 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of 2,6-connectivity in azulene: optical properties and stimuli responsive behavior†
Michael Koch, Olivier Blacque and Koushik Venkatesan*
Institute of Inorganic Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: venkatesan.koushik@aci.uzh.ch; Fax: +41 44-635-68-03; Tel: +41 44-635-46-94
First published on 19th September 2013
Abstract
The possible incorporation of the redox active, small donor acceptor molecule azulene as a core structure for potential optoelectronic applications has been evaluated. The synthesis and characterization of different isomers of di(phenylethynyl)azulene 1–4 have been successfully carried out. The photophysical properties as well as their stimuli responsive behavior reflecting their corresponding electronic properties were also investigated. The experimental observations show that 2,6-connection (3) exhibits intense luminescence and shows the strongest stimuli responsive behavior upon acid treatment. DFT calculations of 3 reveal the highest contributions of the alkyne substituents at the 2- and 6-position of the azulene to the ground- and excited state that makes the 2,6-connectivity at azulene a very promising candidate for low band-gap conjugated materials.
Introduction
Design and synthesis of novel conjugated polymers have been intensely investigated over the last two decades due to their extensive application as optoelectronic materials.1–9 In particular, conjugated polymers have been utilized in a wide variety of transduction schemes to realize sensitive and highly selective detection of chemical and biological analytes by taking advantage of their stimuli responsive behavior.10–17 Due to azulene's unique electronic and optical properties such as large permanent dipole moment, intense blue color and the domination of fluorescence from S2 → S0 with low emission intensity, due to the violation of Kasha's rule,18–20 it has been long considered to be an interesting candidate for application in optoelectronic materials (see Fig. 1).21,22 | ||
| Fig. 1 Mesomeric structure of azulene with the numbering scheme and permanent dipole moment. | ||
However, the evaluation of azulene as a core structure for optoelectronic applications remains scarce, although calculated theoretical results using various aromaticity theories suggest that azulene possesses a much lower delocalization energy (4.2 kcal mol−1) in comparison with benzene (20 kcal mol−1), thiophene (16.1 kcal mol−1) and naphthalene (30.5 kcal mol−1).23 Recently Hawker's group and our group reported on the stimuli responsive behavior of azulene derivatives. The treatment of azulene derivatives with superacids results in an azulenium cation upon protonation at the unsubstituted 1-position with the concomitant increase in luminescence intensity. This is accompanied by a bathochromic shift of the emission wavelength especially in the case of ethynyl spacer bearing groups. The S1 → S0 transition becomes the dominant radiative decay pathway of the protonated species.24–26 Such a response of azulene towards superacids not only allows one to control the emission properties, but also permits to tailor the conducting properties of the azulene as recently demonstrated by Hawker and co-workers.27
Despite quite a large number of theoretical calculations on the influence of the functional bis-substitution pattern of azulenes on their electronic properties, the experimental investigations have been rather limited.28,29 Based on theoretical calculations, the 2,6-connected polyazulenes are expected to be most unique compared with other substitution patterns. The π-electrons can easily delocalize along the direction of the dipolar C2v axis of azulene resulting in a large dipole moment and large hyperpolarizability. This is especially supported by the ability to form an expected quinoid structure. 2,6-connected azulene systems would thus possess a lower band gap than the otherwise connected azulene polymers, which is also highly interesting for preparing low band-gap conducting polymers for application in organic solar cells.29,30 In these polymers, the highest symmetry of C2v will be preserved in the planar form, while in all the other possible polymers of azulene a much lower symmetry is expected because in the latter cases the monomer symmetry axis does not equal the polymeric symmetry axis. Also, the polymer consisting of only a 2,6-connectivity will show a head-to-tail alignment of the monomeric dipole moments.
While there are many examples of mono-alkyne substituted azulenes reported in the literature, dialkyne substituted azulenes remain scarce except for the ones exclusively bearing the alkynes at the 1- and 3-position of the azulene.26,31–38 The only known 2,6-bissubstituted compounds either bear an alkyne or an alkyl chain at the 6 position, or they are connected to a metal center as a bridging ligand or, are part of a 6,6'-connected azulene dimer.39–42
In this work, we report the systematic synthesis of 2,6-diphenyl acetylene substituted azulene and investigation of its stimuli responsive behavior as a model compound for macromolecules. Di(phenylethynyl) substitution was particularly chosen in order to allow for easy correlation of the properties as model compounds. For comparison reasons, we have also prepared the three other isomers of di(phenylethynyl)azulenes 1, 2 and 4 (see Fig. 2).43
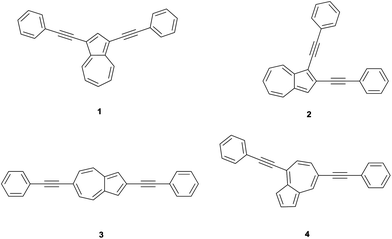 | ||
| Fig. 2 Structures of di(phenylethynyl)azulenes 1, 2, 3 and 4. | ||
Synthesis of specific substitution patterns on azulene is quite challenging and difficult to achieve,44,45 and since there is no common starting material available to prepare the target molecules, different approaches had to be sought to accomplish the desired molecules (see Scheme 1). Given the unique interesting properties expected to arise from the 2,6-connectivity in azulene, we set out to develop a synthetic strategy that would not only allow one to achieve the targeted 2,6-diphenyl substituted azulene, but would also offer considerable modularity in synthesis, starting from 2-bromo-6-phenylethynyl azulene (14). To the best of our knowledge, this work represents the first such experimental study combined with a deeper theoretical insight provided by DFT calculations into the optoelectronic behavior of an azulene core including the targeted synthesis and photophysical evaluation of the elusive 2,6-alkyne substituted azulene 3.
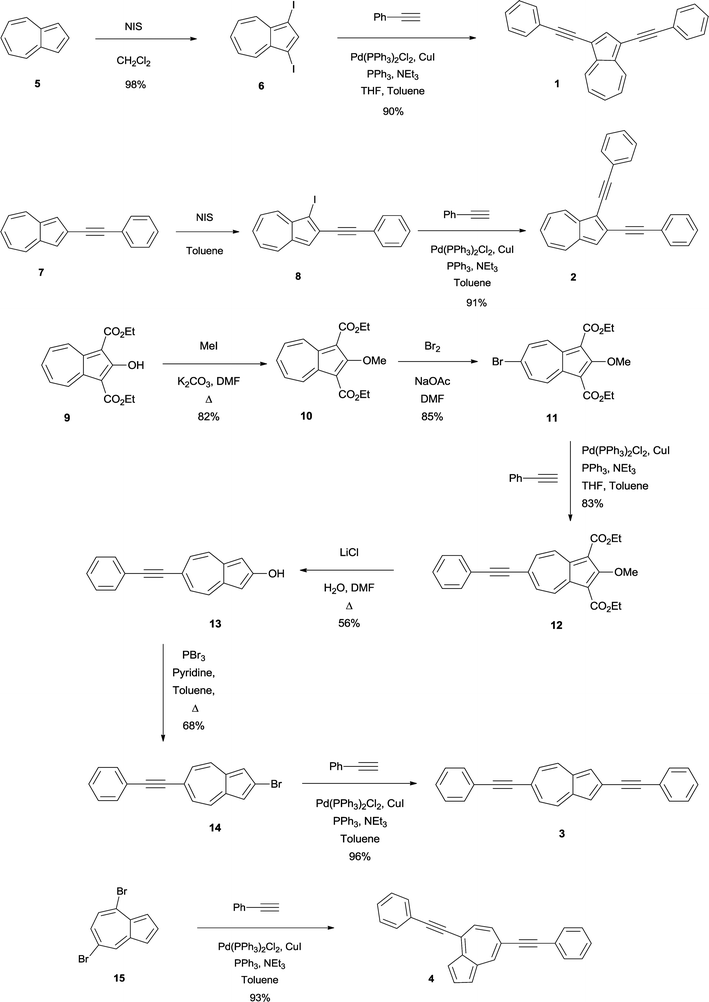 | ||
| Scheme 1 Synthesis of di(phenylethynyl)azulenes 1, 2, 3 and 4. | ||
Synthesis
Since the synthesis of 1 has been already known in the literature,32 our detailed discussion pertains only to the synthesis and properties of the new compounds 2, 3 and 4. As previously mentioned, we utilize different approaches as shown in Scheme 1.461 was synthesized starting from the dark green colored 1,3-diiodoazulene 6, which in turn was prepared in excellent yields (98%) by reacting the commercially available compound 5 with 2 eq. N-iodosuccinimide (NIS) in dry CH2Cl2. Subsequent application of Sonogashira coupling conditions consisting of an excess amount of phenyl acetylene and catalytic amounts of CuI, PPh3 and Pd(PPh3)2Cl2 in the presence of an excess amount of Et3N at elevated temperature gave 1 as a blue green colored solid in very good yield (91%).Compound 2 was prepared from the known 2-phenylethynylazulene 7.26 The mono-alkyne 7 was treated with 1 eq. NIS in dry toluene below 10 °C to minimize diiodination, to give green 1-iodo-2-phenylethynylazulene 8 which decomposes in solid state and is stable in solution only for a couple of days. Therefore the filtered reaction mixture was directly utilized in the Sonogashira coupling reaction leading to green 2 in a good overall yield (91%).
Despite the synthesis of 3 involving many steps due to the challenging substitution pattern, we were able to obtain the targeted compound in an overall yield of 21%. A direct synthesis of the 2,6-pattern is not feasible since it requires an electrophilic attack at the electron rich five membered ring for the substitution of the 2-position which is more favored at the more nucleophilic 1- and 3-positions, while on the other hand a mandatory nucleophilic attack at the electron poor seven membered ring will be directed to the 4- and 8-position and not result in the desired 6-substitution. Therefore the parent azulene 5 cannot be directly used as a precursor for the synthesis of 2,6-dialkyne azulene, but instead starting material (9), based on the work of Nozoe and co-workers and the work of Ito and co-workers,39,47,48 was utilized in which the 1- and 3-positions bearing ethyl ester protected groups shielded the 4- and 8-position from a nucleophilic attack.
Starting from the orange colored diethyl 2-hydroxyazulene-1,3-dicarboxylate 949 the hydroxyl-group was alkylated with methyl iodide to give the known bright red product 10 in very good yields (91%). This product was brominated at the 6-position to result in the known orange product 11 in 85% yield. The ester-functionalities act as protection groups in this step. Due to sterical hindrance, the nucleophilic bromination at 4- and 8-positions is blocked resulting in an exclusive bromination at the 6-position. Sonogashira coupling with phenyl acetylene gave the mono-alkyne product 12 in 83% yield, which was subsequently thermally dealkoxycarboxylated to give 2-hydroxy-6-phenylethynyl-azulene 13 in 56% yield. Conversion of the hydroxyl-group with PBr3 to 14 (68%) followed by Sonogashira coupling gave eventually 3 as a dark green product in 96% yield.
4 was synthesized as a blue colored product in very good yields (93%) starting from the known 4,7-dibromoazulene 15 under Sonogashira conditions.25
UV-Vis studies
The UV-Vis absorption spectra of 1–4 can be classified into three regions: the first region for λ > 500 nm which is composed of the S0 → S1 transitions, the second region for 370 nm < λ < 500 nm showing the S0 → S2 transitions and the third region for λ < 370 nm comprising the S0 → S3 transitions.The UV-Vis absorption spectra of 1–4 exhibit high molar extinction coefficients in the region λ < 500 nm (see Fig. 3). In the region of λ > 500 nm compound 4 possesses the most broad absorption band between 520 nm and 800 nm among the synthesized compounds; however the intensity of the band was low and similar to that found for the parent azulene (5). A similar band could only be detected at much higher concentrations in the spectra of 1, 2 and to an even lesser extent for 3. This difference can be ascribed to the fact that the desired electronic contribution of the substituents to the transition is less pronounced for 4 while it is strongest for 3 (see also DFT calculations). In the region that is assigned to the S0 → S2 transition (ca. 370–500 nm) 1 and 2 reveal only a small molar extinction coefficient in comparison to 3 and 4. It is evident from this observation that the position of the substitution alters the probability of the two possible transitions that govern azulene's absorption behavior. This is especially the case for 3 that shows similar intensities for both transitions. Additionally 3 shows the highest molar extinction coefficient in this red shifted region (44000 M−1 cm−1 compared to 10400 M−1 cm−1 (1), 14000 M−1 cm−1 (2), 34000 M−1 cm−1 (4)). Since azulene's fluorescence results from excited state S2, a transition in this wavelength region with a high molar absorption coefficient is crucial (see Table 1). Furthermore tuning of this band to longer wavelength is desired due to the fact that this correlates with a smaller energy gap of the transition and better polarizability of the molecule. In the region assigned to the S0 → S3 transition (λ < 370 nm), the extinction coefficients as well as the absorption wavelength vary only to a certain extent.
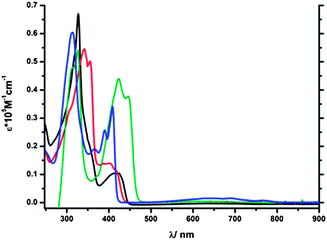 | ||
| Fig. 3 UV-Vis absorption spectra of 1 (black line), 2 (red line), 3 (green line) and 4 (blue line) in CH2Cl2. | ||
Azulene's optical behavior is tremendously changed upon protonation (see Fig. 4 and 5), since the emission of the protonated species results from the S1 states. The corresponding S0 → S1 transition is observed in the UV-Vis absorption spectra in the region of ca. 420–580 nm for 1-H+–4-H+. After protonation, significant changes in the UV-Vis absorption spectra were observed.50
 | ||
| Fig. 4 Plausible structures of di(phenylethynyl)-azulenes after treatment with superacids. | ||
 | ||
| Fig. 5 Left: Photo of 3 (CH2Cl2, treated with MeSO3H, treated with MeSO3H (exc. at 365 nm)). Right: Photo of 4 (CH2Cl2, treated with MeSO3H, treated with MeSO3H (exc. at 365 nm)). | ||
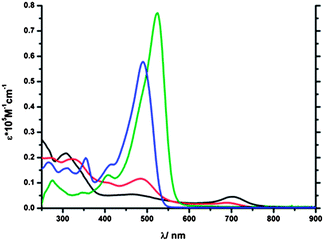
4-H+ showed a very distinct absorption band centered around 490 nm with a high molar extinction coefficient (ε = 57800 M−1 cm−1, see Fig. 6). 3-H+ exhibited an even more distinct absorption band combined with the highest molar extinction coefficient in comparison with the rest of the protonated isomers (525 nm, ε = 77100 M−1 cm−1) and in fact even higher than the extinction coefficients of the untreated isomers 1–4. Although compounds 1-H+ and 2-H+ also display an absorption band in this region, they are not distinct and have significantly lower intensities, especially in the case of 1-H+. 1-H+ and 2-H+ display after protonation an additional broad but low intensity band with a bathochromic shift at 703 nm (1-H+) and 689 nm (2-H+) respectively (see Fig. 6), which is neither observed in the 3-H+ and 4-H+ nor in the protonated 2-phenylethynylazulene (7).
 | ||
| Fig. 6 UV-Vis absorption spectra of 1-H+ (black line), 2-H+ (red line), 3-H+ (green line) and 4-H+ (blue line) in CH2Cl2. | ||
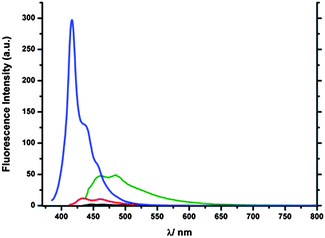
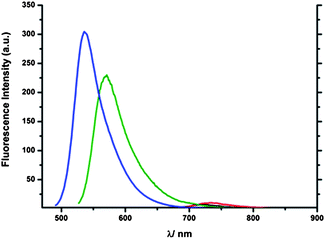
The emission spectra reveal the intensity of 1 to be very weak, while 2 is only slightly stronger; 3 and 4 show a substantial fluorescence intensity although much stronger in the case of 4 but more red shifted in the former case (see Fig. 7). The low emission intensity of 1 and 2 can be caused by the transfer of the additional electron density into the five membered ring and the limited contribution of the phenylethynyl substituents to states relevant for the transition. The emission bands were found to be located in the range of ca. 70 nm (416 nm to 484 nm). In this context 3 showed the most red shifted emission band among all the isomers although it is not as intense as that of 4 but is the most broad band (ca. from 430 nm to 650 nm). 4 shows a more significant hypsochromic shift in the emission wavelength than the mono-alkyne compound 2-phenylethynylazulene 7 (424 nm). Upon protonation the emission intensity of azulene compounds was expected to increase substantially as it was shown for other related compounds.25,26 Nevertheless in the case of 2-H+, only a slightly more intense emission was observed (see Fig. 8) and in the case of 1-H+, the emission was even barely detectable revealing that both connection patterns are not promising candidates for stimuli responsive materials. The emission wavelengths of 2-H+ (733 nm) and 1-H+ (722 nm) are strongly red-shifted. This can be explained based on the dipole-moment of the excited state of the protonated compounds, which is diametrically opposed to the ground-state dipole-moment, and therefore points in the direction of the seven-membered ring. 3-H+ and to a lesser extent 4-H+ display strongly increased emission intensity making both connection patterns promising candidates for stimuli responsive materials. Nevertheless the fluorescence wavelengths were found to be less red shifted than in the case of 1-H+ and 2-H+. In this context 3-H+ is more bathochromically shifted than 4-H+ and exhibits a significantly stronger stimuli response than all other isomers. The smaller shift can be explained by the fact that the distance of the charge-transfer is longer in 1-H+ and 2-H+.
 | ||
| Fig. 7 UV-Vis emission spectra of 1 (black line), 2 (red line), 3 (green line) and 4 (blue line) in CH2Cl2.51 | ||
 | ||
| Fig. 8 UV-Vis emission spectra of 1-H+ (black line), 2-H+ (red line), 3-H+ (green line) and 4-H+ (blue line) in CH2Cl2.50 | ||
DFT calculations
All calculations were performed with the Gaussian 09 program package52 using the hybrid functional PBE1PBE53 in conjunction with the cc-pVDZ basis set.54 Full geometry optimizations without symmetry constraints were carried out in the gas phase for the singlet ground states (S0). The optimized geometries were confirmed to be potential energy minima by vibrational frequency calculations at the same level of theory, as no imaginary frequency was found. The first twenty singlet–singlet transition energies were computed at the optimized S0 geometries by using the time-dependent DFT (TD-DFT) methodology.55–57 Solvent effects were taken into account using the conductor-like polarizable continuum model (CPCM)58,59 with dichloromethane as solvent for the TD-DFT calculations on all optimized gas-phase geometries. The excited state structures were optimized by TD-DFT calculations, and the first four electronic transitions were computed taking into account the state-specific equilibrium solvation of each excited state at its equilibrium geometry.The TD-DFT calculations performed at the PBE1PBE/cc-pVDZ level on the DFT optimized ground state geometries of the di(phenylethynyl)azulene compounds 1–4 (see Fig. S31–S35, ESI†) confirm that the absorption spectra can be divided into three regions:
(1) λ > 550 nm: region with very weak absorption bands corresponding to the S0 → S1 singlet–singlet transition. The S1 excited states are derived from the HOMO → LUMO excitation for 1, 2 and 4, and from the HOMO−1 → LUMO excitation for 3. The calculated oscillator strengths are very small in the range of 0.007–0.025, except for 4 which shows a significant value f of 0.159;
(2) 375 < λ < 550 nm: region with moderate to intense absorption bands (0.292 < f < 1.877) corresponding to the S0 → S2 singlet–singlet transition. The singlet S2 excited states are derived from a combination of the HOMO → LUMO+1 and HOMO−1 → LUMO excitations for 1, 2 and 4, and from the HOMO → LUMO excitation for 3 which shows the highest oscillator strength for this transition;
(3) 300 < λ < 375 nm: region with moderate to intense absorption bands (0.137 < f < 1.805) corresponding to the S0 → S3 singlet–singlet transition. The S3 excited state is derived from the HOMO → LUMO+2 excitation for 1, the HOMO → LUMO+1 excitation for 2, HOMO−2 → LUMO+2 for 3, and mainly from the HOMO−1 → LUMO excitation for 4.
The S0 → S1 transitions are experimentally observed but appear as very weak and broad bands. The wavelength maxima of the S0 → S2 transitions are quite similar for the di(phenylethynyl)azulene compounds except for 3 which shows a significantly red shifted band with the highest molar extinction coefficient. This observation holds well only in the case of 3 where the S2 excited state arises directly from the HOMO and LUMO orbitals. In all cases, the occupied MOs involved in the S0 → S2 transitions are of π character mainly located on the azulene core and to a lesser extent on one or both phenylethynyl substituents with a π bonding character of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond(s) (see Fig. 9 and 10). Except for the LUMO of 2 which is only delocalized over the azulene moiety, LUMO and LUMO+1 show very similar compositions as HOMO−1 and HOMO, respectively, notably with an antibonding π* character of the CC triple bond(s). The absorption data calculated for the protonated cationic species 1-H+–4-H+ are not as accurate as the values obtained for the neutral species (see Table 2). Indeed, the TD-PBE1PBE calculations failed to reproduce the additional broad bathochromic bands at 702 and 688 nm for 1-H+ and 2-H+, respectively. Nevertheless, it appears that upon protonation the S0 → S1 transition exhibits the largest oscillator strength of the first singlet–singlet transitions and corresponds to the HOMO → LUMO excitation (see Fig. 11).
C triple bond(s) (see Fig. 9 and 10). Except for the LUMO of 2 which is only delocalized over the azulene moiety, LUMO and LUMO+1 show very similar compositions as HOMO−1 and HOMO, respectively, notably with an antibonding π* character of the CC triple bond(s). The absorption data calculated for the protonated cationic species 1-H+–4-H+ are not as accurate as the values obtained for the neutral species (see Table 2). Indeed, the TD-PBE1PBE calculations failed to reproduce the additional broad bathochromic bands at 702 and 688 nm for 1-H+ and 2-H+, respectively. Nevertheless, it appears that upon protonation the S0 → S1 transition exhibits the largest oscillator strength of the first singlet–singlet transitions and corresponds to the HOMO → LUMO excitation (see Fig. 11).
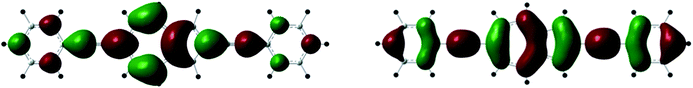 | ||
| Fig. 9 Spatial plots of the HOMO (left) and LUMO (right) orbitals of the ground-state of 3. | ||
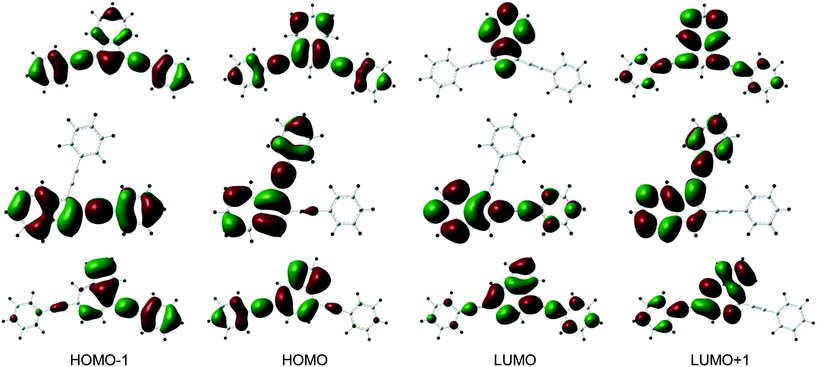 | ||
| Fig. 10 Spatial plots of frontier orbitals of the ground-states of 1 (top), 2 (middle) and 4 (bottom). | ||
| 1 | 2 | 3 | 4 | 1-H+ | 2-H+ | 3-H+ | 4-H+ | |
|---|---|---|---|---|---|---|---|---|
| exp abs λmax | 423 | 418 | 446 | 409 | 703 | 689 | 525 | 490 |
| exp em λmax | 465 | 432 | 484 | 416 | 722 | 733 | 571 | 537 |
| TD-DFT on ground-state structures | ||||||||
| S0 → S1 | 614.0 | 600.2 | 574.6 | 603.7 | 492.6 | 536.7 | 518.4 | 505.6 |
| 0.007 | 0.025 | 0.012 | 0.159 | 0.019 | 0.612 | 2.069 | 1.467 | |
| S0 → S2 | 419.4 | 418.8 | 463.8 | 414.6 | 475.0 | 494.3 | 456.6 | 448.5 |
| 0.292 | 0.331 | 1.877 | 0.972 | 0.026 | 0.039 | 0.022 | 0.170 | |
| S0 → S3 | 353.1 | 358.9 | 347.9 | 360.2 | 445.7 | 394.7 | 398.4 | 386.0 |
| 1.345 | 1.805 | 0.137 | 0.401 | 0.023 | 0.481 | 0.004 | 0.078 | |
| TD-DFT on excited-state structures | ||||||||
| S1 → S0 | 1317.5 | 764.4 | 560.9 | 517.1 | ||||
| 0.004 | 0.131 | 1.006 | 1.257 | |||||
| S2 → S0 | 459.8 | 457.6 | 506.5 | 445.0 | 754.6 | 677.4 | 490.6 | 450.5 |
| 0.222 | 0.424 | 1.680 | 1.244 | 0.038 | 0.019 | 0.945 | 0.097 | |
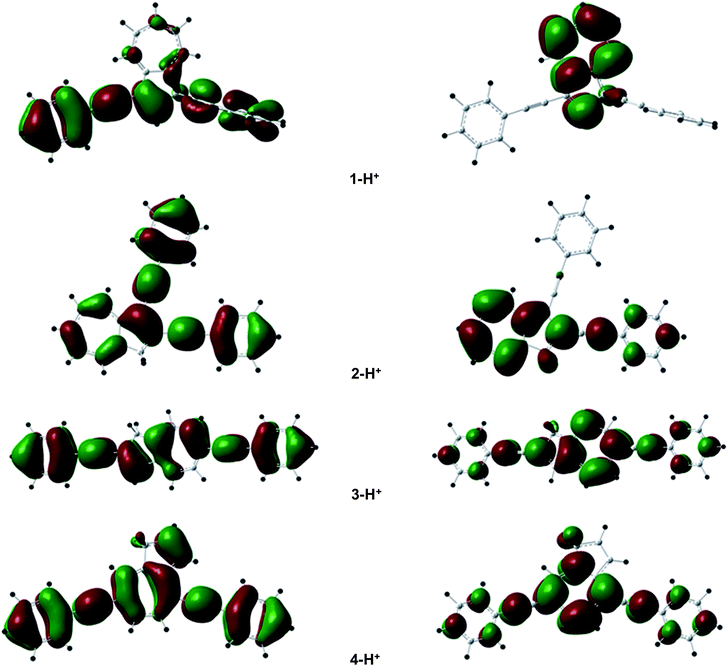 | ||
| Fig. 11 Spatial plots of the HOMO (left) and LUMO (right) of the ground states of 1-H+–4-H+. | ||
Despite the mismatch of both the trends of the calculated absorption wavelengths, the S0 → S1 oscillator strengths mimic well the relative molar extinction coefficients with f = 0.019 (1-H+) < 0.612 (2-H+) < 1.467 (4-H+) < 2.069 (3-H+). The main difference with the unprotonated species is that the LUMOs are of π/π* character on the azulene rings but exhibit mainly nonbonding orbitals on the phenylethynyl substituents, suggesting a lesser participation of the latter in the absorption and fluorescence properties.
The first excited state (S1) for the protonated species and the second excited state (S2) for the neutral compounds were successfully optimized by TD-DFT at the same level of theory used for the DFT calculations (PBE1PBE/cc-pVDZ). Fluorescence wavelengths, energies and oscillator strengths were computed taking into account the state-specific equilibrium solvation of each excited state at its equilibrium geometry (see Table 2). The structural changes between the ground state and excited state structures are especially observed on the five-membered ring as well as on the alkyne substituents for the neutral compounds and, in contrast, only on the seven-membered ring for the protonated species (see ESI†, Fig. S21–S22). The S2 → S0 fluorescence of 1–4 corresponds exactly to the S0 → S2 absorption. The predicted wavelengths of the fluorescence maxima respectively of 460 (−5 nm compared to the exp. data of 465 nm), 458 (+26), 507 (+23), 445 (+29) nm are acceptable and interestingly the calculated oscillator strengths of 0.222, 0.424, 1.6803, and 1.244 reproduce well the relative fluorescence intensity of 1–4, with 3 and 4 showing the most intense emission bands. For the protonated species, the same conclusion can be arrived at with the fluorescence maxima fitting correctly with the experimental data 755 (+33), 764 (+31), 561 (−10) and 517 (−20) for 1-H+ to 4-H+ respectively (except that in the case of 1-H+ it corresponds to the S2 → S0 transition instead of S1 → S0 for the other species) and also the oscillator strengths of 0.038 and 0.131 for 1-H+ and 2-H+, and of 1.006 and 1.257 for 3-H+ and 4-H+, corresponding well with the experimental features (see Fig. 8).
Conclusions
In summary, we have successfully synthesized four di(phenylethynyl)azulene isomers and experimentally demonstrated that 2,6-connection in azulene exhibits quite interesting and unique optical and physical properties in comparison with the other isomers of di(phenylethynyl)azulenes. UV-Vis absorption and luminescence properties were investigated to evaluate possible different incorporation positions of azulene for use as optoelectronic materials. Among the investigated compounds only 3-H+ and to a lesser extent 4-H+ showed a significant increase in emission intensity upon protonation. While 4-H+ displayed a slightly higher intensity, the emission wavelength of 3-H+ was found to be substantially more red-shifted. DFT calculations were carried out to explain the experimental observations. These results led to the conclusion that azulene incorporated as a core structure in optoelectronic materials exhibits very weak S0 → S2 and corresponding S2 → S0 transitions if both substitutions are situated at the five-membered ring. It is required that at least one substitution be present in the seven-membered ring in order to obtain fluorescence materials that display significant stimuli response behavior. Based on these findings, studies involving the incorporation of 2,6-connected azulene into macromolecules and subsequent evaluation of the stimuli responsive behavior as well as its potential in opto-electronic applications are currently under way.Acknowledgements
This work was supported by the Swiss National Science Foundation NRP 62 Smart Materials Program (Grant no. 406240-126142). Support from the University of Zurich and Prof. Heinz Berke is gratefully acknowledged.Notes and references
- V. Kamm, G. Battagliarin, I. A. Howard, W. Pisula, A. Mavrinskiy, C. Li, K. Müllen and F. Laquai, Adv. Energy Mater., 2011, 1, 297–302 CrossRef CAS
.
- R. Mauer, I. A. Howard and F. Laquai, J. Phys. Chem. Lett., 2010, 1, 3500–3505 CrossRef CAS
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed
- A. Heckmann and C. Lambert, Angew. Chem., Int. Ed., 2012, 51, 326–392 CrossRef CAS PubMed
- M. A. Rahman, P. Kumar, D. S. Park and Y. B. Shim, Sensors, 2008, 8, 118–141 CrossRef CAS
- E. J. Feldmeier and C. Melzer, Org. Electron., 2011, 12, 1166–1169 CrossRef CAS PubMed
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed
- I. A. Howard, R. Mauer, M. Meister and F. Laquai, J. Am. Chem. Soc., 2010, 132, 14866–14876 CrossRef CAS PubMed
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed
- S. Malashikhin and N. S. Finney, J. Am. Chem. Soc., 2008, 130, 12846–12847 CrossRef CAS PubMed
- S. A. Malashikhin, K. K. Baldridge and N. S. Finney, Org. Lett., 2010, 12, 940–943 CrossRef CAS PubMed
- A. P. de Silva and T. E. Rice, Chem. Commun., 1999, 163–164 RSC
- B. Esser and T. M. Swager, Angew. Chem., Int. Ed., 2010, 49, 8872–8875 CrossRef CAS PubMed
- J. Bouffard, Y. Kim, T. M. Swager, R. Weissleder and S. A. Hilderbrand, Org. Lett., 2008, 10, 37–40 CrossRef CAS PubMed
- T. Gupta and M. E. van der Boom, J. Am. Chem. Soc., 2006, 128, 8400–8401 CrossRef CAS PubMed
- T. Gupta, R. Cohen, G. Evmenenko, P. Dutta and M. E. van der Boom, J. Phys. Chem. C, 2007, 111, 4655–4660 CAS
- N. J. Turro, Modern Molecular Organic Photochemistry, University Science Books, Sausalito, CA, 1991 Search PubMed
- P. G. Lacroix, I. Malfant, G. Iftime, A. C. Razus, K. Nakatani and J. A. Delaire, Chem.–Eur. J., 2000, 6, 2599–2608 CrossRef CAS
- D. R. Mitchell and G. D. Gillispie, J. Phys. Chem., 1989, 93, 4390–4393 CrossRef CAS
- Y. Yamaguchi, Y. Maruya, H. Katagiri, K. Nakayama and Y. Ohba, Org. Lett., 2012, 14, 2316–2319 CrossRef CAS PubMed
- K. Kurotobi, K. S. Kim, S. B. Noh, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 3944–3947 CrossRef CAS PubMed
- P. Wang, P. Zhu, C. Ye, A. E. Asato and R. S. H. Liu, J. Phys. Chem. A, 1999, 103, 7076–7082 CrossRef CAS
- R. S. H. Liu, R. S. Muthyala, X.-s. Wang, A. E. Asato, P. Wang and C. Ye, Org. Lett., 2000, 2, 269–271 CrossRef CAS
- E. Amir, R. J. Amir, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 10046–10049 CrossRef CAS PubMed
- M. Koch, O. Blacque and K. Venkatesan, Org. Lett., 2012, 14, 1580–1583 CrossRef CAS PubMed
- M. Murai, E. Amir, R. J. Amir and C. J. Hawker, Chem. Sci., 2012, 3, 2721–2725 RSC
- S. Dutta, S. Lakshmi and S. K. Pati, Bull. Mater. Sci., 2008, 31, 353–358 CrossRef CAS PubMed
- J. R. Dias, J. Phys. Org. Chem., 2007, 20, 395–409 CrossRef CAS
- H. Kim, N. Schulte, G. Zhou, K. Müllen and F. Laquai, Adv. Mater., 2011, 23, 894–897 CrossRef CAS PubMed
- N. C. Thanh, M. Ikai, T. Kajioka, H. Fujikawa, Y. Taga, Y. Zhang, S. Ogawa, H. Shimada, Y. Miyahara, S. Kuroda and M. Oda, Tetrahedron, 2006, 62, 11227–11239 CrossRef CAS PubMed
- X.-Y. Chen, C. Barnes, J. R. Dias and T. C. Sandreczki, Chem.–Eur. J., 2009, 15, 2041–2044 CrossRef CAS PubMed
- K. H. H. Fabian, A. H. M. Elwahy and K. Hafner, Eur. J. Org. Chem., 2006, 791–802 CrossRef CAS
- V. V. Filichev, M. C. Nielsen, N. Bomholt, C. H. Jessen and E. B. Pedersen, Angew. Chem., Int. Ed., 2006, 45, 5311–5315 CrossRef CAS PubMed
- S. Ito, H. Inabe, T. Okujima, N. Morita, M. Watanabe and K. Imafuku, Tetrahedron Lett., 2000, 41, 8343–8347 CrossRef CAS
- S. Ito, H. Inabe, T. Okujima, N. Morita, M. Watanabe, N. Harada and K. Imafuku, J. Org. Chem., 2001, 66, 7090–7101 CrossRef CAS PubMed
- K. H. H. Fabian, A. H. M. Elwahy and K. Hafner, Tetrahedron Lett., 2000, 41, 2855–2858 CrossRef CAS
- M. V. Barybin, M. H. Chisholm, N. S. Dalal, T. H. Holovics, N. J. Patmore, R. E. Robinson and D. J. Zipse, J. Am. Chem. Soc., 2005, 127, 15182–15190 CrossRef CAS PubMed
- S. Ito, M. Ando, A. Nomura, N. Morita, C. Kabuto, H. Mukai, K. Ohta, J. Kawakami, A. Yoshizawa and A. Tajiri, J. Org. Chem., 2005, 70, 3939–3949 CrossRef CAS PubMed
- T. R. Maher, A. D. Spaeth, B. M. Neal, C. L. Berrie, W. H. Thompson, V. W. Day and M. V. Barybin, J. Am. Chem. Soc., 2010, 132, 15924–15926 CrossRef CAS PubMed
- K. Nakagawa, T. Yokoyama, K. Toyota, N. Morita, S. Ito, S. Tahata, M. Ueda, J. Kawakami, M. Yokoyama, Y. Kanai and K. Ohta, Tetrahedron, 2010, 66, 8304–8312 CrossRef CAS PubMed
- T. C. Holovics, R. E. Robinson, E. C. Weintrob, M. Toriyama, G. H. Lushington and M. V. Barybin, J. Am. Chem. Soc., 2006, 128, 2300–2309 CrossRef CAS PubMed
- M. Kivala and F. o. Diederich, Acc. Chem. Res., 2009, 42, 235–248 CrossRef CAS PubMed
- D. Balschukat and E. V. Dehmlow, Chem. Ber., 1986, 119, 2272–2288 CrossRef CAS
- S. Carret, A. Blanc, Y. Coquerel, M. Berthod, A. E. Greene and J.-P. Deprés, Angew. Chem., Int. Ed., 2005, 44, 5130–5133 CrossRef CAS PubMed
- See ESI†.
- T. Nozoe, T. Asao and M. Oda, Bull. Chem. Soc. Jpn., 1974, 47, 681–686 CrossRef CAS
- T. Nozoe, K. Takase and N. Shimazaki, Bull. Chem. Soc. Jpn., 1964, 37, 1644–1648 CrossRef CAS
- R. Brettle, D. A. Dunmur, S. Estdale and C. M. Marson, J. Mater. Chem., 1993, 3, 327–331 RSC
- It has to be mentioned that for compound 1 higher concentrations of superacid were needed for full conversion into the protonated species 1-H+.
- For 3 and 4 concentrations resulting in an O.D. of 0.1 a.u. in the UV-vis absorption spectra were chosen for the measurements of the UV-vis emission spectra. In the case of 1 and 2 concentrations resulting in an O.D. of 0.3 a.u. were chosen to determine the fluorescence properties.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS
- J. T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Contains experimental details, characterization data and various NMR spectra. Additional figures showing molecular orbitals, calculated spectra and Cartesian coordinates are also provided. See DOI: 10.1039/c3tc31610f |
| This journal is © The Royal Society of Chemistry 2013 |