 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of structure on the elastic, optical and dielectric properties of nematic phases formed from bent-core molecules
S. Kaur*a, H. Liua, J. Addisa, C. Grecob, A. Ferrarinib, V. Görtz†c, J. W. Goodbyc and H. F. Gleesona
aSchool of Physics and Astronomy, University of Manchester, Manchester M13 9PL, UK. E-mail: sarabjot.kaur@manchester.ac.uk
bDepartment of Chemical Sciences, University of Padua, I-35131 Padua, Italy
cDepartment of Chemistry, University of York, York YO10 5DD, UK
First published on 3rd September 2013
Abstract
The physical properties of the nematic phases formed by four bent-core oxadiazole based materials are reported. In particular, the splay (K11), twist (K22) and bend (K33) elastic constants, the birefringence and the dielectric anisotropy of the materials are described and the effect of chain length and the presence of fluoro-substituents at the outer phenylene group of the aromatic core structure on these parameters is determined. The birefringence and order parameter are found to be independent of the modification of molecular structure. The dielectric anisotropy is quite strongly dependent on molecular structure; the fluoro-substituted material has the largest magnitude of dielectric anisotropy while the alkyl-substituted compound has the smallest. Changes in the molecular length and fluoro-substitution in the bent-core materials are found to have little influence on the splay, twist and bend elastic constants at equivalent reduced temperatures. However, the material substituted with an alkyl terminal chain exhibits both smaller elastic constants and a less marked dependence on temperature than the alkoxy-substituted compounds. A possible insight into the behaviour of the elastic constants relevant to the formation of the dark conglomerate phase, which underlies the nematic phase in one of the compounds studied, is suggested by following the analysis proposed by Berreman and Meiboom. Importantly, using molecular field theory and atomistic modelling, we calculate elastic constants that are in excellent agreement with the experimental values. Our conclusion that the elasticity in the nematic phase formed from bent-core molecules is not strongly influenced by changes to the terminal chains or the presence of fluoro-substituents at the outer phenylene group of the aromatic core structure is in agreement with our previous work showing that the dominant parameter is the bend angle.
1 Introduction
Bent-core nematic liquid crystals (NLCs) are an exciting class of mesogenic systems. In addition to their potential to exhibit a biaxial nematic phase (a still somewhat contentious issue1,2) these systems are reported to possess extraordinary physical properties such as high Kerr constants,3 large flexoelectricity,4 and viscosity and elasticity behaviour quite different from that observed in calamitic NLCs.4–10 Lately, there has been much interest in the particularly intriguing elastic properties of bent-core NLCs. We recently obtained excellent agreement between our experimentally determined elastic constants and calculations based on molecular-field theory and atomistic modelling in an oxadiazole bent-core NLC.6 Subsequently, we established that the bend angle of bent-core molecules plays an extremely important role in determining the elastic properties.10 Our paper demonstrated that in case of bent-core NLCs with a bend angle of ∼164°, the bend (K33) elastic constant is larger than the splay (K11) elastic constant, analogous to the elastic behaviour in calamitic (rod-like) NLCs and in contrast to the expectation that all bent-core materials might have K33 < K11. While the role of bend angle is relatively clear, the difficulty of measuring the physical properties of bent-core nematic liquid crystal materials has ensured that, to date there are no reports on the vital influence of other structural parameters on the elastic behaviour in these materials.The Frank elastic constants are key physical parameters in nematic liquid crystals. The splay (K11), twist (K22) and bend (K33) elastic constants influence both the threshold voltage and the response times of nematic devices. Knowledge of the elastic constants allows an understanding of the microscopic structure of the ordered state, testing mean field theory, and providing insight into the director distortion in devices. Further, information about the elastic constants is needed in measurements of flexoelectricity, a parameter that is currently hotly debated in nematic systems formed from bent-core molecules. The influence of molecular structure variations in calamitic liquid crystals on the elastic behaviour is well-understood. Numerous reports describe the influence of factors such as the change in molecular length and width on the elastic properties of calamitic NLCs.11–15
This paper describes structure–property correlations determined in the nematic phases of four related bent-core materials. We consider the influence of the terminal chain length and the presence of fluoro-substituents at the outer phenylene group of the aromatic core structure on the optical, dielectric and elastic properties of the nematic phase and consider, where possible, the influence of the underlying phases on the elastic properties. We further examine theoretical fits to experimental data, together with calculations of the elastic constants from atomistic modelling.
2 Experimental and computational methods
2.1 Materials and devices
Four bent-core mesogens derived from a 1,3,4-oxadiazole-biphenyl (ODBP) core are shown with their phase transitions in Fig. 1. Although the absolute values of the transition temperatures are slightly higher than those quoted elsewhere,16–18 the nematic range is the same and differences are attributed to the absolute accuracy of hot stages at these high temperatures. All temperatures are, therefore, quoted with respect to the nematic to isotropic phase transition temperature, TNI correcting for differences in absolute temperature accuracy in different apparatus. The rationale to choosing compounds 1–4 is that oxadizoles of similar structure are well-known materials (the first claims of nematic biaxiality were made for compound 1 (ref. 19 and 20)) which have the same core and different alkyl/alkoxy chain lengths, which is a key consideration in the present work. The fact that the materials also have distinct phases underlying the nematic phase offers an additional opportunity to study the behaviour of elasticity near the phase transitions. Compounds 1 and 2 exhibit an underlying smectic-C (SmC) phase, compound 3 an unidentified smectic phase and the ‘dark conglomerate (DC)’ phase is found in compound 4.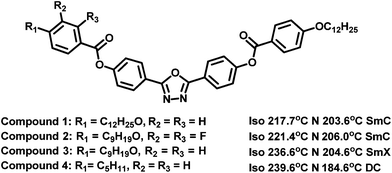 | ||
| Fig. 1 The chemical structure and phase transitions of 1,3,4-oxadiazole bent-core compounds 1–4 (phase transitions other than that directly below the nematic phase are omitted). | ||
All measurements employ devices with glass substrates and a liquid crystal layer approximately 5 μm thick. The transparent indium-tin-oxide electrode area is 30 mm2 (20 Ω □−1) and a high temperature conducting glue (H21D, Epoxy Technology) is used to attach the wires to the electrode surfaces. The electrical properties of the devices were calibrated using air as a standard reference at all temperatures studied and the devices were capillary-filled at 190 °C. All compounds exhibit negative dielectric anisotropy (Δε), across the entire nematic regime and as a consequence the measurement of the splay and bend elastic constants from Freedericksz transition requires homeotropic alignment, which is a challenge in bent-core materials. While excellent alignment was obtained using trichloro-octa-decyl silane in heptane across the entire nematic regime for compounds 2 and 4, the alignment degraded slightly at low temperatures for compounds 1 and 3. We take careful account of this in our results. A homogenously aligned interdigitated electrode device (sometimes known as an in-plane switching, IPS device) is used to measure the twist elastic constant, with the rubbing direction parallel to the field. The IPS device thickness is also ∼5 μm, with an electrode gap and width of 20 μm and 10 μm respectively. The devices were held in a Linkam THMS600 hot stage equipped with a TMS 93 controller which allows temperature control of the samples with a relative accuracy of ±0.1 K.
2.2 Birefringence measurements
The birefringence of the high temperature nematic phases exhibited by the bent-core materials is measured by analysis of the reflection spectra from planar devices via the Berreman methodology.21 Details of both the apparatus used and the fitting procedure are given in ref. 6 and 22. Reflection spectra are first taken for the empty device, allowing the thickness to be measured with an accuracy of ±0.02 μm, before filling with the liquid crystal. The ordinary and extraordinary refractive indices (no and ne) are determined from spectra obtained with the nematic director oriented perpendicular and parallel to the incident light polarization respectively. The birefringence, Δn = ne − no, may then be calculated with an accuracy of ±0.001. All values of refractive index are quoted for light of wavelength 589 nm.2.3 Measurement of the dielectric anisotropy and elastic constants
The methodology employed to determine the splay and bend elastic constants has also been described in detail elsewhere,6,10 and is based on an analysis of the variation in capacitance of a device that results from the electric-field induced Freedericksz transition. For homeotropic devices, the parallel component of dielectric permittivity, ε‖ is observed at fields below the Freedericksz threshold while at high field strength, the director alignment approaches the direction perpendicular to the field. A detailed fit to the capacitance–voltage curve allows the perpendicular component of dielectric permittivity, ε⊥ and hence the dielectric anisotropy, Δε to be deduced with an absolute accuracy that depends on the quality of the alignment, typically ∼5% for excellent homeotropic alignment. The relative accuracy of a particular data set which describes the temperature dependence is however better than this, ∼2%. The threshold voltage is related to the bend modulus while both the splay and bend constants influence the steepness of the threshold behaviour and the fit also allows the splay and bend elastic constants, K11 and K33 to be determined. This methodology allows the elastic constants to be determined straightforwardly, with an accuracy of around 5%, but it does not take account of any contributions from flexoelectric effects.Although measurement of the twist elastic constant, K22, is intricate in comparison with measuring the splay and bend elastic constants in LCs, we have successfully utilized in-plane devices to measure K22 in a positive Δε bent-core NLC material.10 In the present work, similar devices with the rubbing direction perpendicular to the length of the electrodes are used to measure K22.23,24 The voltage is applied initially parallel to the director and the electric-field induced Freedericksz transition occurs at a voltage 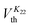 given by:
given by:
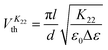 | (1) |
2.4 Atomistic calculation of the order parameters and elastic constants
The approach to calculating the order parameters and elastic constants was presented in ref. 25 and application to bent-core materials has also been described in detail in ref. 6 and 10. Indeed, calculations of the splay (K11) and bend (K33) elastic constants for compound 4 have already been presented,6 but here the study is extended to include compounds 2 and 3, and the twist elastic constant (K22) is also evaluated. All four compounds have the OC12H25 group as one terminal chain. Compounds 1–3 have alkoxy chains as the second terminal group, while compound 4 has an alkyl chain with the least number of carbon atoms (5) at the other end of the molecule. Compounds 2 and 3 are similar in structure as they both have the same terminal alkoxy groups but differ in that the former has two fluoro-substituents in the outer phenylene group of one arm (see Fig. 1). In this paper, compounds 2 and 3 are chosen for the simulation work to investigate the sensitivity of the elastic constants of the bent-core material to the length of the lateral chains and to the presence of substituents in the aromatic rings.As in ref. 6 the Rotational Isomeric State (RIS) approximation was used,26 and Metropolis Monte Carlo (MC)27 sampling of conformers was performed. Conformer geometry and energy were defined on the basis of quantum chemical calculations, carried out for single molecules of molecular fragments in vacuum.28 Thus, the following choices were made:
• Planar geometry of the central three rings (2,5-diphenyl-1,3,4-oxadiazole) was assumed.29
• In phenyl benzoate, a planar benzoate moiety was assumed and two states of the Car–Car–O–CO dihedral were considered (±90°), which correspond to the middle of the highest probability region for this dihedral.6
• The benzoate group of phenyl-2,3-difluoro benzoate was also assumed to be planar, with the CO group on the opposite side of the fluorine (F) atoms; for the Car–Car–O–CO dihedral the same choice as for phenyl benzoate was taken. These assumptions were based on torsional potentials obtained by relaxed scans at the DFT/M062X/6-31+g** level of the theory for the Car–Car–O–CO dihedral, and at the DFT/B3LYP/6-31+g* level for the Car–Car–CO–O dihedral. The torsional potential for the latter has two minima in correspondence to the planar configurations, and the configuration having the CO group pointing in the same direction as the F atoms is higher in energy than the other by about 3 kJ mol−1. The torsional potential for the Car–Car–O–CO dihedral has the same general form as that of phenyl benzoate, but the barriers at 0° and 180° are significantly lower; thus, higher rotational freedom is expected for the Car–O bond.
• The CH2–CH2 and the O–CH2 bonds of the alkyl and alkoxy chains were allowed to jump between the trans (180°), gauche+ (+65°) and gauche− (−65°) states, with relative energies defined as in ref. 6. Two possible equivalent states were assumed for the Car–CH2 bond, with the first CH2–CH2 bond perpendicular to the phenyl ring, on one or the other side. Also for the Car–O bond, two equivalent states were assumed, but in this case with the O–CH2 bond lying on the same plane of the aromatic ring, on either side.6 These two states are equivalent if the aromatic group is a benzoate, whereas in the case of 2,3-difluorobenzoate the conformation having the O–CH2 bond pointing opposite to the F atoms is more stable by about 3 kJ mol−1(DFT/B3LYP/6-31g**).
In MC sampling of conformers, the structures having pairs of atoms closer than a cut-off distance equal to 0.82σ, where σ is the sum of their van der Waals radii, were discarded. Van der Waals radii equal to 0.185 nm (C), 0.15 nm (N and O), and 0.1 nm (H)30 were assumed. In this way sterically hindered conformations were rejected, including those with adjacent gauche+ gauche− (or gauche− gauche+) pairs in the hydrocarbon chains. For each conformer, the molecular surface was generated by the fast molecular surface calculation library (MSMS),31 assuming a rolling sphere radius equal to 0.3 nm and density of vertices equal to 5 Å−2. The same van der Waals radii used for the cut-off distance were assumed for this purpose.
3 Results and discussion
3.1 Birefringence and order parameter
Fig. 2(a) shows the behaviour of the birefringence, Δn as a function of reduced temperature (T − TNI) in compounds 1–4. The birefringence increases monotonically with decreasing temperature for all compounds, behaviour that is entirely consistent with that observed in nematic materials generally. Indeed, the birefringence behaviour of each of the four materials is indistinguishable at the same reduced temperature. All four compounds exhibit Δn ∼ 0.10 near TNI increasing to ∼0.14 (compounds 1 and 2), ∼0.16 (compound 3) and ∼0.18 (compound 4) as the low lying phases are approached (the differences are entirely attributable to the different nematic ranges of each of the compounds). It is clear that the birefringence of the materials is dominated by the oxadiazole-based core. Sathyanarayana et al. report similar values of birefringence in a different bent-core material.7 On the other hand, a shape-persistent thiadiazole bent-core NLC exhibits a much larger birefringence (0.17 just below TNI, gradually increasing to ∼0.39 at lower temperatures); the high birefringence is attributed to the four triple bonds in the thiadiazole mesogen.10 An asymmetric L-shaped molecule has recently been reported to have rather high values of birefringence; Δn > 0.25 at temperatures far from TNI.32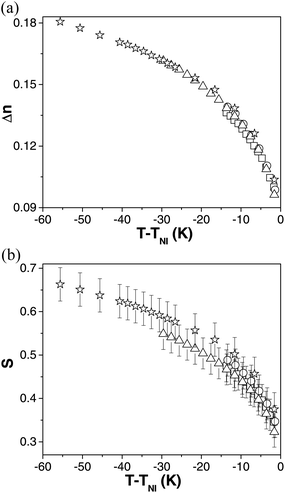 | ||
| Fig. 2 The birefringence, Δn (a) and order parameter, S (b) plotted as a function of reduced temperature, T − TNI for compounds 1 (square), 2 (circle), 3 (triangle) and 4 (star). | ||
As mentioned, the similarity of the birefringence of compounds 1–4 at equivalent reduced temperatures indicates that neither changes in terminal chain length nor the presence of fluoro-substituents on the outer phenylene group of the aromatic core structure has a strong influence on this parameter. This observation can be compared with the behaviour observed in calamitic NLCs such as alkoxy-azoxybenzenes, alkyl cyano-biphenyls (nCB), alkylcyanophenyl cyclohexanes (nPCH) and alkylcyclohexyl isothiocyanato-benzenes (nCHBT). In these systems, a strong odd–even effect is seen for short chain lengths and in general, Δn decreases with increasing molecular length.33–35 However, the variation is not as marked at longer chain lengths, which offer a better comparison with compounds studied here. Indeed the most significant change is seen when the core structure is changed. Further, Avci et al. report the birefringence of two similar L-shaped molecules substituted with C12 and C5 terminal chains; values coincide to around 10 K below TNI, but then appear to diverge at lower temperatures.32
The orientational order parameter, S can be determined from the birefringence data using the well-known Haller methodology36 (details are given in ref. 6 and 10). Fig. 2(b) shows the order parameter as a function of T − TNI for all four compounds. S increases from ∼0.30 near TNI for all of them to ∼0.48 (compounds 1 and 2), ∼0.55 (compound 3) and ∼0.66 (compound 4) near the low lying phases respectively. Excellent agreement is found between these measurements and the order parameter (P200) values obtained via Polarised Raman Spectroscopy (PRS).16Ref. 6 explicitly shows the excellent agreement of the two different approaches for compound 4. Order parameter measurements carried out via NMR on a different bent-core material by Dong et al. also are in excellent agreement with the values obtained here.37 The order parameter values obtained (Fig. 2(b)) at the same reduced temperatures, are very similar to those obtained for calamitic nematics (∼0.6–0.75)16 but are slightly higher than in a thiadiazole10 and another bent-core system.7 It is clear that the variation in molecular structure between compounds 1–4 has a negligible influence on the order parameter. Dong et al. also illustrated (via NMR) that S does not change on increasing the chain length in two carboxylate derived bent-core materials (with ten and eleven C atoms).37
3.2 Dielectric anisotropy and elastic constants
The magnitude of dielectric anisotropy, |Δε| determines the interaction strength of the LC with an electric field, and is therefore a vital parameter that influences the threshold voltage of a material. Fig. 3 shows Δε as a function of T − TNI for compounds 1–4. The magnitude of the dielectric anisotropy is found to increase with decreasing temperature in all compounds as would be expected and as is also observed in calamitic NLCs.38 There is almost no difference in |Δε| between compounds 1 (C12) and 3 (C9) at similar reduced temperatures; again significant changes with chain length are seen in calamitic NLCs only for relatively short chains.35,39 Compound 2 (C9F) exhibits the largest absolute value of Δε varying from −2.8 (T − TNI = −3.4 K) to −4.4 (T − TNI = −14.4 K) and the effect can be attributed to the conjugative mesomeric effect of the phenyl ether oxygen on the lateral fluoro substituents, as is seen for calamitic NLCs.40 Compound 4, which has one terminal alkyl chain replacing the alkoxy chain, has the smallest dielectric anisotropy, varying from just −1.4 (T − TNI = −8.7 K) to −3.2 (T − TNI = −56.7 K); this is approximately half of the value measured in compounds 1–3. While such a variation could be due to the different terminal chain in compound 4, it is to be remembered that the alkyl chain is only C5, i.e., it is somewhat shorter than in the other materials. A comparison of Δε for alkyl (e.g. 8CB) and alkoxy (8OCB) compounds in calamitic NLCs shows little difference in Δε,39,41 while |Δε| is slightly smaller in 8CB than 5CB. However, we suggest that in the oxadiazole materials substituting an alkyl group in place of an alkoxyl terminal chain reduces the lateral dipole of the molecule, hence reducing the dielectric anisotropy.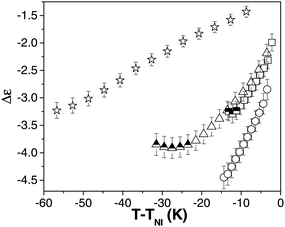 | ||
| Fig. 3 The dielectric anisotropy, Δε as a function of reduced temperature, T − TNI for compounds 1 (square), 2 (circle), 3 (triangle) and 4 (star). The few half filled symbols for compounds 1 and 3 represent the slight deterioration of alignment at low temperatures. | ||
As already mentioned, although excellent homeotropic alignment was obtained in all compounds, the alignment quality deteriorated for compounds 1 and 3 close to the underlying phase transition. The data points affected in Fig. 3 have been denoted by half-filled symbols; the deterioration was noticed within ∼3 K of the underlying SmC phase in compound 1 and within ∼10 K of the underlying phase for compound 3. We have included these data points partly because of the unexpected deterioration of the alignment (which was reproducible in several different cells), but also to illustrate the influence that small changes in alignment can have on the apparent values of the physical parameters being measured. In the case of both compounds 1 and 3, the effect of the reduction in alignment quality is, unsurprisingly, to reduce the dielectric anisotropy measured.
To present a clear view of the elastic behaviour of compounds 1–4 with respect to the molecular structure, the splay (K11), twist (K22) and bend (K33) elastic constants are shown separately in Fig. 4(a)–(c) respectively. Note that data are not included unless the alignment quality is excellent, specifically; it makes no sense to deduce elastic constants for the half-filled data points in Fig. 3. Fig. 4(a) shows that K11 increases monotonically with decreasing temperature for all materials. The data for compounds 1–3 are almost indistinguishable, taking values of ∼3.5 pN close to TNI and increasing to ∼8 pN within 10 K of the low-temperature transition. Both the magnitude and temperature variation of the splay elastic constant is different in compound 4, varying much more gradually from ∼3 pN close to the transition to ∼8.7 pN at ∼57 K below it. Fig. 4(b) also shows a monotonic increase in K22 for all compounds as the temperature is reduced. The magnitude of twist is rather small for the materials (∼1 pN), and again, compounds 1–3 exhibit almost indistinguishable K22 values at equivalent reduced temperatures. Both the magnitude and temperature dependence of K22 are much smaller in compound 4 than in the other materials. Fig. 4(c) displays the behaviour of K33 which generally is seen to increase with decreasing temperature. Once again, the behaviour of K33 is almost indistinguishable for compounds 1–3, changing from ∼2 pN close to TNI, and taking values ∼4 pN, 10 K below the transition. The behaviour of K33 in compound 4 is again very different from the rest of the compounds, increasing only slightly from 2.4 pN (T − TNI = −8.7 K) to 3.2 pN (T − TNI = −44.7 K) and decreasing marginally thereafter. The behaviour of K11 and K33 for compound 4 have been described in detail previously and are found to be in excellent agreement with the calculations obtained from molecular-field theory and atomistic modelling.6
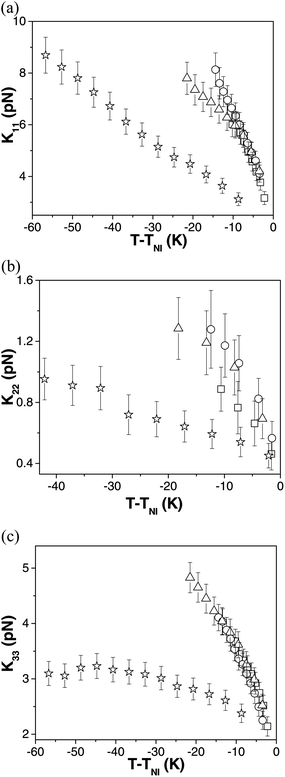 | ||
| Fig. 4 The splay, K11 (a) twist, K22 (b) and bend, K33 (c) elastic constants as a function of reduced temperature, T − TNI for compounds 1 (square), 2 (circle), 3 (triangle) and 4 (star). | ||
Clearly, overall the elastic behaviour is almost indistinguishable between compounds 1–3, while the alkyl substituted compound 4 shows significantly lower values of all three elastic constants, together with weaker temperature dependence. For all of the materials we find K11 > K33 > K22, analogous to the behaviour of most other bent-core NLCs (a thiadiazole bent-core NLC with a bend angle of ∼164° exhibits K33 > K11 > K22 (ref. 10) in common with calamitic NLCs). Indeed, by extracting data from ref. 32 with differently substituted L-shaped molecules, it can also be seen that neither the length of the terminal chains (C12 or C5) nor the presence of fluoro-substituents in the aromatic core structure significantly changes K11 or K33 in the investigated series of compounds.
We now consider the temperature dependence of the elastic constants near the low lying phases for all compounds (Fig. 4(a)–(c)). The simplest materials to consider are compounds 2 and 4, for which excellent homeotropic alignment was achieved across the nematic temperature range. The splay and bend elastic constants of compound 4 have been discussed in detail before (ref. 6); the transition to the underlying DC phase is first order, and no pretransitional divergence of the elastic constants is anticipated. Fig. 4(a) and (c) shows that neither splay nor bend exhibit pretransitional divergence in the nematic phase. The small decrease in the bend constant at lower temperatures is potentially of interest with respect to the discussion of the twist-bend nematic phases.42 No pretransitional divergence is found in either K11 or K33 for compound 2 which has a low lying SmC phase (there are too few data points to draw conclusions about pretransitional behaviour of K22). Such behaviour is in agreement with that reported by Tadapatri et al. who report no pretransitional divergence in either K11 or K33 for a symmetric bent-core benzoate compound (OC12 on both terminal ends) with a low lying SmC phase.8 In contrast, Sathyanarayana et al. report a strong pretransitional divergence in K33 for a bent-core material with a low lying SmC phase.7 Findon and Gleeson43 measured K11 in the nematic and SmC phase of a calamitic mixture, with a continuous variation across the transition.
Any discussion of the pretransitional behaviour in compounds 1 and 3 is complicated by the fact that both materials suffered from deterioration in the otherwise excellent homeotropic alignment as the lower-lying phase was approached. Indeed it is interesting that the deterioration phenomenon was observed in several different cells filled with these two materials, occurring over approximately the same temperature range in every case (∼3 K and ∼10 K for compounds 1 and 3 respectively), but at different temperatures for each compound, ruling out thermal deterioration of the alignment layer. While we cannot, therefore make any comparison of the pretransitional elastic behaviour in these compounds, we can note that such a reproducible change in alignment is unusual and may be of interest given current discussions around anomalous behaviour and texture observations in nematic materials.
3.3 Elastic constants and order parameter
A comparison between the elastic constants and the order parameter not only throws light on the molecular mean field theory but also helps in understanding the microscopic structure of the ordered state. The elastic constants of the few bent-core NLCs6,7,10 and mixtures of calamitic and bent-core NLCs41,44 reported to date do not strictly obey the mean field theory approximation which suggests a square dependence of the order parameter (Kii ∝ S2). Indeed, it has been shown for two different bent-core materials6,10 that the most appropriate way to describe the relationship is to utilize a third order dependency as advocated by Berreman and Meiboom.45 The relationship is given by: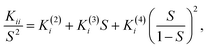 | (2) |
Fig. 5(a) and (b) show Kii/S2 plotted as a function of order parameter for compounds 2 and 4 respectively. The fits to eqn (2), shown as solid lines, have been made using the assumption that K(2–4)i are temperature independent and the requirement that the elastic constants converge at S = 0 is relaxed. The dotted lines in Fig. 5(a) and (b) are the extrapolated values of Kii/S2 calculated using the fitting parameters K(2–4)i which are given in Table 1. Note that fits are not made to the K22 data for compound 2 as there are only 5 experimental data points. Further, as the elastic constants could not be measured close to the underlying phase for compounds 1 and 3, fits to the data are not included as they cannot be used to give any insight to the pretransitional behaviour of the materials.
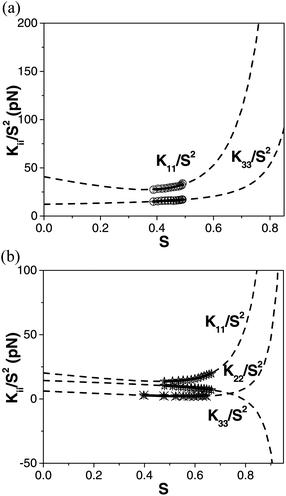 | ||
| Fig. 5 The dependence of Kii/S2 (i = 1,2 or 3 for splay, twist and bend respectively) as a function of order parameter for: (a) compound 2, K11/S2 (open circles) and K33/S2 (circles with a plus sign). (b) The same quantities with star symbols for compound 4 and K22/S2 (star with a cross sign). The solid lines show fits of eqn (2) to the data and the dashed lines extrapolate the fits to low and high order parameter regimes (outside the region of stability of the nematic phase). | ||
| Compounds | K(2)1 (pN) | K(3)1 (pN) | K(4)1 (pN) | K(2)2 (pN) | K(3)2 (pN) | K(4)2 (pN) | K(2)3 (pN) | K(3)3 (pN) | K(4)3 (pN) |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 41 ± 13 | −55 ± 41 | 20 ± 8 | — | — | — | 12 ± 5 | 5 ± 16 | 2.3 ± 3.1 |
| 4 | 20 ± 2 | −19 ± 5 | 3.2 ± 0.3 | 6.2 ± 0.7 | −9.1 ± 1.6 | 0.6 ± 0.1 | 14 ± 1.5 | −7.5 ± 3.3 | −0.6 ± 0.2 |
Berreman and Meiboom successfully applied the theory to three standard calamitic NLCs (MBBA, PAA, E7).45 Their fits all diverge to large (positive) values of elastic constant at high order (low temperature), with the splay curves diverging at higher temperatures (lower order) than those for twist and bend, consistent with a virtual low-lying smectic phase. While we recognise that conclusions drawn from theoretical fits to experimental data depend on both the quality and quantity of the data points, it is nonetheless interesting to apply such an approach to the elastic constants of compounds 2 and 4. Fig. 5(a) indicates that both K11/S2 and K33/S2 for compound 2 diverge to larger values at high S. The splay curve again diverges at a lower order than the bend, consistent with the occurrence of an underlying SmC phase in this material. The behaviour of compound 4 is different with both K11/S2 and K22/S2 diverging to larger values at high S while K33/S2 diverges to negative values. Interestingly, again the splay data begin to diverge at lower order (higher temperatures) than twist or bend even though the lower temperature phase is a DC phase. Further, the bend data clearly diverge to negative values at high order, a point of potential relevance to the twist-bend phases predicted by Dozov et al.46 Currently, little is known about the behaviour of the elastic constants in the DC phase which is the low-lying phase in this material. However, the phase is known to be sponge-like with a local SmC structure and the formation relies on a small (or negative) saddle-splay elastic constant, K24.47 The divergence of K11/S2 at lower order than K33/S2 is again consistent with the formation of a local SmC structure. Perhaps more interestingly, the Ericksen inequalities48 suggest that |K24| < K22 and as K22 takes extremely small values in compound 4 (<1 pN) we can note that the formation of the DC phase is consistent with the elastic behaviour measured. The very low and negatively diverging values of K33 may also be relevant to the formation of the DC phase. Clearly, using eqn (2) to analyse the behaviour of the bent-core materials gives an interesting possible insight into the formation of the underlying phase, though the negative divergence of K33/S2 on approaching the DC phase must be confirmed in other systems before firm conclusions can be drawn.
3.4 Atomistic calculations of the elastic constants
It has already been demonstrated for one material (compound 4) that excellent qualitative and good quantitative agreement (within 2 pN) is found between the experimentally determined splay and bend elastic constants and calculations carried out via simulation and atomistic modelling.6 Indeed, Fig. 6(a) shows the result of calculations of all three elastic constants of compound 4 as a function of the order parameter Szz, with z being the axis passing through the carbon atoms of the oxadiazole ring. In agreement with experiment we find K22 < K33 < K11, with very small K22. Remarkably, the trend of the calculated elastic constants as a function of the order parameter is also in agreement with experiment: K11 and K22 increase with increasing order, whereas K33 first increases and then decreases at high ordering. In the theoretical expressions for the elastic constants25 there is an implicit dependence on the molecular order parameter, which involves higher order terms beyond those proportional to Szz2. Fig. 6(b) shows the ratios Kii/Szz2, for illustrative purposes a wide range of order parameters is considered, including values beyond the experimental ones. It may be worth pointing out that the branches of the curves in Fig. 6(b) and 5(b) at high ordering have a different meaning: in the former case they represent the elastic constants of a hypothetical phase with nematic order extending to very high ordering, whereas in the latter case the fitting curves are meant to reproduce the whole experimental behaviour, including extra-nematic effects. The results reported in Fig. 6(b) show that the splay and twist elastic constants calculated for the nematic phase of compound 4 are not strictly proportional to Szz2, but the ratios K11/Szz2 and K22/Szz2 exhibit a relatively small increase with increasing order. On the contrary, a strong dependence on the order parameter is predicted for the ratio K33/Szz2, which would diverge to negative values at very high ordering. This trend agrees with the behaviour shown in Fig. 5(b).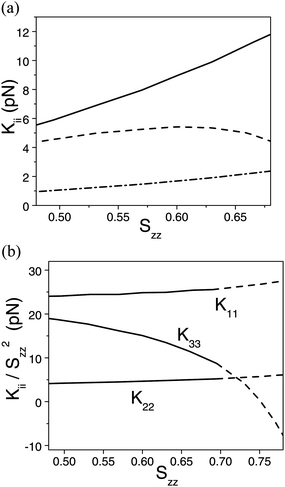 | ||
| Fig. 6 (a) The elastic constants obtained by atomistic calculations for compound 4, as a function of the order parameter (Szz): K11 (solid line), K22 (dash-dotted line) and K33 (dashed line). (b) Ratio of elastic constants (Kii) to the squared order parameter (Szz), as obtained from atomistic calculations for compound 4. Dashed lines are used for points beyond the experimental nematic region. Szz is the order parameter for the axis passing through the carbon atoms of the oxadiazole ring. | ||
Calculations of the elastic constants were also carried out for compounds 2 and 3. Both differ from compound 4 for the replacement of the pentyl with a longer nonyloxy chain. In compound 2 there are also two additional F atoms in the phenyl ring attached to the nonyloxy chain. In the framework of the molecular field model used here, the F atoms are expected to affect the elastic constants if they can induce a change of the conformational preferences, thus a change of the average molecular shape. Quantum chemical calculations show that the main effects of the F atoms are those of lowering the energy barriers opposing the rotation of the CO–O–Car–Car bond and stabilizing the conformations having both the CO group and the O–CH2 bond pointing in the same direction, opposite to the F atoms. We have found that neither these variations, nor those deriving from the change of the lateral chain significantly affect the average molecular shape: the plots of the elastic constants as a function of the Szz order parameter calculated for compounds 2 and 3 are very similar to each other and also not very different from that shown in Fig. 6(a) for compound 4. The small influence of changes in the lateral arms on the elastic constants is in line with the experimental findings for compounds 1–4. Fig. 7 explicitly shows excellent agreement (within 2 pN) between the experimental and calculated elastic constants for compound 2. It is to be noted that though the calculations do not point to any difference in the elastic constants for compound 4 from the rest, the experimental values are slightly lower for compound 4 than the other compounds which could most probably be due to the pentyl/nonyloxy chain difference. Previous calculations for single molecular conformers pointed to the strong sensitivity of elastic constants of bent-core nematics to the conformation of the lateral chains.6 This is not in contradiction to the negligible effect of the terminal chains in compounds 1–4, evidenced here both by experiments and calculations, since the internal flexibility leads to an average molecular shape where the effects of single conformations are washed out.
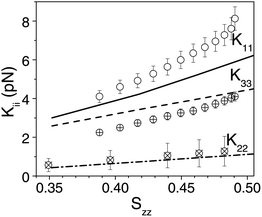 | ||
| Fig. 7 The experimentally deduced splay, K11 (open circles), twist, K22 (circles with a cross) and bend, K33 (circles with a plus sign) elastic constants as a function of order parameter, S for compound 2. Note that the error bars in this figure include the absolute uncertainty in the measurement which is most significant for measurements of K22. The elastic constants calculated by atomistic simulations for compound 2 are denoted by K11 (solid line), K22 (dash-dotted line) and K33 (dashed line). | ||
4 Conclusions
This paper describes the temperature dependence of the splay, twist and bend elastic constants along with the dielectric anisotropy, birefringence and order parameter measurements in four related bent-core oxadiazole compounds. The birefringence, Δn as well as the order parameter, S for all compounds is found to be independent of the change in molecular length or the presence of fluoro-substituents at the outer phenylene group of the aromatic core structure. Excellent agreement is found for the order parameter values derived from Haller method with those reported from PRS16 in all the compounds. The dielectric anisotropy is, however, found to depend strongly on the molecular structure, with the presence of fluoro-substituents at the outer phenylene group of the aromatic core structure (compound 2) resulting in the highest magnitude of dielectric anisotropy while the pentyl-substituted (compound 4) shows the smallest magnitude.All of the bent-core compounds studied show K22 < K33 < K11 analogous to other bent-core compounds with comparable bend angles. The dependence of the elastic constants on temperature is found to be indistinguishable for compounds 1, 2 and 3, all of which have alkoxy terminal chain substitution. The temperature variation of the elastic constants is much less marked in compound 4 than the other materials. A third-order dependence of the elastic constants on order parameter was found to give excellent fits to the experimental data for compounds 2 and 4, but was not used to analyse the elastic behaviour of the other two compounds, both of which showed an unusual but reproducible deterioration in alignment on approaching the underlying higher order phase. Extrapolation of the elastic behaviour was carried out using the fitting parameters obtained for compounds 2 and 4. The insight offered by such an analysis is consistent with what is known about the structure of the DC phase and role of the saddle-splay constant, K24 in its formation. It is possible that the bend elastic constant may also tend to diverge to negative values on approaching the DC phase, a suggestion that merits further experimental study. We have demonstrated that molecular-field theory and atomistic modelling can be used to calculate the temperature dependence of the elastic constants for the materials. The calculations show remarkable agreement with experimentally determined values. Experiments and computation agree in finding that the elastic constants of compounds 1–4 are only weakly affected by changes in the terminal chains. The differences between compound 4 and the other compounds, detected experimentally, are probably beyond the predictive capability of the atomistic calculations. Both approaches show that there is no effect of the presence of fluoro-substituents on the outer phenylene group of the aromatic core structure (compound 2) on the elasticity of the LC materials.
Both the analysis of experimental data and theoretical predictions agree in showing that the elastic constants of bent-core NLCs exhibit significant deviations from the square dependence on the order parameter, which is generally assumed for calamitic NLCs.
In summary, the combination of careful experimentation with calculations has begun to reveal some of the relationships between the molecular structure and the physical properties of nematic phases formed from bent-core molecules. Further, this work has offered a tantalising glimpse into the behaviour of the elastic constants on approaching an underlying DC phase.
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC) under Project no. EP/G023093/1. CG acknowledges the PhD School of Materials Science and Engineering (University of Padova) and Merck Chemicals Ltd. for financial support. HL is grateful to Merck Chemicals Ltd. and the EPSRC for a CASE Award. VG gratefully acknowledges the Royal Society for funding through a Dorothy Hodgkin Fellowship.References
- A. G. Vanakaras and D. J. Photinos, J. Chem. Phys., 2008, 128, 154512 CrossRef PubMed.
- O. Francescangeli, F. Vita, C. Ferrero, T. Dingemans and E. T. Samulski, Soft Matter, 2011, 7, 895 RSC.
- S. Dhara, F. Araoka, M. Lee, K. V. Le, L. Guo, B. K. Sadashiva, K. Song, K. Ishikawa and H. Takezoe, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 050701 CrossRef.
- P. S. Salter, C. Tschierske, S. J. Elston and E. P. Raynes, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 031708 CrossRef CAS.
- M. Majumdar, P. Salamon, A. Jákli, J. T. Gleeson and S. Sprunt, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 031701 CrossRef CAS.
- S. Kaur, J. Addis, C. Greco, A. Ferrarini, V. Görtz, J. W. Goodby and H. F. Gleeson, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 041703 CrossRef CAS.
- P. Sathyanarayana, M. Mathew, Q. Li, V. S. S. Sastry, B. Kundu, K. V. Le, H. Takezoe and S. Dhara, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 81, 010702 CrossRef CAS.
- P. Tadapatri, U. S. Hiremath, C. V. Yelamaggad and K. S. Krishnamurthy, J. Phys. Chem. B, 2010, 114, 1745 CrossRef CAS PubMed.
- P. Salamon, N. Éber, J. Seltmann, M. Lehmann, J. T. Gleeson, S. Sprunt and A. Jákli, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 061704 CrossRef CAS.
- S. Kaur, L. Tian, H. Liu, C. Greco, A. Ferrarini, J. Seltmann, M. Lehmann and H. F. Gleeson, J. Mater. Chem. C, 2013, 1, 2416 RSC.
- H. Gruler and G. Meier, Mol. Cryst. Liq. Cryst., 1973, 23, 261 CrossRef CAS.
- W. H. de Jeu and W. A. P. Claassen, J. Chem. Phys., 1977, 67, 3705 CrossRef CAS.
- F. Leenhouts, H. J. Roebers, A. J. Dekker and J. J. Jonker, J. Phys., Colloq., 1979, 40, 291 CrossRef.
- H. Schad, G. Baur and G. Meier, J. Chem. Phys., 1979, 70, 2770 CrossRef CAS.
- W. H. de Jeu, Physical Properties of Liquid Crystalline Materials, Gordon and Breach Science Publishers, New York, London, Paris, 1980 Search PubMed.
- H. F. Gleeson, C. D. Southern, P. D. Brimicombe, J. W. Goodby and V. Görtz, Liq. Cryst., 2010, 37, 949 CrossRef CAS.
- Y. Xiang, J. W. Goodby, V. Görtz and H. F. Gleeson, Appl. Phys. Lett., 2009, 94, 193507 CrossRef.
- V. Görtz and J. W. Goodby, Chem. Commun., 2005, 3262 RSC.
- B. R. Acharya, A. Primak and S. Kumar, Phys. Rev. Lett., 2004, 92, 145506 CrossRef.
- L. A. Madsen, T. J. Dingemans, M. Nakata and E. T. Samulski, Phys. Rev. Lett., 2004, 92, 145505 CrossRef CAS.
- D. W. Berreman, J. Opt. Soc. Am., 1972, 62, 502 CrossRef CAS.
- H. G. Yoon, N. W. Roberts and H. F. Gleeson, Liq. Cryst., 2006, 33, 503 CrossRef CAS.
- K. Ikeda, H. Okada, H. Onnagawa and S. Sugimori, J. Appl. Phys., 1999, 86, 5413 CrossRef CAS.
- M. Oh-e and K. Kondo, Appl. Phys. Lett., 1996, 69, 623 CrossRef CAS.
- M. Cestari, A. Bosco and A. Ferrarini, J. Chem. Phys., 2009, 131, 054104 CrossRef PubMed.
- P. W. Flory, Statistical Mechanics of Chain Molecules, Wiley-Interscience, New York, 1969 Search PubMed.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087 CrossRef CAS.
- M. J. Frisch, Gaussian 09 (Revision B.01), 2010 Search PubMed.
- J. P. Laguno, PhD Thesis, Durham University, 2007.
- Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, 1996 Search PubMed.
- M. F. Sanner, A. J. Olson and J.-C. Spehner, Biopolymers, 1996, 38, 305 CrossRef CAS.
- N. Avci, V. Borshch, D. D. Sarkar, R. Deb, G. Venkatesh, T. Turiv, S. V. Shiyanovskii, N. V. S. Rao and O. D. Lavrentovich, Soft Matter, 2013, 9, 1066 RSC.
- E. G. Hanson and Y. R. Shen, Mol. Cryst. Liq. Cryst., 1976, 36, 193 CrossRef CAS.
- W. H. de Jeu, Mol. Cryst. Liq. Cryst., 1981, 63, 83 CrossRef CAS.
- P. Sarkar, P. Mandal, S. Paul, R. Paul, R. Dabrowski and K. Czuprynski, Liq. Cryst., 2003, 30, 507 CrossRef CAS.
- I. Haller, Prog. Solid State Chem., 1975, 10, 103 CrossRef.
- R. Y. Dong, J. Phys. Chem. B, 2009, 113, 1933 CrossRef CAS PubMed.
- P. Kula, A. Spadlo, J. Dziaduszek, M. Filipowicz, R. Dabrowski, J. Czub and S. Urban, Opto-Electron. Rev., 2008, 16, 379 CrossRef CAS PubMed.
- B. R. Ratna and R. Shashidhar, Pramana, 1976, 6, 278 CrossRef CAS.
- M. Hird, J. W. Goodby, R. A. Lewis and K. J. Toyne, Mol. Cryst. Liq. Cryst., 2003, 401, 115 CAS.
- B. Kundu, R. Pratibha and N. V. Madhusudana, Phys. Rev. Lett., 2007, 99, 247802 CrossRef.
- V. Görtz, C. Southern, N. W. Roberts, H. F. Gleeson and J. W. Goodby, Soft Matter, 2009, 5, 463 RSC.
- A. Findon and H. F. Gleeson, Ferroelectrics, 2002, 277, 35 CrossRef CAS.
- P. Sathyanarayana, V. S. R. Jampani, M. Skarabot, I. Musevic, K. V. Le, H. Takezoe and S. Dhara, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 011702 CrossRef CAS.
- D. W. Berreman and S. Meiboom, Phys. Rev. A, 1984, 30, 1955 CrossRef CAS.
- I. Dozov, Europhys. Lett., 2001, 56, 247 CrossRef CAS.
- D. Chen, Y. Shen, C. Zhu, L. E. Hough, N. Gimeno, M. A. Glaser, J. E. Maclennan, M. B. Ros and N. A. Clark, Soft Matter, 2011, 7, 1879 RSC.
- J. L. Ericksen, Phys. Fluids, 1966, 9, 1205 CrossRef.
Footnote |
| † Current address: Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. |
| This journal is © The Royal Society of Chemistry 2013 |