 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of tetraselenophenoporphyrazine and its application in transistor devices†
Munazza Shahida,
Nafeezah Padamseya,
John Labramb,
Thomas D. Anthopoulosb and
Martin Heeney*a
aDept of Chemistry and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK. E-mail: m.heeney@imperial.ac.uk
bDept of Physics & Centre for Plastic Electronics, Imperial College, UK
First published on 19th August 2013
Abstract
We report the first synthesis of a soluble selenophene containing porphyrazine derivative, tetraselenophenoporphyrazine (SePz). Thin films of SePz exhibit a broad and strong absorption in the visible to near-IR region, which is red shifted compared to the thiophene analogue, and demonstrate p-type carrier mobilities of 10−2 cm2 V−1 s−1 in bottom gate, bottom contact field effect transistors.
Phthalocyanines (tetrabenzoporphyrazines) are industrially important blue/green dyes and pigments. They have also been extensively investigated for a range of other applications such as catalysts, sensors, singlet oxygen generators for photodynamic therapy, active layer materials for field effect transistors and as photoactive materials for dye-sensitized and bulk heterojunction solar cells.1 For all of these applications control of the photophysical, electrochemical and morphological properties of the phthalocyanine derivative is highly important.2 This has typically been achieved by varying the metal or metalloid at the centre of the phthalocyanine ring, or by varying the nature of the substituents attached to the benzenoid periphery. Another approach is to change the aromatic unit fused to the porphyrazine core, for example changing from benzo(phthalocyanine) to naphtho(napthalocyanine) results in a considerable red shift of the main absorption peak into the near infrared region and significant changes to the redox properties. In an effort to further tune the ring properties, a wide range of other hydrocarbon aromatics such as azulene,3 pyrene,4 triphenylene5 and helicene6 as well as six membered electron deficient heterocycles like pyridine7 and pyrazine8 have been fused to the porphyrazine core.
Encouraged by the promising charge carrier mobility of soluble substituted phthalocyanines,9 we were interested in exploring phthalocyanine analogues for possible application in field effect transistor devices. In order to further facilitate charge transport, we reasoned that the substitution of the benzenoid ring system with an electron rich heterocycle containing a chalcogen atom would be a promising approach. Indeed the thiophene analogue (tetrathienoporphyrazine, TPz) containing four solubilising side chains has recently been reported by Takimiya and co-workers to show promising transistor performance for films fabricated from solution.10 We were interested to investigate analogous porphyrazines in which the thiophene was replaced with selenophene. We anticipated that the inclusion of the larger, more polarisable selenium atoms might improve intermolecular contacts, as observed in other molecular species, and therefore might result in promising charge transport.11
To the best of our knowledge, selenophene containing porphyrazines have not previously been reported. In fact there are relatively few examples of porphyrazines fused with five membered heterocycles described.12 Although electron deficient examples like thiadiazole,13 selenodiazole14 and tellurodiazole15 have been reported, the only well characterised examples containing electron rich heterocycles have been based on thiophene.10,16 The attempted incorporation of furan or pyrrole has been reported to result in the formation of unstable species.17
In theory selenophene could be fused to the porphyrazine ring via either the 2,3-positions or the 3,4-positions. Here we concentrate exclusively on derivatives fused via the 2,3-positions, since in the alternative case, the structure cannot be represented by a usual bonding system and is likely to be less stable. Such systems, in which one heteroatom has an unusual tetravalent bonding within the ring, have recently been reported for thiophene containing bulky substituents at the 2,5-position, but were only isolated in very low yield.18 Fusing selenophene via the 2,3-positions potentially affords a mixture of four structural isomers, differing in the relative positioning of the heteroatoms (Fig. 1). However in all structural isomers the substituent in the 5-position (peripheral position) of the selenophene has a similar relative position with respect to other isomers, which should facilitate intermolecule packing. Therefore the required solubilising side chain was included in the peripheral position of the tetraselenophenoporphyrazine ring.
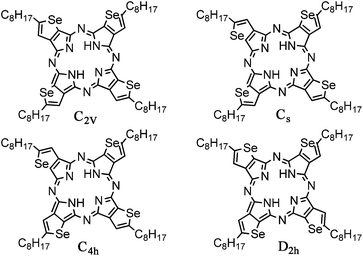 | ||
| Fig. 1 Possible isomers of tetraselenoporphyrazine. | ||
The synthetic route to tetraselenophenoporphyrazine (SePz) involves the preparation of 2,3-dicyano-5-octylselenophene, which can be readily converted to a macrocycle by reaction under Linstead conditions (Scheme 1). The route is similar to that reported by Takimiya for the thiophene derivative;10 however the reduced aromaticity and higher reactivity of selenophene over thiophene required some careful optimisation of reaction conditions. Beginning with commercially available selenophene, the octyl side chain was introduced in the 2-position by treatment with one equivalent of butyllithium, followed by reaction of the resulting anion with octyl bromide. Relatively low yields of 40% were obtained after purification by vacuum distillation. The major impurities were the 2,5-dialkylated product and unreacted starting material. Treatment of the resulting 2-octylselenophene with NBS in THF afforded 2-bromo-5-octylselenophene in 88% yield. Usage of a slight excess of NBS was found to result in a dibrominated impurity. Although this could not be separated by chromatography, it was identified as 2,4-dibromo-5-octylselenophene by comparison of the crude NMR with a sample of 2,3-dibromo-5-octylselenophene (vide infra). As expected the second electrophilic addition occurred in the more active 4-position, ortho to the alkyl chain, indicating that direct bromination was not a feasible route to the desired 2,3-dibromo-5-octylthiophene. Instead the ability of selenophene to undergo the so-called halogen dance was exploited. Thus treatment of 2-bromo-5-octylselenophene with one equivalent of freshly prepared LDA resulted in lithiation in the 3-position. Upon stirring at −78 °C the anion rearranged over time, most likely via an intermolecular mechanism, to afford 3-bromo-5-octylselenophene in 91% yield after quenching with methanol. Reaction with NBS resulted in bromination in the vacant 2-position to afford 2,3-dibromo-5-octylthiophene. Treatment with CuCN in hot DMF resulted in the formation of the desired 2,3-dicyano-5-octylselenophene in 60% yield. The metal free SePz was formed in 18% yield by reaction with lithium pentoxide in refluxing pentanol followed by acidic work-up.
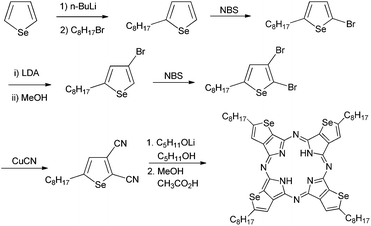 | ||
| Scheme 1 Synthesis of tetraselenoporphyrazine (SePz). | ||
The resulting SePz was readily soluble in a range of organic solvents, and was purified by chromatography over alumina to afford SePz as a mixture of isomers. No separation of the individual isomers was observed by normal or reverse phase chromatography. The existence of a mixture of isomers was confirmed by 1H and 77Se NMR (see ESI†). Assuming no steric or electronic factors favour the formation of any particular isomer, a statistical mixture would be expected with a relative ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:4 for C4h, D2h, C2V and Cs respectively. For the aromatic protons, the higher symmetry isomers (C4h and D2h) would be expected to give rise to a singlet each in the 1H NMR, whereas the lower symmetry Cs and C2v isomers could be expected to afford up to 6 singlets. Inspection of the 1H NMR, however, shows that the aromatic protons are split into two broad signals centered at 8.33 and 8.29 ppm respectively. The proton decoupled 77Se NMR also showed two broadly similar environments at 558 and 555 ppm, however in this case each broad peak could be resolved into four peaks suggesting that all isomers are present.
:1:2:4 for C4h, D2h, C2V and Cs respectively. For the aromatic protons, the higher symmetry isomers (C4h and D2h) would be expected to give rise to a singlet each in the 1H NMR, whereas the lower symmetry Cs and C2v isomers could be expected to afford up to 6 singlets. Inspection of the 1H NMR, however, shows that the aromatic protons are split into two broad signals centered at 8.33 and 8.29 ppm respectively. The proton decoupled 77Se NMR also showed two broadly similar environments at 558 and 555 ppm, however in this case each broad peak could be resolved into four peaks suggesting that all isomers are present.
The UV-Vis absorption spectrum of SePz in chloroform and as a spun cast thin film is shown in Fig. 2. The spectrum shows the characteristic split Q-band of a metal free phthalocyanine, with an intense maxima at 701 nm (ε 219000 M−1 cm−1) and a less intense broader peak at 653 nm, along with a broad Soret absorption around 360 nm. Compared to the data reported for the thiophene analogue,19 the incorporation of selenophene results in a red shift of 14 nm and a significant increase in the molar absorption coefficient of 35%. Upon film formation, the Q-band shifts bathochromically to 760 nm with a significant broadening of the absorption bands, suggestive of intermolecular interactions in the solid state. The optical band gap in the solid state, measured by the onset of absorption, is 1.51 eV, a reduction of approximately 0.05 eV compared to the thiophene analogue. SePz demonstrated good temperature stability by TGA in dry air, with the onset of decomposition (5% wt loss) occurring at 250 °C.
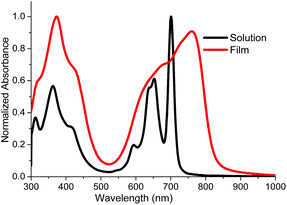 | ||
| Fig. 2 UV-Vis absorption spectra of SePz in CHCl3 and as a spun cast film. | ||
The ionization potential of a thin film of SePz was measured by photoelectron spectroscopy in air (PESA) and was found to be 5.27 (±0.05) eV. This is similar to the value reported for the thiophene analogue (TPz, 5.25 eV),19 which suggests that the reduction in band gap for SePz is mainly the result of stabilisation of the LUMO, rather than destabilisation of the HOMO similar to selenophene containing polymers.11 These results suggest that SePz may be a promising alternative to the thiophene analogue for solar cell applications, since the smaller optical band gap may potentially enhance device photocurrent, whilst the similar ionisation potential to TPz suggests no detrimental loss of the open circuit voltage.
The electronic performance of SePz was investigated in bottom gate, bottom contact field effect transistors. Gold (Au) was used as drain–source electrodes and SiO2 as the gate dielectric treated with HMDS. The as-spun films of SePz exhibited clear p-type behavior with mobilities on the order of 10−4 cm V−1 s−1. Annealing of the devices resulted in an improvement in hole mobility, with the best performance observed for transistors annealed at 50 °C which exhibited hole mobilities around 10−2 cm V−1 s−1 with channel current on/off ratios in excess of 104 (Fig. 3). Annealing at higher temperatures up to 150 °C did not result in any further improvement in device performance. Comparison of Fig. 3 with the performance characteristics of other phthalocyanine-based transistors is not straightforward due to the significant impact of device geometry as well as the non-optimized film morphology of SePz. Despite this, however, the present results show that SePz is amongst the best performing solution processed phthalocyanines reported to date.9,10
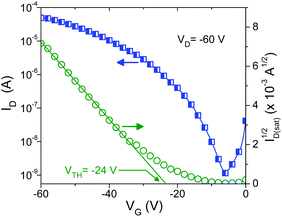 | ||
| Fig. 3 Transfer characteristics of a bottom gate, bottom contact transistor based on solution processed SePz after annealing at 50 °C (transistor channel = 10 μm, transistor width = 2.5 mm). | ||
In conclusion we have reported the synthesis of the first selenophene containing porphyrazine derivative, tetraselenophenoporphyrazine, as an inseparable mixture of isomers. The macrocycle exhibits a strong and sharp absorption in the visible region in solution, with a significant broadening and red shift upon film formation, indicative of ordering in the solid state. Field effect transistors fabricated from solution exhibit promising mobilities on the order of 10−2 cm V−1 s−1.
This work was supported by the EPRSC (EP/I002936/1 and EP/G060738/1). We thank Scott E. Watkins (CSIRO Materials Science and Engineering, Victoria, Australia) for performing PESA measurements.
Notes and references
- (a) G. de la Torre, C. G. Claessens and T. Torres, Chem. Commun., 2007, 2000 RSC; (b) M. V. Martinez-Diaz, G. de la Torrea and T. Torres, Chem. Commun., 2010, 46, 7090 RSC.
- J. Mack and N. Kobayashi, Chem. Rev., 2011, 111, 281 CrossRef CAS PubMed.
- A. Muranaka, M. Yonehara and M. Uchiyama, J. Am. Chem. Soc., 2010, 132, 7844 CrossRef CAS PubMed.
- (a) A. N. Cammidge and H. Gopee, Chem.–Eur. J., 2006, 12, 8609 CrossRef CAS PubMed; (b) S. Shimizu, S. Nakano, T. Hosoya and N. Kobayashi, Chem. Commun., 2011, 47, 316 RSC; (c) L. Zophel, K. S. Mali, P. S. Reddy, M. Wagner, S. De Feyter, W. Pisula and K. Mullen, Chem.–Eur. J., 2012, 18, 3264 CrossRef PubMed.
- A. N. Cammidge and H. Gopee, Chem. Commun., 2002, 966 RSC.
- T. Sooksimuang and B. K. Mandal, J. Org. Chem., 2002, 68, 652 CrossRef PubMed.
- M. J. Cook and A. JafariFini, J. Mater. Chem., 1997, 7, 2327 RSC.
- S. V. Kudrevich and J. E. van Lier, Coord. Chem. Rev., 1996, 156, 163 CrossRef CAS.
- (a) N. B. Chaure, J. L. Sosa-Sanchez, A. N. Cammidge, M. J. Cook and A. K. Ray, Org. Electron., 2010, 11, 434 CrossRef CAS PubMed; (b) S. Dong, H. Tian, D. Song, Z. Yang, D. Yan, Y. Geng and F. Wang, Chem. Commun., 2009, 3086 RSC; (c) N. B. Chaure, A. N. Cammidge, I. Chambrier, M. J. Cook, M. G. Cain, C. E. Murphy, C. Pal and A. K. Ray, Sci. Technol. Adv. Mater., 2011, 12, 025001 CrossRef; (d) Y. Miyake, Y. Shiraiwa, K. Okada, H. Monobe, T. Hori, N. Yamasaki, H. Yoshida, M. J. Cook, A. Fujii, M. Ozaki and Y. Shimizu, Appl. Phys. Express, 2011, 4, 021604 CrossRef.
- E. Miyazaki, A. Kaku, H. Mori, M. Iwatani and K. Takimiya, J. Mater. Chem., 2009, 19, 5913 RSC.
- (a) M. Al-Hashimi, M. A. Baklar, F. Colleaux, S. E. Watkins, T. D. Anthopoulos, N. Stingelin and M. Heeney, Macromolecules, 2011, 44, 5194 CrossRef CAS; (b) A. Patra and M. Bendikov, J. Mater. Chem., 2010, 20, 422 RSC.
- M. S. Rodriguez-Morgade and P. A. Stuzhin, J. Porphyrins Phthalocyanines, 2004, 8, 1129 CrossRef CAS.
- (a) E. M. Bauer, D. Cardarilli, C. Ercolani, P. A. Stuzhin and U. Russo, Inorg. Chem., 1999, 38, 6114 CrossRef CAS PubMed; (b) M. P. Donzello, C. Ercolani, K. M. Kadish, G. Ricciardi, A. Rosa and P. A. Stuzhin, Inorg. Chem., 2007, 46, 4145 CrossRef CAS PubMed; (c) P. A. Stuzhin, E. M. Bauer and C. Ercolani, Inorg. Chem., 1998, 37, 1533 CrossRef CAS.
- E. M. Bauer, C. Ercolani, P. Galli, I. A. Popkova and P. A. Stuzhin, J. Porphyrins Phthalocyanines, 1999, 3, 371 CrossRef CAS.
- P. A. Stuzhin, M. S. Mikhailov, E. S. Yurina, M. I. Bazanov, O. I. Koifman, G. L. Pakhomov, V. V. Travkin and A. A. Sinelshchikova, Chem. Commun., 2012, 48, 10135 RSC.
- (a) M. J. Cook and A. Jafari-Fini, Tetrahedron, 2000, 56, 4085 CrossRef CAS; (b) D. M. Knawby and T. M. Swager, Chem. Mater., 1997, 9, 535 CrossRef CAS; (c) M. J. Cook and A. Jafari-Fini, J. Mater. Chem., 1997, 7, 5 RSC.
- P. A. Stuzhin and C. Ercolani, Porphyrazines with Annulated Heterocycles, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2003, vol. 15, pp. 263–365 Search PubMed.
- T. Kimura, T. Iwama, T. Namauo, E. Suzuki, T. Fukuda, N. Kobayashi, T. Sasamori and N. Tokitoh, Eur. J. Inorg. Chem., 2011, 2011, 888 CrossRef.
- H. Mori, E. Miyazaki, I. Osaka and K. Takimiya, Org. Electron., 2012, 13, 1975 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and MALDI. See DOI: 10.1039/c3tc31584c |
| This journal is © The Royal Society of Chemistry 2013 |