 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis of rhodium substituted ε-iron oxide exhibiting super high frequency natural resonance†
Asuka
Namai
a,
Marie
Yoshikiyo
a,
Sayaka
Umeda
a,
Takayuki
Yoshida
b,
Tatsuro
Miyazaki
b,
Makoto
Nakajima
c,
Keita
Yamaguchi
d,
Tohru
Suemoto
d and
Shin-ichi
Ohkoshi
*ae
aDepartment of Chemistry, School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: ohkoshi@chem.s.u-tokyo.ac.jp
bDowa Electronics Materials Co., Ltd., 1-3-1 Kaigandori, Minami-ku, Okayama 702-8506, Japan
cDepartment of Physics, Chiba University, 1-33, Yayoicho, Inage Ward, Chiba-shi, Chiba 263-8522, Japan
dInstitute for Solid State Physics, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8581, Japan
eCREST, JST, K's Gobancho, 7 Gobancho, Chiyoda-ku, Tokyo 102-0076, Japan
First published on 17th July 2013
Abstract
In this study, we demonstrate a synthesis of rhodium substituted ε-iron oxide, ε-RhxFe2−xO3 (0 ≤ x ≤ 0.19), nanoparticles in silica. The synthesis features a sol–gel method to coat the metal hydroxide sol containing Fe3+ and Rh3+ ions with a silica sol via hydrolysis of alkoxysilane to form a composite gel. The obtained samples are barrel-shaped nanoparticles with average long- and short-axial lengths of approximately 30 nm and 20 nm, respectively. The crystallographic structure study using X-ray diffraction shows that ε-RhxFe2−xO3 has an orthorhombic crystal structure in the Pna21 space group. Among the four non-equivalent substitution sites (A–D sites), Rh3+ ions mainly substitute into the C sites. The formation mechanism of ε-RhxFe2−xO3 nanoparticles is considered to be that the large surface area of the nanoparticles increases the contribution from the surface energy to Gibbs free energy, resulting in a different phase, ε-phase, becoming the most stable phase compared to that of bulk or single crystal. The measured electromagnetic wave absorption characteristics due to natural resonance (zero-field ferromagnetic resonance) using terahertz time domain spectroscopy reveal that the natural resonance frequency shifts from 182 GHz (ε-Fe2O3) to 222 GHz (ε-Rh0.19Fe1.81O3) upon rhodium substitution. This is the highest natural resonance frequency of a magnetic material, and is attributed to the large magnetic anisotropy due to rhodium substitution. The estimated coercive field for ε-Rh0.19Fe1.81O3 is as large as 28 kOe.
Introduction
As electronic devices and telecommunication technologies advance, the demand for electromagnetic wave absorbing materials to prevent electromagnetic interference is on the rise. Hence, the search for appropriate materials is an important materials science issue.1–3 Electromagnetic wave absorbing materials include magnetic materials and composite materials composed of electrically conductive and dielectric materials. Magnetic materials absorb electromagnetic waves by magnetic loss, and include spinel ferrite such as NixZn1−xFe2O4 (1 kHz to 10 GHz),4–6 soft magnetic metal such as Fe powder (1 kHz to 10 GHz),7,8 and hexagonal ferrite such as BaFe12O19 (1 GHz to 80 GHz).9–11 Composite materials between electrically conductive and dielectric materials absorb electromagnetic waves through dielectric loss, and include composite materials between carbon materials and urethane foam (1 MHz to 10 GHz).12,13 However, the prevalent use of wireless communications and radar systems in recent years, which use high frequency electromagnetic waves called millimeter waves (30–300 GHz),14–17 requires the development of materials that absorb millimeter waves exceeding 100 GHz.Our research group has recently demonstrated electromagnetic wave absorption of ε-iron oxide (ε-Fe2O3), which exhibits a large coercive field (Hc) of 20 kOe,18–28 due to magnetic loss at 182 GHz.29 Furthermore, we found that substituting ε-iron oxide with non-magnetic ions, such as Al3+ and Ga3+, can control the absorption frequency from 182 GHz to 35 GHz,29–31 and in 2012, we achieved absorption frequencies above 182 GHz up to 209 GHz with rhodium-substituted ε-iron oxide (ε-RhxFe2−xO3).32
Herein we successfully develop a synthesis method of ε-RhxFe2−xO3 suitable for large-scale synthesis, and achieve an electromagnetic wave absorption frequency of 222 GHz, which exceeds the previously reported frequency and corresponds to the highest frequency for windows of air in millimeter waves (220 GHz).
Results and discussion
Previously, we synthesized ε-RhxFe2−xO3 nanoparticles in a SiO2 matrix using mesoporous silica as a template.32 In addition to being labor intensive, this method produces small quantities of ε-RhxFe2−xO3. In contrast, the present method employs a sol–gel method33,34 to synthesize ε-RhxFe2−xO3 by coating a metal hydroxide sol with a silica sol via hydrolysis of alkoxysilane to form a composite gel (Fig. 1a). This sol–gel synthesis has four steps: (i) generation of the metal hydroxide, (ii) formation of the silica matrix, (iii) sintering, and (iv) removal of the silica matrix (see the Experimental section). Nine samples (samples 1 to 9) with varying concentrations of Fe and Rh were synthesized. | ||
| Fig. 1 (a) Schematic diagram of ε-RhxFe2−xO3 nanoparticle synthesis using the sol–gel method. (i) Aqueous ammonia is added to an aqueous solution containing both rhodium nitrate and ferric nitrate, yielding the sol of rhodium and iron hydroxide, (RhxFe2−x)(OH)6 (shown in brown). (ii) TEOS (Si(OC2H5)4) is added to the reaction solution to yield a complex gel, (RhxFe2−x)(OH)6/SiO2, coated with a sol of silica (shown in light blue) via hydrolysis. (iii) Sintering the obtained gel in air yields RhxFe2−xO3 in silica. Sintering at 1080 °C, which is higher than the glass transition temperature, causes nanoparticle aggregation and accelerates crystallization. (iv) SiO2 coated material is added to a NaOH solution to react and remove the SiO2 matrix as a Na2SiO3 solution. (b) TEM images of the final products after removing the silica matrix (samples 1, 3, 5, 7, and 9). | ||
Fig. 1b shows the transmission electron microscope (TEM) images of the obtained samples. The TEM images indicate that the samples are composed of barrel-shaped nanoparticles. The average long- and short-axial lengths of each sample are: (33 ± 15) × (24 ± 11) nm (1), (35 ± 16) × (25 ± 11) nm (2), (36 ± 17) × (26 ± 12) nm (3), (35 ± 16) × (27 ± 12) nm (4), (36 ± 18) × (27 ± 14) nm (5), (29 ± 17) × (21 ± 13) nm (6), (27 ± 18) × (19 ± 12) nm (7), (23 ± 14) × (16 ± 9) nm (8), and (22 ± 13) × (16 ± 9) nm (9), respectively. The average aspect ratio (i.e., [long axis length]/[short axis length]) of each sample is approximately 1.4 (Fig. S2 and Table S2†).
The crystallographic structures of the obtained samples were studied using powder X-ray diffraction (XRD). As shown in Fig. 2a, the XRD patterns of the obtained samples indicated that the ε-phase was formed in all the samples. Besides the ε-phase, each sample contains the α-phase or γ-phase. Fig. 2b shows the phase ratios of the samples. The compositions for the ε-phase are ε-Fe2O3 (1), ε-Rh0.03Fe1.97O3 (2), ε-Rh0.05Fe1.95O3 (3), ε-Rh0.08Fe1.92O3 (4), ε-Rh0.10Fe1.90O3 (5), ε-Rh0.14Fe1.86O3 (6), ε-Rh0.15Fe1.85O3 (7), ε-Rh0.18Fe1.82O3 (8), and ε-Rh0.19Fe1.81O3 (9) (Table 1).35 The lattice constants of the ε-phase are roughly the same regardless of the increases in the concentration of rhodium substitution, i.e., a-axis: 5.0917 ± 0.0003 Å (1) → 5.1110 ± 0.0009 Å (9), b-axis: 8.7857 ± 0.0005 Å (1) → 8.7980 ± 0.0015 Å (9), and c-axis: 9.4783 ± 0.0008 Å (1) → 9.4722 ± 0.0031 Å (9) (Table 1), because the ionic radius of Rh3+ (0.67 Å) is approximately the same as that of Fe3+ (0.65 Å).36 Among the four non-equivalent substitution sites (A–D sites), Rh3+ ions mainly substitute into the C sites (Fig. 3). Additionally, as the amount of Rh substitution increases, the A- and B-sites also begin to be substituted. The details of the crystal structure analysis are described in the ESI.†
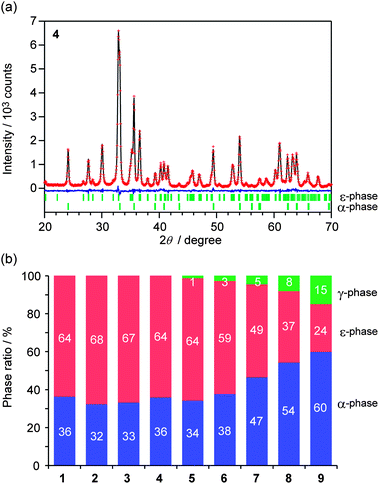 | ||
| Fig. 2 (a) Powder X-ray diffraction pattern and Rietveld analysis result of sample 4. Red dots, black line, and blue line represent the observed values, calculated values, and the residual error, respectively. Bars denote Bragg peak positions of the ε- and α-phases. (b) Phase ratios of the samples obtained from the Rietveld analyses. Red, blue, and green represent ε-, α-, and γ-phases, respectively. | ||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| x(ε) | 0 | 0.03 | 0.05 | 0.08 | 0.10 | 0.14 | 0.15 | 0.18 | 0.19 | |
| W(ε)/wt% | 64 | 68 | 67 | 64 | 64 | 59 | 49 | 37 | 24 | |
| a/Å | 5.0917 (3) | 5.0945 (3) | 5.0966 (3) | 5.0999 (3) | 5.1020 (3) | 5.1055 (3) | 5.1067 (4) | 5.1095 (5) | 5.1110 (9) | |
| b/Å | 8.7857 (5) | 8.7867 (5) | 8.7881 (5) | 8.7900 (5) | 8.7906 (5) | 8.7941 (6) | 8.7932 (7) | 8.7951 (9) | 8.798 (2) | |
| c/Å | 9.4783 (8) | 9.4773 (7) | 9.4761 (8) | 9.4745 (7) | 9.4735 (8) | 9.4734 (9) | 9.4699 (11) | 9.471 (2) | 9.472 (3) | |
| V/Å3 | 424.00 (5) | 424.24 (5) | 424.43 (5) | 424.72 (5) | 424.88 (5) | 425.34 (6) | 425.24 (7) | 425.59 (10) | 425.9 (2) | |
| Rh occupancy/% | A | 0 | 0 | 0 | 0 | 0 | 0 (1) | 0 (1) | 2 (2) | 3 (3) |
| B | 0 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (1) | 2 (1) | 2 (3) | 5 (4) | |
| C | 0 | 6 (0) | 11 (0) | 17 (0) | 21 (0) | 26 (1) | 29 (1) | 32 (2) | 31 (3) | |
| D | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
 | ||
| Fig. 3 (a) Crystallographic structure of ε-RhxFe2−xO3. (b) Rh occupancies versus substitution ratios of x(ε) at the A–D sites. Dotted lines are a guide to the eye. | ||
Next we discuss the formation mechanism of ε-RhxFe2−xO3 nanoparticles in the present synthesis. In general, the bulk or single-crystal Fe2O3 transforms from γ-Fe2O3 to α-Fe2O3 when the temperature is elevated.37 However, the large surface area of the nanoparticles increases the contribution from the surface energy to the Gibbs free energy (G),38 which is responsible for a different phase, ε-phase, becoming the most stable phase compared to that of bulk or single crystals.39–41Fig. 4 depicts the particle size (d) dependence of the free energy per molar volume (G/Vm) for the γ-phase, ε-phase, and α-phase.42,43 The G/Vmversus d curve of the ε-phase intercepts the G/Vmversus d curves of the γ-phase and α-phase, indicating that particle sizes in between these intercept points make the ε-phase as the most stable phase. In our method, synthesizing RhxFe2−xO3 as nanoparticles yields particle sizes that make the ε-phase most stable.
 | ||
| Fig. 4 Schematic curves of Gibbs free energy per unit volume for each phase of Fe2O3 (solid lines) and RhxFe2−xO3 (dotted lines), (Gi/Vm,i), versus particle size (d) where i = γ, ε, and α.42,43 Green, red, and blue lines represent the γ-, ε-, and α-phases, respectively. If rhodium substitution elevates the chemical potential of RhxFe2−xO3, and the increase of the chemical potential of the ε-phase is greater than those of the other two phases, the particle size range where the ε-phase is most stable becomes narrower. This explains why the α-phase and γ-phase co-exist. | ||
The reason for the difficulty to obtain a single ε-phase in the Rh-substituted system can be explained by the fact that the chemical potential of RhxFe2−xO3 increases due to rhodium substitution. If the chemical potential of the ε-phase increases largely compared to the other two phases, the G/Vmversus d curve of the ε-phase becomes relatively higher, narrowing the particle size range where the ε-phase is most stable. A narrower range explains the co-existence of the α- and γ-phases.
The following possibility can be cited as the reason that the particles have a barrel-shaped morphology. In (RhxFe2−x)(OH)6/SiO2, which is a precursor, a small amount of Fe3+ and Rh3+ ions is included in silica. Their presence reduces the glass transition temperature compared to pure silica.44 Thus, aggregation of nanoparticles progresses in the silica matrix, which increases the crystallization degree and forms the barrel-shaped nanoparticles.
A terahertz time domain spectroscopy (THz-TDS) system was set up to measure electromagnetic wave absorption properties in the millimeter wave region (Fig. 5). Fig. 6 shows the measured millimeter wave absorption spectra of ε-RhxFe2−xO3. As we previously reported,29 sample 1, which is ε-Fe2O3, shows an absorption peak centered at 182 GHz. Rhodium substitution increases the resonance absorption frequency: 187 GHz (2), 192 GHz (3), 197 GHz (4), 202 GHz (5), 207 GHz (6), 213 GHz (7), 217 GHz (8), and 222 GHz (9). Sample 9 breaks our previously reported record of 209 GHz for ε-Rh0.14Fe1.86O3,32 and currently has the highest electromagnetic wave resonance frequency for a magnetic material.
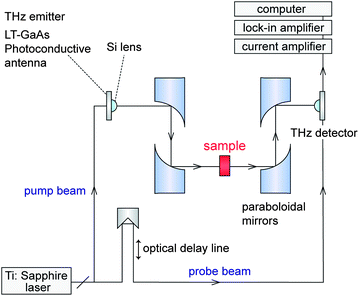 | ||
| Fig. 5 Schematic diagram of the THz-TDS measurement system. | ||
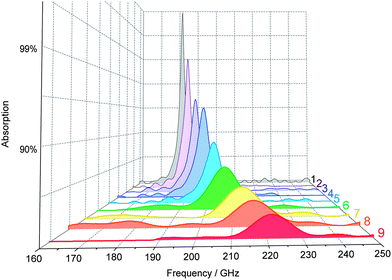 | ||
| Fig. 6 Millimeter wave absorption spectra measured at room temperature. | ||
The mechanism of electromagnetic wave absorption in magnetic materials is mainly caused by magnetic loss due to magnetic domain wall motion or natural resonance (zero-field ferromagnetic resonance).45 The observed high-frequency millimeter wave absorption in the present material is attributed to the natural resonance phenomenon. The mechanism is described below. When an electromagnetic wave is irradiated into a magnetic material, the magnetic component of the electromagnetic wave tilts the magnetization from the anisotropy field (Ha). Once the magnetization is tilted, it precesses around Ha due to the gyromagnetic effect. A resonance occurs at the frequency matching this precession and causes electromagnetic wave absorption. The resonance frequency fr can be expressed as: fr = νHa/2π, where ν is the gyromagnetic constant. Because a uniaxial magnetic anisotropic sample exhibits a proportional relationship between Ha and the coercive field (Hc), we investigated the Hc values of the samples.
Fig. 7 shows the magnetization versus external magnetic field curve at room temperature (300 K). Although samples 1 to 4 show an increase in Hc as the amount of rhodium substitution increases, i.e., 21.7 kOe (1), 22.5 kOe (2), 22.9 kOe (3), and 23.9 kOe (4), samples 5 to 9 exhibit a distortion in the hysteresis loop with a negative effect on Hc, i.e., 23.9 kOe (5), 22.6 kOe (6), 15.5 kOe (7), 4.2 kOe (8), and 0.9 kOe (9) (Fig. 8a, black open squares). To estimate the true Hc value of the ε-phase, we applied a correction that considered the contributions of the α-phase and the γ-phase. The estimated Hc values for ε-RhxFe2−xO3 were 21.9 kOe (ε-Fe2O3), 22.5 kOe (ε-Rh0.03Fe1.97O3), 22.9 kOe (ε-Rh0.05Fe1.95O3), 24.0 kOe (ε-Rh0.08Fe1.92O3), 25.2 kOe (ε-Rh0.10Fe1.90O3), 26.3 kOe (ε-Rh0.14Fe1.86O3), 26.7 kOe (ε-Rh0.15Fe1.85O3), 27.7 kOe (ε-Rh0.18Fe1.82O3), and 28.1 kOe (ε-Rh0.19Fe1.81O3) (Fig. 8a, red solid circles). The last two Hc values exceed our previously reported value, and are the highest values among metal oxides to date. ESI contains detailed magnetic characteristics with respect to their temperature dependences.†
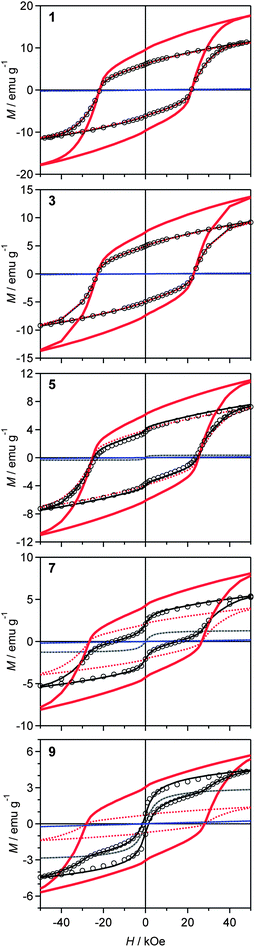 | ||
| Fig. 7 Magnetization versus external magnetic field curves at 300 K. Open circles are the observed values. Dotted lines represent contributions from each phase (red, blue, and gray dotted lines represent the ε-, α-, and γ-phases, respectively). Black solid lines are their sum. Red solid lines denote the estimated value for a single-phase ε-RhxFe2−xO3. | ||
 | ||
| Fig. 8 (a) Hcversus x(ε) plots. Solid red circles and black open squares represent the estimated Hc values and observed Hc values, respectively. Red dotted lines are a guide to the eye. (b) frversus Hc plots. | ||
The correlation between Hc and fr demonstrates a monotonic increase in fr with Hc (Fig. 8b). This means that rhodium substitution increases Hc, which indicates an increase in the magnetic anisotropy, Ha, and achieves a high fr of 222 GHz.
Conclusions
Herein the highest natural resonance frequency to date of 222 GHz is reported for a high frequency electromagnetic wave absorption material, ε-RhxFe2−xO3. This material was synthesized via the sol–gel method, which uses a liquid-phase reaction where water is the only solvent. This method is simple and is possible for industrial scale synthesis. Additionally, the lack of organic solvents and surfactants makes this method economical and environmentally friendly.220 GHz is the highest-frequency window of air. Around this frequency, electromagnetic wave absorption by air is small. Consequently, this frequency band is expected as a carrier frequency for wireless communications.46–49 The materials developed herein should be useful as millimeter wave absorbing materials for unnecessary electromagnetic waves, which cause electromagnetic interference.
Experimental section
Synthesis
Samples 1–9 were prepared by the sol–gel method. First, 10.2 ml of aqueous ammonia (13 mol dm−3) was added while stirring 240 ml of an aqueous solution with the following Fe(NO3)3 and Rh(NO3)3 concentrations: (Fe(NO3)3, Rh(NO3)3) = (62.5 × 10−3, 0 mol dm−3) (1), (61.3 × 10−3, 1.25 × 10−3 mol dm−3) (2), (60.0 × 10−3, 2.50 × 10−3 mol dm−3) (3), (58.8 × 10−3, 3.75 × 10−3 mol dm−3) (4), (57.5 × 10−3, 5.00 × 10−3 mol dm−3) (5), (56.3 × 10−3, 6.25 × 10−3 mol dm−3) (6), (54.7 × 10−3, 7.81 × 10−3 mol dm−3) (7), (53.1 × 10−3, 9.38 × 10−3 mol dm−3) (8), and (51.6 × 10−3, 10.9 × 10−3 mol dm−3) (9). Successively, 22.6 ml of tetraethoxysilane (TEOS) was added to this solution, and stirred for 20 hours. Then, the obtained gel was separated by centrifugation, washed with distilled water, and dried. The obtained dry powder was sintered in air for four hours at 1080 °C. An aqueous solution of sodium hydroxide was added to the sintered powder, and stirred for one day at 60 °C. The solution was centrifuged and washed to obtain the final products. We adopted the sintering temperature of 1080 °C, since it had a high ε-phase ratio among the various sintering temperatures in the range of 1020–1300 °C.Characterization
Elemental analysis of the prepared samples was performed using an inductively coupled plasma mass spectroscopy (ICP-MS) system, Agilent Technologies 7700. The TEM images were collected with a JEOL JEM-2000EX transmission electron microscope, where the accelerating voltage was 200 kV. The XRD patterns were measured at 294 K using a Rigaku Ultima IV and a Rigaku RINT2100 with Cu Kα radiation (λ = 1.5418 Å), and Rietveld analyses were performed using the RIETAN-FP program.50Millimeter wave absorption measurement
Electromagnetic wave absorption properties in the millimeter wave region were measured by a THz-TDS system, where a pump–probe measurement system was used. Ultra short pulse with a time duration of 20 fs and a center photon energy of 1.55 eV generated from a mode-locked Ti:sapphire femtosecond pulse laser at a repetition rate of 76 MHz was divided into a pump and probe beam. The pump pulse was irradiated into a dipole type low-temperature-grown GaAs photoconductive antenna, which generated a THz pulse. The THz pulse was condensed with a Si lens and a set of paraboloidal mirrors and was irradiated into the sample. The electric field of the transmitted THz pulse wave formed in the time domain was obtained by changing the delay time between the pump and the probe pulses. Powder-form samples were filled in paper cells (6.5 mm × 6.5 mm × 4.8 mm for samples 1–8 and 6.5 mm × 6.5 mm × 8 mm for sample 9) with filling ratios of 29–31 vol%. In the present millimeter wave absorption study, the sample thickness of at least 3 mm was necessary to avoid interference from the edges of the cell. The obtained spectra were calibrated as spectra of 30 vol% of the ε-phase, 4.8 mm thickness.Magnetic property measurement
The magnetic properties were measured using a superconducting quantum interference device (SQUID) magnetometer, Quantum Design, MPMS 7. Field cooled magnetization (FCM) curves were measured in the temperature range of 4–550 K, where the cooling rate was −1 K min−1. Magnetic hysteresis loops were measured at 300 K.Acknowledgements
The present research was supported partly by the Core Research for Evolutional Science and Technology (CREST) program of the Japan Science and Technology Agency (JST); a Grant-in-Aid for Young Scientists (S) from the Japan Society for the Promotion of Science (JSPS); DOWA Technofund; the Asahi Glass Foundation; a Grant for the Global COE Program “Chemistry Innovation through Cooperation of Science and Engineering”; Advanced Photon Science Alliance (APSA) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT); JSPS KAKENHI Grant Number 24850004; Office for Gender Equality, the University of Tokyo; the Cryogenic Research Center, the University of Tokyo; the Center for Nano Lithography & Analysis, the University of Tokyo, supported by MEXT. M. Y. is grateful to Advanced Leading Graduate Course for Photon Science (ALPS) and JSPS Research Fellowships for Young Scientists. We are grateful to Ms Y. Kitano for her technical support and Mr Y. Kakegawa and Mr H. Tsunakawa for collecting the TEM images.References and notes
- K. J. Vinoy and R. M. Jha, in Radar Absorbing Materials, Kluwer, Boston, 1996 Search PubMed.
- S. Celozzi, R. Araneo and G. Lovat, in Electromagnetic Shielding, Wiley-Interscience: IEEE Press, New Jersey, 2008 Search PubMed.
- C. M. Watts, X. Liu and W. J. Padilla, Adv. Mater., 2012, 24, OP98 Search PubMed.
- Y. Naito and K. Suetake, IEEE Trans. Magn., 1971, 19, 65 Search PubMed.
- R. K. Selvan, V. Krishnan, C. O. Augustin, H. Bertagnolli, C. S. Kim and A. Gedanken, Chem. Mater., 2008, 20, 429 Search PubMed.
- F. Wang, J. Liu, J. Kong, Z. Zhang, X. Wang, M. Itoh and K. Machida, J. Mater. Chem., 2011, 21, 4314 RSC.
- M. Matsumoto and Y. Miyata, IEEE Trans. Magn., 1997, 33, 4459 CrossRef.
- Z. Yu, Z. Yao, N. Zhang, Z. Wang, C. Li, X. Han, X. Wub and Z. Jiang, J. Mater. Chem. A, 2013, 1, 4571 RSC.
- S. Sugimoto, S. Kondo, K. Okayama, H. Nakamura, D. Book, T. Kagotani, M. Homma, H. Ota, M. Kimura and R. Sato, IEEE Trans. Magn., 1999, 35, 3154 Search PubMed.
- Y. Chen, T. Sakai, T. Chen, S. D. Yoon, A. L. Geiler, C. Vittoria and V. G. Harris, Appl. Phys. Lett., 2006, 88, 062516 Search PubMed.
- K. A. Korolev, J. S. McCloy and M. N. Afsar, J. Appl. Phys., 2012, 111, 07E113 Search PubMed.
- Y. Yang, M. C. Gupta, K. L. Dudley and R. W. Lawrence, Adv. Mater., 2005, 17, 1999 CrossRef CAS.
- Z. Liu, G. Bai, Y. Huang, Y. Ma, F. Du, F. Li, T. Guo and Y. Chen, Carbon, 2007, 45, 821 CrossRef CAS.
- M. J. W. Rodwell, in High Speed Integrated Circuit Technology, towards 100 GHz Logic, World Scientific, Singapore, 2001 Search PubMed.
- K. C. Huang and Z. Wang, in Millimeter Wave Communication Systems, Wiley-IEEE Press, New Jersey, 2011 Search PubMed.
- J. Capmany and M. Novak, Nat. Photonics, 2007, 1, 319 Search PubMed.
- D. Dawson, L. Samoska, A. K. Fung, K. Lee, R. Lai, R. Grundbacher, P.-H. Liu and R. Raja, IEEE Microw. Wireless Compon. Lett., 2005, 15, 874 Search PubMed.
- J. Jin, S. Ohkoshi and K. Hashimoto, Adv. Mater., 2004, 16, 48 CrossRef CAS.
- E. Tronc, C. Chanéac and J. P. Jolivet, J. Solid State Chem., 1998, 139, 93 CrossRef CAS.
- M. Popovici, M. Gich, D. Niznansky, A. Roig, C. Savii, L. Casas, E. Molins, K. Zaveta, C. Enache and J. Sort, Chem. Mater., 2004, 16, 5542 CrossRef CAS.
- J. Jin, K. Hashimoto and S. Ohkoshi, J. Mater. Chem., 2005, 15, 1067 RSC.
- K. Kelm and W. Mader, Z. Anorg. Allg. Chem., 2005, 631, 2383 Search PubMed.
- E. Tronc, C. Chaneac, J. P. Jolivet and J. M. Greneche, J. Appl. Phys., 2005, 98, 053901 Search PubMed.
- M. Kumoo, J. Rehspringer, A. Hutlova, C. D'Orleans, S. Vilminot, C. Estoumes and D. Niznansky, Chem. Mater., 2005, 17, 1106 Search PubMed.
- T. Nakamura, Y. Yamada and K. Yano, J. Mater. Chem., 2006, 16, 2417 RSC.
- M. Hermanek and R. Zboril, Chem. Mater., 2008, 20, 5284 CrossRef CAS.
- Y. Kusano, T. Fujii, J. Takada, M. Fukuhara, A. Doi, Y. Ikeda and M. Takano, Chem. Mater., 2008, 20, 151 CrossRef CAS.
- J. Tucek, R. Zboril, A. Namai and S. Ohkoshi, Chem. Mater., 2010, 22, 6483 CrossRef CAS.
- A. Namai, S. Sakurai, M. Nakajima, T. Suemoto, K. Matsumoto, M. Goto, S. Sasaki and S. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 1170 Search PubMed.
- S. Ohkoshi, S. Kuroki, S. Sakurai, K. Matsumoto, K. Sato and S. Sasaki, Angew. Chem., Int. Ed., 2007, 46, 8392 CrossRef CAS.
- S. Sakurai, S. Kuroki, H. Tokoro, K. Hashimoto and S. Ohkoshi, Adv. Funct. Mater., 2007, 17, 2278 Search PubMed.
- A. Namai, M. Yoshikiyo, K. Yamada, S. Sakurai, T. Goto, T. Yoshida, T. Miyazaki, M. Nakajima, T. Suemoto, H. Tokoro and S. Ohkoshi, Nat. Commun., 2012, 3, 1035 Search PubMed.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33 CrossRef CAS.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893 CrossRef CAS.
- The values of x for each phase of RhxFe2−xO3 [x(ε), x(α), and x(γ)] were obtained as follows. Sintering precursors at 1180 °C (x = 0), 1160 °C (0.12), 1140 °C (0.30), and 1140 °C (0.36) gave single-phase nanoparticles of α-RhxFe2−xO3. Rietveld analysis on the obtained α-RhxFe2−xO3 nanoparticles revealed a linear increase in the lattice constants (a and c) and lattice volume (V) as x(α) increased. This linear relationship was used to obtain x(α) in α-RhxFe2−xO3 for samples 1–9. For samples 1–4, which were composed of the ε- and α-phases, the x(ε) values in the ε-RhxFe2−xO3 phase were obtained by considering the compositions of the α-phase, the weight ratio between the phases obtained from the Rietveld analysis, and the quantity of Rh in the entire sample obtained from the ICP-MS measurement. This revealed a linear relationship between the lattice constants (a, b, and c) and lattice volume (V) and x(ε) for the ε-RhxFe2−xO3 phase. This linear relationship was used to obtain x(ε) in ε-RhxFe2−xO3 of samples 5–9. For samples 5–9, which were composed of the ε, α, and γ-phases, the x(γ) values in the γ-RhxFe2−xO3 phase were obtained by considering the compositions of the ε- and α-phases, the weight ratio between the phases obtained from the Rietveld analysis, and the quantity of Rh in the entire sample. The details are described in the ESI†.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- K. J. Standley, in Oxide Magnetic Materials, Clarendon Press, Oxford, 1972 Search PubMed.
- H. Zhang and J. F. Banfield, J. Mater. Chem., 1998, 8, 2073 RSC.
- J. M. McHale, A. Auroux, A. J. Perrotta and A. Navrotsky, Science, 1997, 277, 788 CrossRef CAS.
- H. L. Wen, Y. Y. Chen, F. S. Yen and C. H. Huang, Nanostruct. Mater., 1999, 11, 89 Search PubMed.
- S. Ohkoshi, Y. Tsunobuchi, T. Matsuda, K. Hashimoto, A. Namai, F. Hakoe and H. Tokoro, Nat. Chem., 2010, 2, 539 CrossRef CAS.
- S. Sakurai, A. Namai, K. Hashimoto and S. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 18299 CrossRef CAS.
- S. Ohkoshi, S. Sakurai, J. Jin and K. Hashimoto, J. Appl. Phys., 2005, 97, 10K312 Search PubMed.
- S. N. Houde-Walter, J. M. Inman, A. J. Dent and G. N. Greaves, J. Phys. Chem., 1993, 97, 9330 Search PubMed.
- S. Chikazumi, in Physics of Ferromagnetism, Oxford University Press, New York, 1997 Search PubMed.
- J. W. Archer, R. Lai, R. Grundbacher, M. Barsky, R. Tsai and P. Reid, IEEE Microw. Wireless Compon. Lett., 2001, 11, 4 Search PubMed.
- M. Urteaga, D. Scott, S. Krishnan, Y. Wei, M. Dahlstrom, Z. Griffith, N. Parthasarathy and M. J. W. Rodwell, IEEE J. Solid-State Circuits, 2003, 38, 1451 Search PubMed.
- D. Dawson, L. Samoska, A. K. Fung, K. Lee, R. Lai, R. Grundbacher, P.-H. Liu and R. Raja, IEEE Microw. Wireless Compon. Lett., 2005, 15, 874 Search PubMed.
- I. Minin, in Microwave and Millimeter Wave Technologies: from Photonic Bandgap Devices to Antenna and Applications, Intech Press, New York, 2010 Search PubMed.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The detailed information of synthesis, investigation of the sintering temperature, TEM study, crystal structure analysis, and magnetic properties. See DOI: 10.1039/c3tc30805g |
| This journal is © The Royal Society of Chemistry 2013 |