 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent materials for pH sensing and imaging based on novel 1,4-diketopyrrolo-[3,4-c]pyrrole dyes†
Daniel
Aigner
a,
Birgit
Ungerböck
a,
Torsten
Mayr
a,
Robert
Saf
b,
Ingo
Klimant
a and
Sergey M.
Borisov
*a
aInstitute of Analytical Chemistry and Food Chemistry, Graz University of Technology, Stremayrgasse 9, Graz, Austria. E-mail: sergey.borisov@tugraz.at; Tel: +43 316 873 32516
bInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, A-8010 Graz, Austria
First published on 17th July 2013
Abstract
New optical pH-sensors relying on 1,4-diketopyrrolo-[3,4-c]pyrroles (DPPs) as fluorescent pH-indicators are presented. Different polymer hydrogels are useful as immobilization matrices, achieving excellent sensitivity and good brightness in the resulting sensor. The operational pH can be tuned over a wide range (pH 5–12) by selecting the fine structure of the indicator and the matrix. A ratiometric sensor in the form of nanoparticles is also presented. It is suitable for RGB camera readout, and its practical applicability for fluorescence imaging in microfluidic systems is demonstrated. The indicators are synthesized starting from the commercially available DPP pigments by a straightforward concept employing chlorosulfonation and subsequent reaction with amines. Their sensitivity derives from two distinct mechanisms. At high pH (>9), they exhibit a remarkable alteration of both absorption and fluorescence spectra due to deprotonation of the lactam nitrogen atoms. If a phenolic group is introduced, highly effective fluorescence quenching at near-neutral pH occurs due to photoinduced electron transfer (PET) involving the phenolate form.
Introduction
pH is one of the key parameters in medical, environmental and life sciences. Despite the strong performance of electrochemical pH-sensors, optical pH-sensors have proved invaluable in numerous important applications. They possess numerous advantages including greater ease of miniaturization and the possibility of contactless measurement. Moreover, optical probes enable imaging applications.1–5 These features are particularly attractive in high-throughput screening and for probing small samples such as living cells or sub-cellular structures.6,7Optical pH-sensors typically consist of a pH-sensitive dye (i.e. a pH-indicator) immobilized in a polymer matrix which has to provide suitable mechanical and adhesive properties, together with sufficient water uptake. Although most optical pH-indicators are essentially (de)protonatable chromophores,8–10 those with proton receptors separated from the chromophore have also found numerous applications. They are the most flexible in terms of rational dye design since the chromophore and the receptor can be selected independently. Most frequently, they take advantage of the photoinduced electron transfer (PET)11–13 process. Though PET is an extensively investigated effect, in most publications it is introduced by amine functionalities,14–17 while phenolic groups have attracted comparatively little attention. In 1997, Gareis et al.18 presented a boron-dipyrromethene (BODIPY) pH-indicator with a phenolic proton receptor. Most comparable systems have relied on the BODIPY chromophore since then.19–21
Derivatives of 1,4-diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole, often referred to as DPPs, are chemically stable, brightly fluorescent22 molecules that have found a variety of applications. While the parent compounds are commonly used pigments, the attachment of suitable substituents yields readily soluble fluorescent dyes. Alkylation of the lactam nitrogen atoms is most effective in this regard since hydrogen bonding interactions are suppressed. DPP-based dyes and pigments have been used as high-performance colorants in prints and inks, as components of solid-state dye lasers23–27 and more recently in the field of organic optoelectronics. Particularly, DPP-containing conjugated polymers28–30 and small molecules31 have found extensive use in Organic Field Effect Transistors (OFETs)32,33 and organic solar cells.34,35 DPP dyes are also particularly promising for the design of two-photon excitable fluorophores.36,37 A few DPP-based fluorescent probes and sensors for fluoride,38 cyanide,39 thiols40 and molecular hydrogen41 have been developed. The DPP-based probe presented by Yamagata et al.42 is suitable for detecting strong acids in organic solvents, rather than measuring near-neutral pH in aqueous solution. Recently, we presented carbon dioxide sensors that exploit the deprotonation of the lactam nitrogen atoms in DPPs.43 The same mechanism is useful for the determination of comparatively high pH (>9), as will be demonstrated in this work. Furthermore, we present – to the best of our knowledge for the first time – DPP-based pH-sensors that operate at near-neutral pH. They rely on PET from phenolate groups to the chromophore.
Results and discussion
The preparation of the new pH-indicators and sensors is shown in Fig. 1. The indicators 2 and 3 feature phenolic PET groups. 4 carries a morpholino group for solubilization and is an example of a DPP pH-indicator relying on deprotonation of the lactam nitrogen.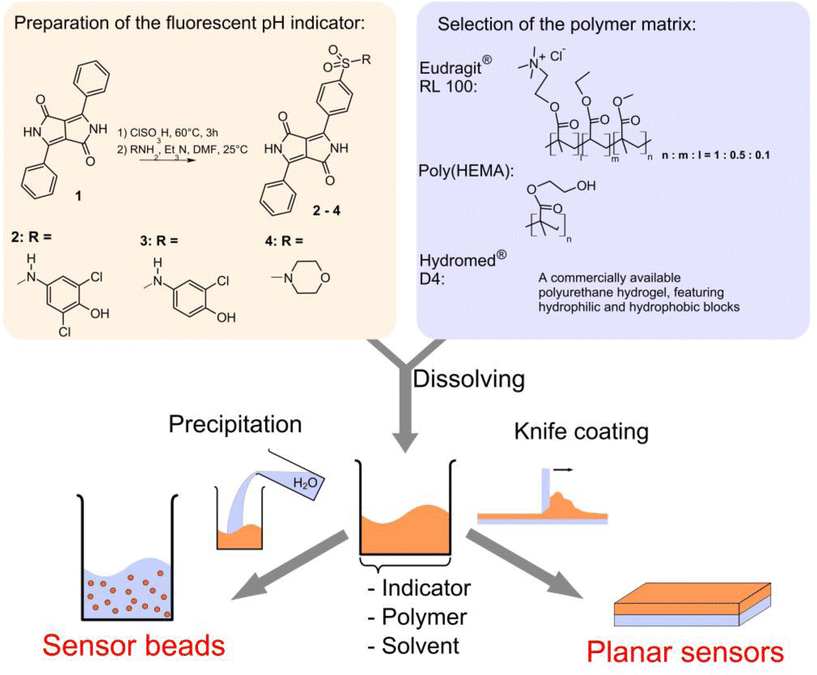 | ||
| Fig. 1 Scheme for the preparation of the fluorescence pH-sensors in this work. | ||
Indicator syntheses
The indicators 2–4 can be easily prepared in a single step starting from commercially available 1,4-diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole. Notably, the reaction conditions applied afforded only monosulfonated products. Doubly sulfonated products are formed under harsher reaction conditions.43 The intermediate, a sulfonyl chloride, can yield a large variety of sulfonamides, depending on the amine it is reacted with. The synthetic concept employed is simple and versatile as it is applicable for any class of chromophores that can withstand chlorosulfonation. It is useful for tagging a large variety of structures and is not limited to the preparation of pH-indicators, which has been the main focus of this work.The modest yields (13–22%) are due to difficulties in purifying the products by column chromatography. They strongly bind to the stationary phase and are hard to elute completely. Nevertheless, all products could be easily isolated in sufficient amounts starting from the cheap commercial pigment.
Indicator properties
Spectral properties of the DPP sulfonamides 2–4 in comparison with the starting material are listed in Table 1. The attachment of a sulfonamide group results in bathochromically shifted and less structured absorption bands, while the Stokes shifts are significantly enlarged. Despite that 2–4 are not N-substituted, their solubilities exceed 2 g l−1 in N,N-dimethylformamide and tetrahydrofuran. This is dramatically higher than for pigment 1 which is virtually insoluble in these solvents at 25 °C. Note that for the majority of applications requiring soluble DPPs, N-substituted derivatives are used. However, our preliminary experiments indicated that chlorosulfonation of a N,N′-dialkylated DPP (N,N′-di(2-ethylhexyl)-1,4-diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole) and subsequent reaction with 4-amino-2,6-dichlorophenol did not yield a pH-indicator.| Dye | λ max abs(ε × 10−4)/nm (M−1 cm−1), acidic/basic | λ max em/nm, acidic/basic | Φ F, acidic/basic |
|---|---|---|---|
| a The bathochromically shifted spectra correspond to the dianionic form (both phenol and lactam are deprotonated). The absorption of the monoanionic form (only phenol is deprotonated) is not shifted with respect to the acidic form (Fig. 3). | |||
| 1 | 468(2.95), 502(3.91) | 514, 552 | n.d. |
| 2 | 509(2.23), 543(2.40)/575(1.88), 606(2.16)a | 580/n.m. | 0.70/n.m. |
| 3 | 508(1.74), 541(1.86)/575(1.60), 606(1.86)a | 577/n.m. | 0.66/n.m. |
| 4 | 509(2.02), 541(2.14)/391(0.95), 584(1.89), 619(2.39) | 576/679 | 0.71/0.08 |
The pH-sensitivity of the DPP indicators is associated with two distinct mechanisms, as illustrated in Fig. 2. 2 and 3 are subject to photoinduced electron transfer (PET) when the phenolic group is deprotonated. The result is fluorescence quenching around the pKa of the phenolic group (that is, pH 5.9–9.3, Table 2). No alteration of the absorption spectra at all is observed (Fig. 3), indicating that the effect is solely PET. Importantly, the quenching is extremely efficient (virtually no fluorescence from the deprotonated form is detected) which indicates that phenolates are suitable proton receptor groups for designing fluorescent pH-indicators. Thus they represent a very promising alternative to the much more common amino-based receptors.
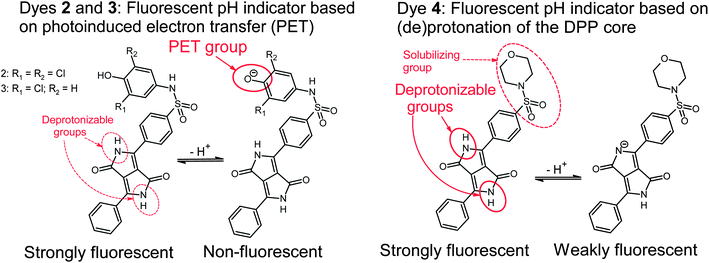 | ||
| Fig. 2 Mechanisms causing pH-sensitivity in the DPP-based indicators. | ||
| Dye | EtOH–H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) :1 (v/v) |
D4 | poly(HEMA) | RL100 | ||
|---|---|---|---|---|---|---|
| pH1/2 (Fluo.) | pH1/2 (Abs.) | pH1/2 (Fluo.) | pH1/2 (Abs.) | pH1/2 (Fluo.) | pH1/2 (Fluo.) | |
| a Corresponds to deprotonation of the phenolic PET group. b Corresponds to deprotonation of the lactam nitrogen. c The doubly charged basic form is quickly leached out of the sensor. | ||||||
| 2 | 6.49a | 11.3b | 7.76a | n.d.c | 7.08a | 5.88a |
| 3 | 7.63a | 11.3b | 9.34a | n.d.c | 8.36a | 7.62a |
| 4 | 9.75b | 9.88b | 11.1b | 11.6b | 11.1b | 9.65b |
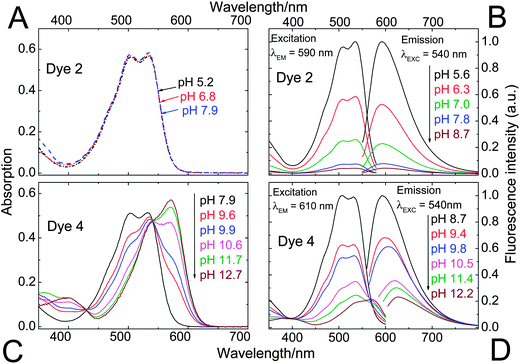 | ||
| Fig. 3 pH-dependent absorption (A and C) and fluorescence (B and D) spectra of the DPPs 2 and 4. Since 2 and 3 feature virtually identical spectral properties and differ only by their sensitive pH ranges, 3 has been included in the electronic supplementary information (ESI; Fig. S1†) only. Spectra were recorded in ethanol–aqueous buffer (ionic strength 100 mM) solution 1:1 (v/v). pH values are those of the aqueous buffer used. DPP concentration was 20 μM for absorption and 4 μM for fluorescence measurements. | ||
Fluorescence quenching of 4 occurs at higher pH (9.7–11.6, Fig. 4) and is clearly accompanied by a bathochromic shift in absorption and fluorescence spectra. This effect is caused by deprotonation of the lactam nitrogen within the chromophore.43 Similar changes in the absorption spectra can also be observed for 2 and 3 at higher pH. The absorption spectra shown in Fig. 3C correspond to the neutral and the monoanionic form of 4. Note that the monoanion exhibits weaker but clearly detectable fluorescence. Under more basic conditions, a further bathochromic shift is observable, which originates from partial deprotonation of the second lactam nitrogen, resulting in the dianion.
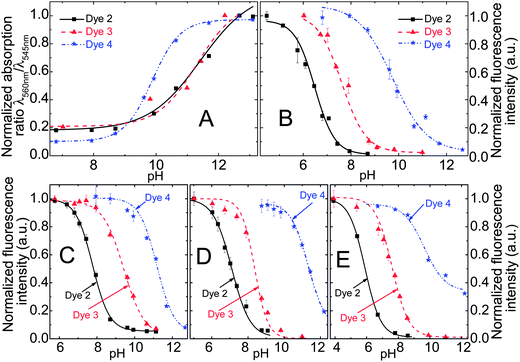 | ||
| Fig. 4 (A) pH calibration curves of the DPP indicators in ethanol–aqueous buffer (ionic strength 100 mM) solution 1:1 (v/v); pH values are those of the aqueous buffer used. (B) Corresponding fluorescence calibration curves for the solution (λEXC = 540 nm, observed at 595 nm); (C and D) pH calibration curves based on fluorescence in planar sensors (dye content 0.25%); (C) in D4® hydrogel; (D) in poly(2-hydroxyethylmethacrylate), (E) calibration curves in RL100 sensor beads dispersed in aqueous buffer (dye content 0.5% (w/w), bead concentration 0.2 mg ml−1). | ||
Notably, the sulfonamide moiety itself can also undergo deprotonation. Typical pKa values would be in the range of 8–11 for structures comparable to 2 and 3.44,45 Such a deprotonation mechanism may contribute to their pH-sensitivity to some extent. The anion formed, a sulfonimide, is in protolytic equilibrium with the phenolate form shown in Fig. 2. Note that the sulfonamide group in 4 cannot be deprotonated and its pH-sensitivity is thus related exclusively to lactam deprotonation.
The photostability of the DPPs has been investigated and compared to reference dyes which represent two of the most commonly used types of pH-indicators. It is vastly superior to that of fluorescein octadecyl ester (Fig. 5). Compared to HPTS (8-hydroxypyrene-1,3,6-trisulfonate), a highly photostable pH-indicator, DPPs are degraded 3–4 times faster under the same illumination conditions. Photostability is greatly increased (dye degraded >20 times slower) in deoxygenated samples which implies an oxidative photo-degradation pathway or may involve photosensitized singlet oxygen. No degradation at all was observed for the phenolate form of 2. This could be because PET causes fast quenching, leaving little time for degradation in the excited state.
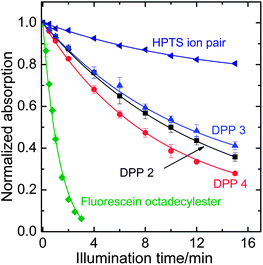 | ||
| Fig. 5 Photodegradation profiles of the DPP dyes, compared to fluorescein octadecyl ester and 1-hydroxypyrene-3,6,8-trisulfonate (HPTS) in the form of an ion pair with tetraoctylammonium. The solutions in tetrahydrofuran (for HPTS, 0.02 mM tetraoctylammonium hydroxide in the form of a 1 M methanolic solution was added) were illuminated with a 458 nm high-power LED array. The decay rates compared to 2 are 0.92 for 3, 1.12 for 4, 0.31 for the HPTS ion pair and 6.1 for FODE. | ||
pH-sensing materials
The pH-indicators have been immobilized in polymer matrices to obtain pH-sensors. The host materials are Hydromed® D4 (a commercially available polyurethane-based hydrogel) and poly(2-hydroxyethylmethacrylate) (p(HEMA)) in the form of planar sensors and Eudragit® RL100 (a positively charged acrylate polymer) in the form of sensor nanobeads (typical average size 30 nm). The sensors are promising not only due to their high brightness and sensitivity, but in particular because their sensitive range can be tuned by selecting the indicator structure and the matrix (Table 2, Fig. 4). The PET-based indicators 2 and 3 can tackle pH 5–10. Monochlorinated 3 covers higher pH than dichlorinated 2. The basic form of the indicator is negatively charged and thus most effectively stabilized by the cationic RL100 matrix and least effectively stabilized by the comparatively hydrophobic D4; this results in pH1/2 values increasing in the same order. The pH-range covered by 2 and 3 in those matrices meets the pH-range of interest for the most relevant applications of pH-sensors, namely medical applications (physiological pH 7–7.5), marine science (optimal pH 7.5–8.5) and biotechnological process monitoring (pH 5–7, depending on the process). 4 enables measurement at pH 9–12, a range that is more rarely addressed but is of importance to applications such as concrete quality testing. In this pH-range, the choice of already available pH-indicators is rather limited.In practical applications, not only the sensitive range of a fluorescence sensor is of importance but also referencing possibilities. Referencing can make the signal independent of the optical path length, the efficiency of the light source and the guidance of light to the detector. Therefore, a ratiometric pH-sensor employing the commercially available coumarin Macrolex Yellow (3-(5-chloro-2-benzoxazolyl)-7-(diethylamino)-2H-1-benzopyran-2-one) as a reference dye has been developed. In this approach, the reference dye is excited, its excitation energy is partially transferred to the DPP pH-indicator dye by Förster resonance energy transfer (FRET) and the emission ratio between both dyes is detected. The referenced system is particularly promising since it can be read out with a simple RGB-CCD camera (Fig. S2 in the ESI†). Imaging with RGB cameras is becoming increasingly popular.46–48 Its sensitive range is very similar to that of the non-referenced analog, as demonstrated in Fig. S3.†
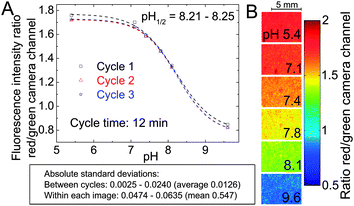 | ||
| Fig. 6 Fluorescence imaging with a planar ratiometric pH-sensor (7 μm thick) containing Macrolex Yellow (1.5% (w/w)) and pH-indicator 2 (0.5% (w/w)), read out with a RGB-CCD camera. The sensor was placed in a home-made flow-through cell and buffer solutions (ionic strength 100 mM) were applied (flow rate 1 ml min−1, 2 min for each buffer). A blue (458 nm) LED array in combination with a Schott BG 12 bandpass filter (350–465 nm) was used for excitation. Excitation light was excluded from the camera employing a Schott OG 515 nm long-pass filter. (A) Calibration curve, based on the ratio between red and green color channels; (B) corresponding false color images. | ||
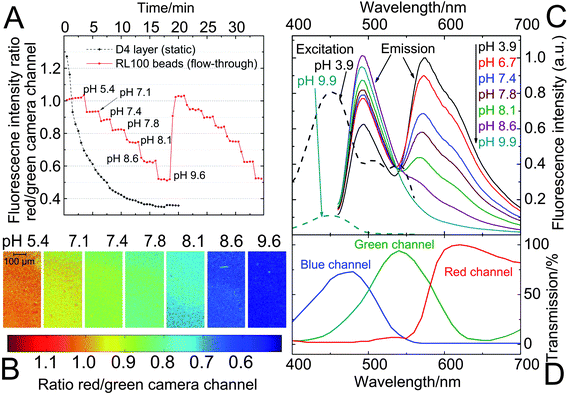 | ||
| Fig. 7 pH-imaging in a microfluidic system using a fluorescence microscope, employing ratiometric pH-nanosensor beads containing indicator 3 (1% w/w) and Macrolex® yellow (1.25% w/w) in RL100 polymer. The sensors were read out employing a RGB-CCD camera; (A) response curve of the bead suspensions; each measurement point was recorded after illumination for 2 s (the corresponding calibration curve can be found in Fig. S6 in the ESI†). They are compared to the performance of a planar sensor (static, placed on a microscope slide; composition as specified in Fig. 6) under the same measurement conditions; (B) images of the sensor bead suspensions corresponding to measurement A (cycle 1) – the ratio between red and green channels is visualized; (C) pH-dependent fluorescence spectra (emission excited at 450 nm; excitation observed at 570 nm) of the sensor beads; (D) spectral characteristics of the RGB-CCD camera. | ||
 | ||
| Fig. 8 Example of a possible application of the DPP-based ratiometric pH-nanosensor beads in high-throughput measurements, using pH-imaging in a microfluidic system. pH can be determined simultaneously in every chip compartment. The pH-sensor beads (as specified in Fig. 7) in aqueous buffer of defined pH were read out employing a RGB-CCD camera. Excitation source and filters were the same as in Fig. 6. (A) Photographic image; (B) false color image of the chip. At the channel inlets, local higher optical pathlengths cause deviations in fluorescence measurements due to overexposure. When red and green color channels exhibit maximum intensities, the ratio of these channels is 1. For the sake of clarity, the inlets are not shown in (B). | ||
Conclusions
In conclusion, new pH-sensors have been presented that exclusively rely on fluorescent dyes and host polymers which are either commercially available or can be prepared from commercially available compounds in a single, simple reaction step. By appropriate selection of the sensor components, one can tune the sensitive pH-range of the material to cover a broad region (pH 5–12). This pH-region meets the requirements of a vast majority of pH-sensor applications. While sensors in the planar format are limited to short-time applications, sensitive nanobeads in Eudragit® RL100 polymer are most promising for practical applications in (micro)fluidic systems and even for fluorescence imaging and microscopy. By taking advantage of a ratiometric approach, good compatibility with a simple RGB camera for readout is accomplished.Beyond the presented new pH-sensors, this work highlighted the usefulness of the tools employed for rational dye design. The phenolate proton receptors have been demonstrated to be very efficient quenchers operating via photoinduced electron transfer and represent a promising alternative to the more commonly used amino-receptors. They can be used in combination with other fluorophores in order to match the requirements of a particular application considering spectral properties or indicator stability. The receptors have been attached by a simple concept, i.e. chlorosulfonation and subsequent reaction with amines. Following this concept, a variety of functionalities can be tagged to DPPs, making them suitable for probing analytes other than pH or targeting particular biomolecular structures.
Experimental
Materials and methods
1,4-Diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole (Irgazin Scarlet) was purchased from Kremer Pigmente (http://kremer-pigmente.de/en), and Macrolex Fluorescent Yellow from Simon and Werner GmbH (http://www.simon-und-werner.de). 4-Amino-2-chlorophenol was from TCI Europe (http://www.tcichemicals.com), and 4-amino-2,6-dichlorophenol from ABCR (http://www.abcr.de). Solvents used for work-up and purification (synthesis grade) as well as buffer salts were supplied by Carl Roth (http://www.roth.de). Deuterated solvents were obtained from Eurisotop (http://www.eurisotop.com), and silica gel from Acros (http://www.fishersci.com). Polyurethane hydrogel D4® was from CardioTech (http://www.cardiotech-inc.com), poly(2-hydroxyethylmethacrylate) (MW = 150000 g mol−1) from Polysciences Inc. (http://www.polysciences.com), and Eudragit® RL100 from Evonik Industries (http://corporate.evonik.de). All other chemicals were from Sigma-Aldrich (http://www.sigmaaldrich.com). The poly(ethylene glycol terephthalate) support (Mylar®) was from Goodfellow (http://www.goodfellow.com). Fluorescein octadecyl ester was prepared according to the literature procedure.50
NMR spectra were recorded on a 300 MHz instrument (Bruker) with TMS as a standard. MALDI-TOF mass spectra were taken on a Micromass TofSpec 2E in reflectron mode at an accelerating voltage of +20 kV. Absorption measurements were performed on a Cary 50 UV-Vis spectrophotometer from Varian (http://www.varianinc.com). Fluorescence spectra were recorded on a Hitachi F-7000 spectrofluorimeter (http://www.hitachi.com). Relative fluorescence quantum yields were determined at 25 °C using rhodamine 101 (ΦF = 0.98 in ethanol51) as a standard. Photostability measurements were performed by irradiating the samples with the light of a 458 nm high-power LED array (10 W input power, http://www.led-tech.de) focused through a lens purchased from Edmund Optics. The photodegradation profiles were obtained by monitoring the absorption spectra.
pH-imaging was performed using a RGB-CCD camera (Marlin F201C, Allied Vision Technologies, http://www.stemmer-imaging.de) equipped with a Xenoplan 1.4/23 objective lens (http://www.schneiderkreuznach.com). For images taken on the fluorescence microscope (Zeiss Axiovert 25 CFL, http://corporate.zeiss.com), a blue ultrabright LED with emission maximum at λ = 450 nm (Luxeon lambert emitter, blue, 5 W) was applied as the excitation light source and combined with a filter set-up consisting of Linos DT blue/Linos DC blue/Schott OG 515 (LINOS Photonics, Göttingen, Germany; Schott, http://www.schott.com) as the excitation filter/dichromatic mirror/barrier filter, respectively. Image acquisition was performed with the software AVT SmartView (http://www.alliedvisiontec.com). Matlab R2008a (http://www.mathworks.com) was used for image processing. The color channels of the obtained images were separated and the ratiometric images were obtained by dividing the red by the green channel.
Microfluidic flow-through experiments were performed using a custom made flow cell or a 6 channel μ-Slide (ibidi μ-Slide VI 0.4, http://ibidi.com), which was connected to a syringe pump (model 540060, TSE systems, http://www.tse-systems.com).
The pH of the buffer solutions was controlled by a digital pH-meter (InoLab pH/ion, WTW GmbH & Co. KG, http://www.wtw.com) calibrated at 25 °C with standard buffers of pH 7.0 and 4.0. The buffers were adjusted to a constant ionic strength of 100 mM using sodium chloride as the background electrolyte.
Indicator synthesis
000), 291 (32100), 509 (22300) and 543 (24000). IR absorption: νmax/cm−1 3426 and 3320 (NH), 3222 (OH), 3030–3160 and 2800–2980 (CH), 1676, 1630 and 1595 (CO), 1555, 1488, 1395, 1331, 1283, 1219, 1156, 1088, 986, 893, 843, 811, 757, 726, 701, 645, 605, 552, 470. NMR (300 MHz, DMSO-d6, TMS): δH = 11.47 (1H, s, Ar-H), 10.44 (1H, s, ArOH), 10.11 (1H, s, SO2NH), 8.3–8.7 (2H, br s, CONH), 8.33 (3H, d, J = 8.4 Hz, Ar-H), 8.26 (1H, dd, J1 = 7.7 Hz, J2 = 1.1 Hz, Ar-H), 7.70–7.86 (4H, m, Ar-H), 7.09 (2H, s, Ar-H). δC = 175.67, 166.14 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 147.20, 146.29, 139.39, 139.02, 135.80, 133.54, 132.49, 131.34, 130.49, 130.16, 129.17, 127.65, 126.25, 124.73, 122.64, 120.99, 110.81, 100.35 (aromatic). MALDI-TOF: m/z [MH+] 528.0190 found, 528.0188 calcd.
200), 291 (23800), 508 (17400) and 541 (18600). νmax/cm−1 3407 and 3314 (NH), 3225 (OH), 3030–3160 and 2800–2980 (CH), 2360, 2339, 1677, 1635 and 1593 (CO), 1557, 1507, 1487, 1405, 1332, 1287, 1202, 1156, 1088, 1053, 953, 895, 843, 820, 757, 726, 701, 614, 597, 544, 496. NMR (300 MHz, DMSO-d6, TMS): δH = 11.45 (1H, s, Ar-H), 10.10 (2H, d, ArOH, SO2NH), 8.3–8.7 (2H, br s, CONH), 8.31 (3H, dd, J1 = 8.1 Hz, J2 = 2.1 Hz, Ar-H), 8.26 (1H, dd, J1 = 7.7 Hz, J2 = 1.4 Hz, Ar-H), 7.70–7.86 (4H, m, Ar-H), 7.06 (1H, d, J = 2.3 Hz, Ar-H), 6.89 (1H, dd, J1 = 8.7 Hz, J2 = 2.4 Hz, Ar-H), 6.85 (1H, d, J = 8.5 Hz, Ar-H). δC = 175.64, 166.15 (CO); 150.50, 147.15, 139.79, 139.16, 135.81, 133.26, 132.46, 131.31, 130.48, 129.24, 128.99, 127.65, 126.67, 124.71, 123.23, 121.78, 119.54, 116.90, 110.75, 100.36 (aromatic). MALDI-TOF: m/z [MH+] 494.0600 found, 494.0577 calcd.
000), 292 (27900), 509 (20000) and 541 (21400). IR absorption: νmax/cm−1 3419 and 3309 (NH), 3030–3170 and 2780–2980 (CH), 2368, 2339, 1665, 1627 and 1597 (CO), 1554, 1486, 1449, 1436, 1340, 1325, 1284, 1260, 1161, 1114, 1095, 938, 839, 747, 701, 668, 612, 595, 536, 469. NMR (300 MHz, DMSO-d6, TMS): δH = 11.58 (1H, s, Ar-H), 8.3–8.8 (2H, CONH), 8.50 (2H, d, J = 8.7 Hz, Ar-H), 8.36 (1H, d, J = 7.8 Hz, Ar-H), 8.30 (1H, dd, J1 = 7.8 Hz, J2 = 1.1 Hz, Ar-H), 7.85 (3H, dt, J1 = 8.4 Hz, J2 = 1.9 Hz, Ar-H), 7.77 (1H, t, J = 7.5 Hz, Ar-H), 3.67 (4H, t, J = 4.2 Hz, OCH2), 2.95 (4H, t, J = 4.1 Hz, ArNCH2). δC = 175.71, 166.19 (CO); 147.27, 138.99, 135.81, 135.01, 133.76, 132.49, 131.34, 130.48, 129.15, 127.66, 127.26, 124.73, 110.90, 100.41 (aromatic); 65.29 (C–O); 45.92 (C–N). MALDI-TOF: m/z [MH+] 438.10 found, 438.11 calcd.
O); 147.20, 146.29, 139.39, 139.02, 135.80, 133.54, 132.49, 131.34, 130.49, 130.16, 129.17, 127.65, 126.25, 124.73, 122.64, 120.99, 110.81, 100.35 (aromatic). MALDI-TOF: m/z [MH+] 528.0190 found, 528.0188 calcd.
200), 291 (23800), 508 (17400) and 541 (18600). νmax/cm−1 3407 and 3314 (NH), 3225 (OH), 3030–3160 and 2800–2980 (CH), 2360, 2339, 1677, 1635 and 1593 (CO), 1557, 1507, 1487, 1405, 1332, 1287, 1202, 1156, 1088, 1053, 953, 895, 843, 820, 757, 726, 701, 614, 597, 544, 496. NMR (300 MHz, DMSO-d6, TMS): δH = 11.45 (1H, s, Ar-H), 10.10 (2H, d, ArOH, SO2NH), 8.3–8.7 (2H, br s, CONH), 8.31 (3H, dd, J1 = 8.1 Hz, J2 = 2.1 Hz, Ar-H), 8.26 (1H, dd, J1 = 7.7 Hz, J2 = 1.4 Hz, Ar-H), 7.70–7.86 (4H, m, Ar-H), 7.06 (1H, d, J = 2.3 Hz, Ar-H), 6.89 (1H, dd, J1 = 8.7 Hz, J2 = 2.4 Hz, Ar-H), 6.85 (1H, d, J = 8.5 Hz, Ar-H). δC = 175.64, 166.15 (CO); 150.50, 147.15, 139.79, 139.16, 135.81, 133.26, 132.46, 131.31, 130.48, 129.24, 128.99, 127.65, 126.67, 124.71, 123.23, 121.78, 119.54, 116.90, 110.75, 100.36 (aromatic). MALDI-TOF: m/z [MH+] 494.0600 found, 494.0577 calcd.
000), 292 (27900), 509 (20000) and 541 (21400). IR absorption: νmax/cm−1 3419 and 3309 (NH), 3030–3170 and 2780–2980 (CH), 2368, 2339, 1665, 1627 and 1597 (CO), 1554, 1486, 1449, 1436, 1340, 1325, 1284, 1260, 1161, 1114, 1095, 938, 839, 747, 701, 668, 612, 595, 536, 469. NMR (300 MHz, DMSO-d6, TMS): δH = 11.58 (1H, s, Ar-H), 8.3–8.8 (2H, CONH), 8.50 (2H, d, J = 8.7 Hz, Ar-H), 8.36 (1H, d, J = 7.8 Hz, Ar-H), 8.30 (1H, dd, J1 = 7.8 Hz, J2 = 1.1 Hz, Ar-H), 7.85 (3H, dt, J1 = 8.4 Hz, J2 = 1.9 Hz, Ar-H), 7.77 (1H, t, J = 7.5 Hz, Ar-H), 3.67 (4H, t, J = 4.2 Hz, OCH2), 2.95 (4H, t, J = 4.1 Hz, ArNCH2). δC = 175.71, 166.19 (CO); 147.27, 138.99, 135.81, 135.01, 133.76, 132.49, 131.34, 130.48, 129.15, 127.66, 127.26, 124.73, 110.90, 100.41 (aromatic); 65.29 (C–O); 45.92 (C–N). MALDI-TOF: m/z [MH+] 438.10 found, 438.11 calcd.
Preparation of planar sensors
A “cocktail” containing the indicator dye (0.16 mg), hydrogel D4/p(HEMA) (41 mg) and ethanol–water 9:1 (v/v) (500 μl) was knife-coated on a dust-free Mylar support to obtain an ≈7 μm thick layer after solvent evaporation. Ratiometric sensors were prepared analogously, using 0.2 mg of 2, 0.6 mg of Macrolex Yellow and tetrahydrofuran (461 μl) as a solvent.
Preparation of sensor nanoparticles
Eudragit RL100 (100 mg) was dissolved in acetone (50 ml), and indicator dye (1 mg) and Macrolex Yellow (1.25 mg) were added. Water (300 ml) was added quickly (5 s). Acetone was removed on a rotary evaporator and the particle suspension was concentrated to a volume of 50 ml.Acknowledgements
Financial support from the Austrian Science Fund FWF (project P 21192-N17) is gratefully acknowledged. We thank Martin Thonhofer, Institute of Organic Chemistry, and Stefan Müller, Institute of Inorganic Chemistry, for their support in measuring melting points and IR spectra.Notes and references
- M. Schäferling, Angew. Chem., Int. Ed., 2012, 51, 3532–3554 CrossRef
.
- M. Kühl and L. Polerecky, Aquat. Microb. Ecol., 2008, 53, 99–118 CrossRef
- H. Stahl, A. Glud, C. R. Schröder, I. Klimant, A. Tengberg and R. N. Glud, Limnol. Oceanogr.: Methods, 2006, 4, 336–345 CrossRef CAS
- G. Liebsch, I. Klimant, C. Krause and O. S. Wolfbeis, Anal. Chem., 2001, 73, 4354–4363 CrossRef CAS
- S. Hulth, R. C. Aller, P. Engström and E. Selander, Limnol. Oceanogr., 2002, 47, 212–220 CrossRef CAS
- J. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef CAS
- A. Almutairi, S. J. Guillaudeu, M. Y. Berezin, S. Achilefu and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 444–445 CrossRef CAS
- C. Munkholm, D. R. Walt, F. P. Milanovich and S. M. Klainer, Anal. Chem., 1986, 58, 1427–1430 CAS
- N. Strömberg, E. Mattsson and A. Hakonen, Anal. Chim. Acta, 2009, 636, 89–94 CrossRef
- J. E. Whitaker, R. P. Haugland and F. G. Prendergast, Anal. Biochem., 1991, 194, 330–344 CrossRef CAS
- A. P. de Silva, T. Gunnlaugsson and T. E. Rice, Analyst, 1996, 121, 1759–1762 RSC
- A. P. de Silva, T. P. Vance, M. E. S. West and G. D. Wright, Org. Biomol. Chem., 2008, 6, 2468–2480 CAS
- R. A. Bissell, A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, G. E. M. Maguire and K. R. A. S. Sandanayake, Chem. Soc. Rev., 1992, 21, 187–195 RSC
- R. A. Bissell, E. Calle, A. P. de Silva, S. A. de Silva, H. Q. N. Gunaratne, J.-L. Habib-Jiwan, S. L. A. Peiris, R. A. D. D. Rupasinghe, T. K. Shantha, D. Samarasinghe, K. R. A. S. Sandanayake and J.-P. Soumillion, J. Chem. Soc., Perkin Trans. 2, 1992, 1559–1564 RSC
- K. Zhou, Y. Wang, X. Huang, K. Luby-Phelps, B. D. Sumer and J. Gao, Angew. Chem., 2011, 50, 6109–6114 CAS
- N. Saleh, Y. A. Al-Soud and W. M. Nau, Spectrochim. Acta, Part A, 2008, 71, 818–822 CrossRef
- T. Werner, C. Huber, S. Heinl, M. Kollmannsberger, J. Daub and O. S. Wolfbeis, Fresenius' J. Anal. Chem., 1997, 359, 150–154 CrossRef CAS
- T. Gareis, C. Huber, O. S. Wolfbeis and J. Daub, Chem. Commun., 1997, 1717–1718 RSC
- M. Baruah, W. Qin, N. Basarić, W. M. De Borggraeve and N. Boens, J. Org. Chem., 2005, 70, 4152–4157 CrossRef CAS
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC
- C. N. Baki and E. U. Akkaya, J. Org. Chem., 2001, 66, 1512–1513 CrossRef CAS
- T. Potrawa and H. Langhals, Chem. Ber., 1987, 120, 1075–1078 CrossRef CAS
-
W. Herbst and K. Hunger, Industrial Organic Pigments, Wiley-VCH, Weinheim, 3rd edn, 2004, pp. 487–494 Search PubMed
- Z. Hao and A. Iqbal, Chem. Soc. Rev., 1997, 26, 203–213 RSC
- S. Luňák Jr, M. Vala, J. Vyňuchal, I. Ouzzane, P. Horáková, P. Možíšková, Z. Eliáš and M. Weiter, Dyes Pigm., 2011, 91, 269–278 CrossRef
- S. Luňák Jr, J. Vyňuchal, M. Vala, L. Havel and R. Hrdina, Dyes Pigm., 2009, 82, 102–108 CrossRef
- J. S. Zambounis, Z. Hao and A. Iqbal, Nature, 1997, 388, 131–132 CrossRef CAS
- J. C. Bijleveld, A. P. Zoombelt, S. G. J. Mathijssen, M. M. Wienk, M. Turbiez, D. M. de Leeuw and R. A. J. Janssen, J. Am. Chem. Soc., 2009, 131, 16616–16617 CrossRef CAS
- Y. Zhu, A. R. Rabindranath, T. Beyerlein and B. Tieke, Macromolecules, 2007, 40, 6981–6989 CrossRef CAS
- K. Zhang and B. Tieke, Macromolecules, 2008, 41, 7287–7295 CrossRef CAS
- B. Walker, A. B. Tamayo, X.-D. Dang, P. Zalar, J. H. Seo, A. Garcia, M. Tantiwiwat and T.-Q. Nguyen, Adv. Funct. Mater., 2009, 19, 3063–3069 CrossRef CAS
- M. Tantiwiwat, A. Tamayo, N. Luu, X.-D. Dang and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 17402–17407 CAS
- L. Bürgi, M. Turbiez, R. Pfeiffer, F. Bienewald, H.-J. Kirner and C. Winnewisser, Adv. Mater., 2008, 20, 2217–2224 CrossRef
- M. M. Wienk, M. Turbiez, J. Gilot and R. A. J. Janssen, Adv. Mater., 2008, 20, 2556–2560 CrossRef CAS
- Y. Zou, D. Gendron, R. Badrou-Aïch, A. Najari, Y. Tao and M. Leclerc, Macromolecules, 2009, 42, 2891–2894 CrossRef CAS
- Y. Jiang, Y. Wang, J. Hua, S. Qu, S. Qian and H. Tian, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4400–4408 CrossRef CAS
- E. Q. Guo, P. H. Ren, Y. L. Zhang, H. C. Zhang and W. J. Yang, Chem. Commun., 2009, 5859–5861 RSC
- Y. Qu, J. Hua and H. Tian, Org. Lett., 2010, 12, 3320–3323 CrossRef CAS
- Y.-H. Jeong, C.-H. Lee and W.-D. Jang, Chem.–Asian J., 2012, 7, 1562–1566 CrossRef
- L. Deng, W. Wu, H. Guo, J. Zhao, S. Ji, X. Zhang, X. Yuan and C. Zhang, J. Org. Chem., 2011, 76, 9294–9304 CrossRef CAS
- J. Mizuguchi, T. Imoda, H. Takahashi and H. Yamakami, Dyes Pigm., 2006, 68, 47–52 CrossRef CAS
- T. Yamagata, J. Kuwabara and T. Kanbara, Tetrahedron Lett., 2010, 51, 1596–1599 CrossRef CAS
- S. Schutting, S. M. Borisov and I. Klimant, Anal. Chem., 2013, 85, 3271–3279 CrossRef CAS
- M. Thakur, A. Thakur, P. V. Khadikar and C. T. Supuran, Bioorg. Med. Chem. Lett., 2005, 15, 203–209 CrossRef CAS
- C. Caltagirone, G. W. Bates, P. A. Gale and M. E. Light, Chem. Commun., 2008, 61–63 RSC
- M. Larsen, S. M. Borisov, B. Grunwald, I. Klimant and R. N. Glud, Limnol. Oceanogr.: Methods, 2011, 9, 348–360 CrossRef CAS
- R. J. Meier, L. H. Fischer, O. S. Wolfbeis and M. Schäferling, Sens. Actuators, B, 2013, 177, 500–506 CrossRef CAS
- X. Wang, H. H. Gorris, J. A. Stolwijk, R. J. Meier, D. B. M. Groegel, J. Wegener and O. S. Wolfbeis, Chem. Sci., 2011, 2, 901 RSC
- S. Jezierski, D. Belder and S. Nagl, Chem. Commun., 2013, 49, 904–906 RSC
- B. M. Weidgans, C. Krause, I. Klimant and O. S. Wolfbeis, Analyst, 2004, 129, 645–650 RSC
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455–462 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: NMR and MS spectra, further sensor characteristics and sensor long-time performance. See DOI: 10.1039/c3tc31130a |
| This journal is © The Royal Society of Chemistry 2013 |