, 3712–3717 CrossRef CAS PubMed.
- Z. Wang, S. Qu, X. Zeng, J. Liu, F. Tan, L. Jin and Z. Wang, Appl. Surf. Sci., 2010, 257, 423–428 CrossRef CAS PubMed.
- L. Martínez, A. Stavrinadis, S. Higuchi, S. L. Diedenhofen, M. Bernechea, K. Tajima and G. Konstantatos, Phys. Chem. Chem. Phys., 2013, 15, 5482–5487 RSC.
- Y. Y. Wang, K. F. Cai and X. Yao, J. Nanopart. Res., 2012, 14, 848 CrossRef.
- G. Konstantatos, L. Levina, J. Tang and E. H. Sargent, Nano Lett., 2008, 8, 4002–4006 CrossRef CAS PubMed.
- H. Jung, C.-M. Park and H.-J. Sohn, Electrochim. Acta, 2011, 56, 2135–2139 CrossRef CAS PubMed.
- K. Yao, Z. Y. Zhang, X. L. Liang, Q. Chen, L.-M. Peng and Y. Yu, J. Phys. Chem. B, 2006, 110, 21408–21411 CrossRef CAS PubMed.
- K. Yao, W. W. Gong, Y. F. Hu, X. L. Liang, Q. Chen and L. M. Peng, J. Phys. Chem. C, 2008, 112, 8721–8724 CAS.
- M. Ahmad, M. A. Rafiq, K. Rasool, Z. Imran and M. M. Hasan, J. Appl. Phys., 2013, 113, 043704 CrossRef.
- H. Li, J. Yang, J. Zhang and M. Zhou, RSC Adv., 2012, 2, 6258–6261 RSC.
- J. Ma, J. Yang, L. Jiao, T. Wang, J. Lian, X. Duan and W. Zheng, Dalton Trans., 2011, 40, 10100–10108 RSC.
- R. Chen, M. H. So, C.-M. Che and H. Sun, J. Mater. Chem., 2005, 15, 4540–4545 RSC.
- Q. Han, J. Chen, X. Yang, L. Lu and X. Wang, J. Phys. Chem. C, 2007, 111, 14072–14077 CAS.
- W. Lou, M. Chen, X. Wang and W. Liu, Chem. Mater., 2007, 19, 872–878 CrossRef CAS.
- Y. W. Koh, C. S. Lai, A. Y. Du, E. R. T. Tiekink and K. P. Loh, Chem. Mater., 2003, 15, 4544–4554 CrossRef CAS.
- M. E. Rincón, H. Hu and G. Martínez, Synth. Met., 2003, 139, 63–69 CrossRef.
- Y. Li, Y. Zhang, Y. Lei, P. Li, H. Jia, H. Hou and Z. Zheng, Mater. Sci. Eng., B, 2012, 177, 1764–1768 CrossRef CAS PubMed.
- T. Rath, M. Edler, W. Haas, A. Fischereder, S. Moscher, A. Schenk, R. Trattnig, M. Sezen, G. Mauthner, A. Pein, D. Meischler, K. Bartl, R. Saf, N. Bansal, S. A. Haque, F. Hofer, E. J. List and G. Trimmel, Adv. Energy Mater., 2011, 1, 1046–1050 CrossRef CAS.
- T. Rath and G. Trimmel, Hybrid Mater., 2013, 1, 15–36 Search PubMed.
- H. C. Leventis, S. P. King, A. Sudlow, M. S. Hill, K. C. Molloy and S. A. Haque, Nano Lett., 2010, 10, 1253–1258 CrossRef CAS PubMed.
- S. Dowland, T. Lutz, A. Ward, S. P. King, A. Sudlow, M. S. Hill, K. C. Molloy and S. A. Haque, Adv. Mater., 2011, 23, 2739–2744 CrossRef CAS PubMed.
- M. Arar, A. Pein, W. Haas, F. Hofer, K. Norrman, F. C. Krebs, T. Rath and G. Trimmel, J. Phys. Chem. C, 2012, 116, 19191–19196 CAS.
- N. Bansal, F. T. F. O'Mahony, T. Lutz and S. A. Haque, Adv. Energy Mater., 2013, 3, 986–990 CrossRef CAS.
- T. Rath, V. Kaltenhauser, W. Haas, A. Reichmann, F. Hofer and G. Trimmel, Sol. Energy Mater. Sol. Cells, 2013, 114, 38–42 CrossRef CAS PubMed.
- M. A. Khwaja, T. J. Cardwell and R. J. Magee, Anal. Chim. Acta, 1973, 64, 9–17 CrossRef CAS.
- M. R. Snow and E. R. T. Tiekink, Aust. J. Chem., 1987, 40, 743–750 CrossRef CAS.
- B. F. Hoskins, E. R. T. Tiekink and G. Winter, Inorg. Chim. Acta, 1985, 99, 177–182 CrossRef CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef.
- G. M. Sheldrick, SADABS, Version 2.10, Siemens Area Detector Correction, Universitaet Goettingen, Germany, 2003 Search PubMed.
- G. M. Sheldrick, SHELXTL, Version 6.1, Bruker AXS, Inc., Madison, WI, 2002 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. L. Raston and A. H. White, J. Chem. Soc., Dalton Trans., 1976, 791–794 RSC.
- N. Pradhan, B. Katz and S. Efrima, J. Phys. Chem. B, 2003, 107, 13843–13854 CrossRef CAS.
- X.-P. Shen, G. Yin, W.-L. Zhang and Z. Xu, Solid State Commun., 2006, 140, 116–119 CrossRef CAS PubMed.
- K. Trentelman, J. Raman Spectrosc., 2009, 40, 585–589 CrossRef CAS.
- Y. Zhao, K. T. E. Chua, C. K. Gan, J. Zhang, B. Peng, Z. Peng and Q. Xiong, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 205330 CrossRef.
- D. Barreca, A. Gasparotto, C. Maragno, R. Seraglia, E. Tondello, A. Venzo, V. Krishnan and H. Bertagnolli, Appl. Organomet. Chem., 2005, 19, 1002–1009 CrossRef CAS.
- E. Martinez-Ferrero, J. Albero and E. Palomares, J. Phys. Chem. Lett., 2010, 1, 3039–3045 CrossRef CAS.
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425–2427 CrossRef CAS PubMed.
- R. Zhou, R. Stalder, D. Xie, W. Cao, Y. Zheng, Y. Yang, M. Plaisant, P. H. Holloway, K. S. Schanze, J. R. Reynolds and J. Xue, ACS Nano, 2013, 7, 4846–4854 CrossRef CAS PubMed.
- J. Tang and A. P. Alivisatos, Nano Lett., 2006, 6, 2701–2706 CrossRef CAS PubMed.
- H.-C. Liao, M.-C. Wu, M.-H. Jao, C.-M. Chuang, Y.-F. Chen and W.-F. Su, CrystEngComm, 2012, 14, 3645–3652 RSC.
- W. Yue, S. Han, R. Peng, W. Shen, H. Geng, F. Wu, S. Tao and M. Wang, J. Mater. Chem., 2010, 20, 7570–7578 RSC.
- H. Geng, C. M. Hill, S. Pan and L. Huang, Phys. Chem. Chem. Phys., 2013, 15, 3504–3509 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 949783. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3tc31684j |
| This journal is © The Royal Society of Chemistry 2013 |