 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Linear oligofluorene-BODIPY structures for fluorescence applications†
Neil J.
Findlay
a,
Clara
Orofino-Peña
a,
Jochen
Bruckbauer
b,
Saadeldin E. T.
Elmasly
a,
Sasikumar
Arumugam
a,
Anto R.
Inigo
a,
Alexander L.
Kanibolotsky
a,
Robert W.
Martin
*b and
Peter J.
Skabara
*a
aWestCHEM, Department of Pure and Applied Chemistry, Thomas Graham Building, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: peter.skabara@strath.ac.uk
bDepartment of Physics, SUPA, University of Strathclyde, Glasgow, G4 0NG, UK
First published on 13th February 2013
Abstract
A family of linear oligofluorene-BODIPY structures, containing either a ter- or quaterfluorene unit, have been prepared, in which the attachment of the oligofluorene chain to the BODIPY unit is switched between the meso- and beta-positions. Each member of this family was investigated by UV-vis absorption and photoluminescence spectroscopy, cyclic voltammetry and thermal studies (TGA and DSC) to determine their suitability as emissive layers in hybrid luminescent devices. One candidate was then successfully deployed as a down converter to convert UV to visible light.
Introduction
The 4,4-difluoro-4-borata-3a-azonia-4a-aza-s-indacene unit, hereafter referred to as BODIPY, has attracted significant interest in recent years owing to its desirable blend of properties such as excellent stability, high absorption and fluorescence profiles and good solubility in common solvents.1–3 Such properties have meant BODIPY has found widespread application in a variety of roles, including biological labelling,4,5 luminescence6–8 and as sensors for pH and ions.9–13 The use of BODIPY as part of an emissive layer in luminescent devices is an attractive prospect due to its strong and tunable emissive properties. However, although strong, the absorption band for the BODIPY unit is narrow and confined to around 500 nm.1 As such, coupling of a BODIPY unit with a partner absorbing component, providing a more complex architecture, is a useful tactic in the development of designer organic molecules.Linear extended and monodisperse π-conjugated oligomers are materials of interest as they provide a comparison to conjugated polymeric materials, whilst being able to offer certain advantages over their polymeric analogues, such as knowledge of the precise molecular structure and complete synthetic reproducibility.14 We have previously produced molecules incorporating the truxene15–20 and 1,4-diketo-2,3,5,6-tetraphenyl-pyrrolo[3,4-c]pyrrole (DPP) cores,21 bonded to two, three or four oligofluorene arms. The DPP-based molecules showed that the antennae-like arms acted in concert with the emissive core, resulting in light-harvesting from the oligofluorene arms and energy transfer to the DPP core. Bo and co-workers have also shown efficient energy transfer in a series of star-shaped, monodisperse oligofluorene-porphyrins, bearing oligofluorene chains of between 1 and 4 units bound to the meso-position. All these compounds absorbed in the blue region, but emitted in the red, due to efficient energy transfer.22 Ziessel and co-workers have described the synthesis and properties of a unique molecule, consisting of a truxene core surrounded by three different BODIPY units, providing four separate chromophores within one molecule. Both Dexter and Förster energy transfer mechanisms contributed to the overall energy migration process with the BODIPY luminescence dominating.23 Several accounts of BODIPY-fluorene co-polymers, where the ratios of fluorene and BODIPY can vary greatly, have been reported recently,24–27 with a BODIPY-fluorene co-polymer being used in the imaging of rat liver cells. Burgess and co-workers found that the brightest co-polymers were formed when approximately four fluorene units were present for every BODIPY motif.27
Here, we present four novel, linear oligofluorene-BODIPY systems (Fig. 1), differing in both the length of oligofluorene chain (either three or four fluorenes) and the substitution position on the BODIPY (meso- vs. beta-). The synthetic route for each will be detailed, and their photophysical properties discussed to determine their suitability as emissive layers in hybrid luminescent devices. Hybrid inorganic/organic light-emitting diode (LED) structures offer the potential to combine the advantages of each technology, for example, the excellent electrical properties of inorganic LEDs and the broad emission properties of organic semiconductors.28–30 Such a structure would provide a means of absorbing higher energy, shorter wavelength light and, through energy transfer, converting it to lower energy, longer wavelength visible light.
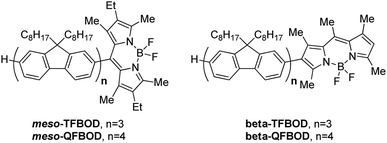 | ||
| Fig. 1 The structures of linear oligofluorene-BODIPYs mesomesomesomeso-TFBOD, mesomeso-QFBOD, beta-TFBOD and beta-QFBOD. | ||
Results and discussion
Synthesis
Initially, formation of the oligofluorene arms was achieved via an iterative approach, whereby the number of fluorene units in the chain was increased stepwise (see ESI for compounds 1–10, Scheme S1†).15 This provided sufficient quantities of bi-, ter- and quaterfluoreneboronic acids (11–13) suitable for coupling to functionalised BODIPY cores.Formation of the meso-substituted analogues, mesomesomesomeso-TFBOD and mesomeso-QFBOD, began by initial formation of novel BODIPY molecule 15, where a dialkylated fluorene molecule has been substituted with a BODIPY motif at the 2-position (at the meso-position of the BODIPY) and a bromine at the 7-position, providing a synthetic handle for further functionalisation (Scheme 1). To form the BODIPY motif, the requisite aldehyde functionality was necessary for the initial condensation with pyrrole. Starting from commercially available 9,9′-dioctyl-2,7-dibromofluorene, monolithiation and reaction of the subsequent lithiated species with DMF furnished monoaldehyde 14 in good yield.22 Formation of 15 was then attempted in a “one-pot” reaction; however, the resultant mixture was difficult to purify and several by-products were identified by crude 1H NMR. As such, a “pseudo-one-pot” protocol was developed, whereby the reaction mixture was subjected to a mini work-up after each of the three synthetic steps. As such, purification was only attempted after the third and final step, furnishing the required intermediate 15 in good yield (54% over three steps). Since intermediate 15 contained a suitable handle for transition metal catalysed cross-coupling, formation of the meso-substituted linear BODIPYs was realised via Suzuki-Miyaura cross-coupling with the complementary boronic acids. Now, with each coupling partner in hand, linear oligofluorene-BODIPYs mesomesomesomeso-TFBOD and mesomeso-QFBOD were isolated in good yields using the Pd2(dba)3/PtBu3·HBF4 catalyst system, with K3PO4 as the base. Each was isolated as a bright red-orange powder that, when in solution, strongly fluoresced under UV irradiation.
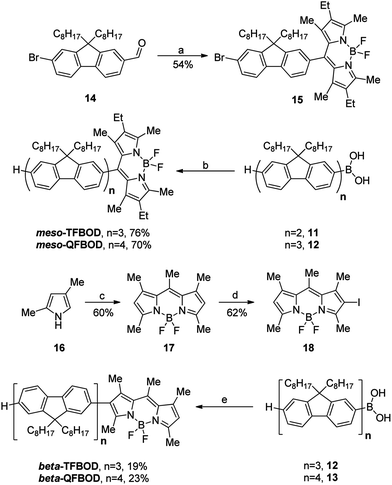 | ||
Scheme 1 Formation of oligofluorene-BODIPYs – reaction conditions: (a) (i) 3-ethyl-2,4-dimethylpyrrole, cat. TFA, CH2Cl2, 18 h, rt; (ii) DDQ, CH2Cl2, 18 h, rt; (iii) Et3N, CH2Cl2, 15 min; BF3·OEt2, 18 h, Ar; (b) 15, Pd2(dba)3, PtBu3·HBF4, K3PO4, THF/H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), reflux, 72 h; (c) (i) CH3COCl, CH2Cl2, 1.5 h, reflux, Ar; (iii) Et3N, CH2Cl2, 15 min; BF3·OEt2, 18 h, Ar; (d) ICl, MeOH/DMF (1:1), rt, 21 h, Ar; (e) 18, Pd2(dba)3, PtBu3·HBF4, K3PO4, THF/H2O (10:1), reflux, 72 h. :1), reflux, 72 h; (c) (i) CH3COCl, CH2Cl2, 1.5 h, reflux, Ar; (iii) Et3N, CH2Cl2, 15 min; BF3·OEt2, 18 h, Ar; (d) ICl, MeOH/DMF (1:1), rt, 21 h, Ar; (e) 18, Pd2(dba)3, PtBu3·HBF4, K3PO4, THF/H2O (10:1), reflux, 72 h. | ||
To form the beta-substituted analogues, beta-TFBOD and beta-QFBOD, a slightly modified strategy was required (Scheme 1). Owing to the ease of halogenation of BODIPY at the beta-position, it was believed that simple monohalogenation of the BODIPY core would be facile, allowing direct coupling to the appropriate oligofluorene boronic acid. With this in mind, BODIPY 18 was synthesised in two steps from 2,4-dimethylpyrrole 16 through initial condensation of 16 with acetyl chloride, forming the intermediate hydrochloride salt, that was then deprotonated and cyclised under standard conditions forming 17.31 To provide the requisite halogen at the beta-position, monoiodination was attempted with carefully controlled stoichiometry to avoid significant formation of the di-iodinated by-product, furnishing mono-iodinated BODIPY 18 in moderate yield.32 Formation of the targeted linear beta-oligofluorene-BODIPYs was then achieved through use of ter- and quaterfluorene boronic acids (12 and 13) using the conditions detailed above. Beta-substituted oligofluorene-BODIPYs, beta-TFBOD and beta-QFBOD, were isolated in low yields (19% and 23%) despite the previous success under the same conditions for the meso-substituted analogues. One possible reason for this is that the halogen site of 18 is sterically hindered relative to that in 15 due to the two methyl groups on the adjacent carbons.
Absorption, fluorescence, electrochemical and thermal properties
UV-vis absorption spectra for all oligofluorene-BODIPY compounds, recorded as dilute solutions in dichloromethane, are shown in Fig. 2, with details also shown in Table 1. All compounds exhibited similar HOMO–LUMO gaps of between 2.20 and 2.26 eV, reflecting the significant structural similarities across all four compounds. Within the series, it was clearly evident that separate peaks corresponding to the absorbance band for the oligofluorene fragment and the BODIPY fragment were present. For example, the oligofluorene band was observed at 354–370 nm, whereas the BODIPY band is present as a lower energy band from 513–527 nm. When comparing the meso-substituted compounds and their beta-substituted counterparts, the longer oligofluorene chain in the quaterfluorene analogues mesomeso-QFBOD and beta-QFBOD results in a slight red shift of 12–13 nm, compared to the terfluorene compounds mesomesomesomeso-TFBOD and beta-TFBOD, as well as a larger absorbance, consistent with a longer effective conjugation length. The low energy absorption bands of the meso-substituted compound have a distinct shoulder at the short wavelength region representative of vibronic splitting. In the spectra of the beta-substituted analogues this band is more symmetric, without a clear signature of vibronic splitting, and the peak is blue-shifted by 13–14 nm; this is due to the effects of the different substitution pattern33 and contributions from various vibronic modes in beta- and meso-substituted analogues (vide infra).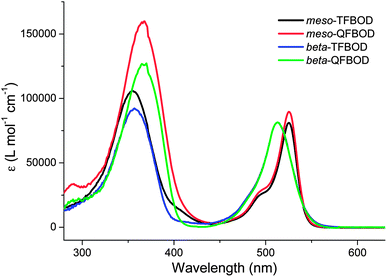 | ||
| Fig. 2 UV-vis absorption graphs for oligofluorene-BODIPYs mesomesomesomeso-TFBOD, mesomeso-QFBOD, beta-TFBOD and beta-QFBOD as dichloromethane solutions (∼10−5 M). | ||
Key to the employment of such molecules as effective down-converters for white light emission is efficient energy transfer between the absorbing and emitting units within the molecule. Whilst absorbance of the requisite wavelength of light ensures that materials can be useful in organic solar cells for example, both absorbance and emission of light, of the appropriate wavelength, is necessary for down-converter materials for hybrid LED applications.28–30 In order to determine whether the synthesised linear oligofluorene-BODIPYs were effective in this role, photoluminescence measurements were recorded in dilute dichloromethane solutions by excitation at the wavelength corresponding to the maximum absorption for the oligofluorene fragment for each compound respectively. The results are shown in Fig. 3 and summarised in Table 1. Our hypothesis that efficient energy transfer from the oligofluorene chain to the BODIPY unit would occur was proven to be correct, with each compound exhibiting fluorescence from the BODIPY unit whilst under excitation of the oligofluorene chain. Once more, comparison within each substitution series (either meso- or beta-) is useful. For example, the meso-substituted compounds, mesomesomesomeso-TFBOD and mesomeso-QFBOD, exhibited an intense, narrow emission band at 537 nm. In contrast, the beta-substituted series exhibited a broad red-shifted emission from the BODIPY fragments at 563–564 nm, with a Stokes shift of 50–51 nm vs. 10–11 nm for the meso-substituted analogues. The aforementioned featureless long wavelength absorption bands of the beta-substituted compounds, as well as the increased Stokes shifts, are evidence of a significant difference in the structure of the ground and the first excited state after structural relaxation. Recently, beta-substitution of BODIPY has been used as an approach for creating materials with a large Stokes shift due to geometry relaxation of the excited state.34 However, in the case of each compound it is clear that, despite a change in the substitution position on the BODIPY core, efficient and effective energy transfer from the oligofluorene absorber to the BODIPY emitter occurs readily. To support this further, photoluminescence quantum yield measurements on mesomeso-QFBOD gave a value of 0.84 ± 0.04, when referenced to the known BODIPY dye 17 (ref. 35) (see Fig. S9 and S10 in ESI†), a value which compares favourably with other highly emissive BODIPY-containing compounds.1
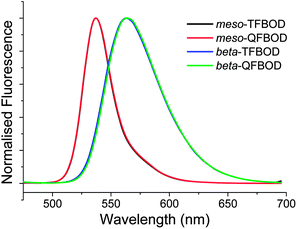 | ||
| Fig. 3 Normalised fluorescence curves for linear oligofluorene-BODIPYs mesomesomesomeso-TFBOD, mesomeso-QFBOD, beta-TFBOD and beta-QFBOD as dichloromethane solutions (∼10−6 M). Samples were excited at the λmax corresponding to the oligofluorene chain. | ||
Further information on the performance of these molecules was investigated by cyclic voltammetry to comprehend the redox properties of each series. The information is summarised below in Table 2 (with the spectra available in the ESI, Fig. S1–S8†). The redox properties of all four compounds are similar, principally in that they all exhibit three reversible oxidations and a single reversible reduction. Differences are revealed however, upon closer inspection. The first oxidation potential for the meso-substituted series was observed at E1/2 = +0.65 to 0.67 V, whereas the first oxidation potential for the beta-substituted series occurred at slightly higher potentials (E1/2 = +0.75 V). This first oxidation wave corresponds to the formation of a radical cation on the oligofluorene,36 so it is likely that the BODIPY fragment influences the position that the wave appears. The second and third oxidation waves are likely to correspond to the formation of a dication on the oligofluorene chain36 and oxidation of the BODIPY unit.31,37 Once more, comparisons between the series can be drawn. The terfluorene compounds, mesomesomesomeso-TFBOD and beta-TFBOD, showed values more positive for both the second and the third oxidation waves, compared with the quaterfluorene analogues, mesomeso-QFBOD and beta-QFBOD, reflecting the increased oligofluorene effective conjugation length present in the latter. All four compounds revealed a reversible reduction wave at E1/2 = −1.74 to ¬1.16 resulting from formation of a radical anion on the BODIPY fragment.33 No further reductive processes were observed within the analysis window. All the oligomers studied have a HOMO–LUMO gap of approximately 2.2 eV, with little difference between the electrochemical and optical values.
| mesomesomesomeso-TFBOD | mesomeso-QFBOD | beta-TFBOD | beta-QFBOD | |
|---|---|---|---|---|
| a HOMO and LUMO levels are calculated from the onset of the first peak of the corresponding redox wave and are referenced to ferrocene which has a HOMO of −4.8 eV. b Electrochemical HOMO–LUMO gap is the energy gap between the HOMO and LUMO levels. | ||||
| E 1ox (V) | +0.69/+0.64 | +0.67/+0.63 | +0.77/+0.72 | +0.77/+0.73 |
| E 2ox (V) | +0.94/+0.87 | +0.86/+0.81 | +0.93/+0.88 | +0.89/+0.85 |
| E 3ox (V) | +1.20/+1.10 | +1.00/+0.99 | +1.20/+1.10 | +1.08/+1.00 |
| E red (V) | −1.77/−1.71 | −1.66/−1.58 | −1.65/−1.57 | −1.68/−1.59 |
| HOMOa (eV) | −5.23 | −5.43 | −5.47 | −5.49 |
| LUMOa (eV) | −3.00 | −3.25 | −3.26 | −3.23 |
| HOMO–LUMO gapb (eV) | 2.23 | 2.18 | 2.21 | 2.26 |
Thermogravimetric analysis (TGA) of each of the linear oligofluorene-BODIPY samples revealed that all were stable at temperatures up to ca. 400 °C (Table 3). Furthermore, each sample showed two distinct decompositions, reflecting the fact that each compound combines two individual structural motifs. In both sets of compounds, the length of the oligomeric chain is reflected in the percentage decomposition; the larger the molecule, the lower the percentage decomposition. This is again shown in the temperature at which 5% decomposition is observed; with higher temperatures for beta-QFBOD and mesomeso-QFBODversusbeta-TFBOD and mesomesomesomeso-TFBOD.
| Temperature at which 5% decomposition occurs (°C) | Total % decomposition up to 550 °C | T g (°C) | T c/Tm (°C) | |
|---|---|---|---|---|
| mesomesomesomeso-TFBOD | 408 | 71.9 | 47.97 | 115.04/171.71 |
| mesomeso-QFBOD | 411 | 60.4 | 53.12 | — |
| beta-TFBOD | 394 | 58.6 | 42.63 | — |
| beta-QFBOD | 403 | 53.0 | 46.63 | — |
Differential scanning calorimetry (DSC) revealed all compounds had a rather low glass transition temperature (Tg) due to the high numbers of lengthy alkyl chains on the oligofluorene fragments. A difference of 4–5 °C was observed between the terfluorene and quaterfluorene members of each substitution series. The values for the meso-substituted compounds were approximately 5–6 °C higher than the beta-substituted compounds. Additionally, mesomesomesomeso-TFBOD is unique in showing both a crystallisation temperature and a melt temperature in contrast to the other members of the family (see Fig. S11 in ESI†).
Demonstration of down conversion of light
In order to demonstrate the down conversion process, mesomeso-QFBOD was deposited on a commercially available ultra-violet (UV) LED. The UV LED is based on the AlGaN material system and emits around 365 nm. The LED die is mounted in a TO46 metal can and is contacted by gold wire bonds. mesomeso-QFBOD was chosen as the candidate molecule as its absorption maximum almost coincides with the emission peak of the UV LED. For the deposition process 30 mg of mesomeso-QFBOD was dissolved in 1 ml of toluene. A 10 μl drop of this solution was deposited on top of the LED. Optical microscope images of the LED with and without the mesomeso-QFBOD and under a forward current are shown in Fig. 4(a)–(c). As seen in Fig. 4(b) most of the molecule has accumulated around and between the two contacts.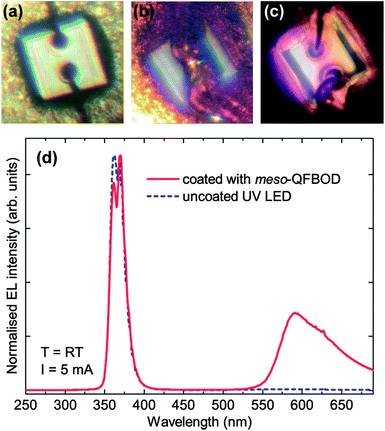 | ||
| Fig. 4 Optical microscope image of the (a) uncoated, (b) coated with mesomeso-QFBOD and (c) coated UV LED under forward current of 1 mA. The edge of the LED is 280 μm in length. Part (d) shows the electroluminescence spectra of the uncoated and coated LED under a forward current of 5 mA. | ||
Electroluminescence (EL) spectra under a forward current of 5 mA of the uncoated and mesomeso-QFBOD coated UV LED are displayed in Fig. 4(d). A weak defect band is observed around 585 nm. The emission from the LED with mesomeso-QFBOD is also plotted in Fig. 4(d). The emission from mesomeso-QFBOD is clearly visible as a broad peak in the region of 600 nm and dominates over the LED defect band. Additionally, subtraction of the spectrum of the uncoated LED from the spectrum of the LED coated with mesomeso-QFBOD shows almost no change, suggesting that the emission peak at 585 nm originates from the organic material, with a negligible contribution from the defect band of the LED (Fig. S12†). The two spectra are normalised to the 365 nm emission to see the relative ratio between the two emission peaks. The LED main emission peak is unchanged, whereas the maximum of the higher wavelength peak is shifted to approximately 590 nm and its intensity is increased relative to the main LED emission. This change can be associated with emission from mesomeso-QFBOD, which is pumped by the 365 nm line of the UV LED. The steeper drop on the lower wavelength side is due to self-absorption by the second absorption peak of mesomeso-QFBOD at 526 nm (see Fig. 2).
Conclusion
Here, we have presented the synthesis and photophysical study of a novel series of linear oligofluorene-BODIPYs, consisting of two distinct substitutions on the BODIPY core. All of the molecules synthesised exhibited high absorption of short wavelength light and successful conversion to longer wavelength light. The thermal properties of both series of compounds suggest that they would prove stable under device operating conditions. Finally, the suitability of such molecules as organic down converters has been demonstrated in light emitting devices. In summary, attachment of the oligofluorene chain at either the meso- and beta-position results in a useful down converter molecule capable of absorbing higher energy light and converting it into lower energy, visible light. Research is currently ongoing in our laboratory in order to achieve a more diverse range of absorption and emission characteristics towards the ultimate goal of achieving white light emitting organic materials for hybrid lighting applications.Experimental
General experimental
Tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) was prepared prior to use and stored under nitrogen. Unless otherwise stated, all other reagents were sourced commercially and used without further purification. Dry solvents were obtained from a solvent purification system (SPS 400 from Innovative Technologies) using alumina as the drying agent. 1H and 13C NMR spectra were recorded on either a Bruker DRX 500 apparatus at 500.13 and 125.76 MHz, or a Bruker Avance DPX400 apparatus at 400.13 and 100.61 MHz. Chemical shifts are given in ppm; all J values are in Hz. Elemental analyses were obtained on a Perkin-Elmer 2400 analyser. MS LDI-TOF spectra were run on a Shimadzu Axima-CFR spectrometer (mass range 1–150000 Da). Thermogravimetric analysis (TGA) was performed using a Perkin-Elmer Thermogravimetric Analyzer TGA7 under a constant flow of helium. Melting points were taken using a TA instruments DSC QC1000 Differential Scanning Calorimeter, or a Stuart Scientific apparatus, and are uncorrected.
Cyclic voltammetry (CV) measurements were performed on a CH Instruments 660A electrochemical workstation with iR compensation using anhydrous dichloromethane as the solvent. The electrodes were glassy carbon, platinum wire and silver wire as the working, counter and reference electrodes, respectively. All solutions were degassed (Ar) and contained monomer substrates in concentrations of ca. 10−4 M, together with n-Bu4NPF6 (0.1 M) as the supporting electrolyte. All measurements are referenced against the E1/2 of the Fc/Fc+ redox couple. Absorption spectra and CIE coordinates were recorded on a Unicam UV 300 instrument. Photoluminescence measurements were recorded using a Perkin-Elmer LS 50 B fluorescence spectrometer in a quartz cuvette (path length 10 mm). For PLQY measurements, absorption and photoluminescence measurements were performed using a Jasco V-660 spectrophotometer and a Jasco FP 6500 spectroflurometer, respectively.
Electroluminescence measurements of the LEDs were carried out at room temperature using a Keithley source measure unit. The light was dispersed by an Oriel 1/8 m spectrograph before being collected by an Andor charge-coupled device. The UV LEDs (XSL-365 class) were purchased from Roithner Lasertechnik GmbH and were used with the glass lens removed.
8-(7-Bromo-9,9′-dioctylfluorene-2)-1,3,5,7-tetramethyl-2,6-diethyl-4,4′difluoro-4-bora-3a,4a-diaza-s-indacene (15)
To a solution of 7-bromo-9,9′-dioctylfluorene-2-carbaldehyde 14 (2.4 g, 4.52 mmol, 1.0 eq.) in anhydrous dichloromethane (120 ml) was added 3-ethyl-2,4-dimethylpyrrole (1.63 ml, 12.06 mmol, 2.5 eq.). The solution was stirred at room temperature for 15 min then a catalytic amount of trifluoroacetic acid (3 drops) was added. The reaction mixture rapidly darkened and was stirred under Ar at room temperature for 18 h. The solution was then diluted with further dichloromethane and washed sequentially with sat. NaHCO3 (2 × 150 ml) and brine (200 ml). The dichloromethane layer was dried (MgSO4), filtered and concentrated under vacuum to a dark brown gum that was subsequently re-dissolved in anhydrous dichloromethane (75 ml) and DDQ (1.88 g, 8.27 mmol, 1.5 eq.) added in one portion. The dark solution was stirred under Ar at room temperature for 18 h then diluted with dichloromethane and washed with water (200 ml) and brine (200 ml), then dried (MgSO4) and concentrated to a deep red residue. The residue was re-dissolved in anhydrous dichloromethane (120 ml) and triethylamine (14.44 ml, 103.6 mmol, 15.0 eq.) added. The reaction mixture changed colour from red to dark green and was stirred under Ar at room temperature for 15 min. Boron trifluoride diethyl etherate (12.79 ml, 103.6 mmol, 15.0 eq.) was then added, causing a further colour change to dark red. The resulting solution was stirred at room temperature under Ar for 18 h before being diluted with dichloromethane and washed with water (3 × 150 ml) and brine (200 ml). The resulting dichloromethane layer was dried (MgSO4), filtered and concentrated under vacuum to afford a dark red residue. Purification on silica gel, eluting with 20% dichloromethane in hexane, afforded the title compound15 as a dark red powder (1.9 g, 54%); Tg = 42.33 °C; δH (400.13 MHz, CDCl3) 0.55–0.72 (m, 4H), 0.80–0.85 (m, 6H), 1.00–1.30 (m, 26H), 1.35 (s, 6H), 1.95–1.99 (m, 4H), 2.33 (q, J 7.6, 4H), 2.57 (s, 6H), 7.26–7.30 (m, 2H), 7.50–7.53 (m, 2H), 7.63 (d, J 8.0, 1H), 7.80 (d, J 8.0, 1H); δC (125.76 MHz, CDCl3) 12.1, 12.5, 14.0, 14.5, 17.1, 22.6, 23.8, 29.2, 29.4, 30.0, 31.8, 40.5, 55.9, 120.4, 121.4, 121.8, 123.0, 126.2, 127.3, 130.2, 130.9, 132.8, 134.9, 138.2, 139.3, 140.6, 140.9, 151.3, 153.0, 153.8; m/z (ESI) 769 (79Br, 100), 771 (81Br, 100); anal. calculated for C46H62BBrF2N2: C, 71.59; H, 8.10; N, 3.63; found: C, 71.92; H, 7.94; N, 3.21%.mesomesomesomeso-TFBOD
Compound 15 (199 mg, 0.26 mmol, 1.0 eq.), bifluorene boronic acid 11 (382 mg, 0.46 mmol, 1.8 eq.), potassium phosphate (123 mg, 0.58 mmol, 2.25 eq.), tris-tert-butylphosphonium tetrafluoroborate (15 mg, 0.05 mmol, 0.2 eq.) and tris(dibenzylideneacetone)dipalladium(0) (24 mg, 0.026 mmol, 0.1 eq.) were charged to a reaction flask, evaporated, then purged with Ar. This was repeated twice to ensure an oxygen free environment. Anhydrous tetrahydrofuran (40 ml) was then added, followed by degassed water (4 ml). The reaction mixture was then heated to 65 °C under Ar for 72 h. After this time, the reaction mixture was cooled to room temperature and diluted with dichloromethane (100 ml). The organic layer was washed with water (75 ml) and this aqueous wash extracted with further dichloromethane (75 ml). The organic layers were combined and washed sequentially with water (2 × 100 ml) and brine (150 ml), before being dried (MgSO4), filtered and concentrated under vacuum. Silica gel column chromatography, eluting with first 20% dichloromethane in hexane, then 1:1 dichloromethane/hexane, afforded the title compoundmesomesomesomeso-TFBOD as a bright orange powder (288 mg, 76%); Tg = 47.97 °C, Tc = 115.04 °C, Tm = 171.71 °C; δH (500.13 MHz, CDCl3) 0.65–0.90 (m, 30H), 1.00–1.27 (m, 66H), 1.39 (s, 6H), 2.03–2.14 (m, 12H), 2.33 (q, J 7.5, 4H), 2.57 (s, 6H), 7.27–7.39 (m, 5H), 7.64–7.76 (m, 9H), 7.79–7.87 (m, 5H); δC (125.76 MHz, CDCl3) 12.3, 12.7, 14.3, 14.8, 17.3, 22.8, 24.0, 24.1, 24.2, 29.4, 29.6, 30.3, 32.0, 40.6, 40.8, 55.4, 55.6, 55.9, 119.9, 120.1, 120.2, 120.5, 120.6, 121.6, 121.7, 123.2, 126.3, 126.4, 126.5, 127.0, 127.2, 127.3, 131.2, 133.0, 134.6, 138.6, 139.7, 140.1, 140.4, 140.6, 140.7, 140.9, 141.0, 141.2, 141.5, 141.9, 151.2, 151.7, 152.0, 152.09, 152.15, 153.9; m/z (MALDI) 1469 (100); anal. calculated for C104H144BF2N2: C, 84.97; H, 9.80; N, 1.91; found: C, 85.01; H, 9.77; N, 1.94%.
mesomeso-QFBOD
Compound 15 (192 mg, 0.25 mmol, 1.0 eq.), terfluorene boronic acid 12 (543 mg, 0.45 mmol, 1.8 eq.), potassium phosphate (119 mg, 0.56 mmol, 2.25 eq.), tris-tert-butylphosphonium tetrafluoroborate (26 mg, 0.05 mmol, 0.2 eq.) and tris(dibenzylideneacetone)dipalladium(0) (41 mg, 0.025 mmol, 0.1 eq.) were charged to a reaction flask, evaporated, then purged with Ar. This was repeated twice to ensure an oxygen free environment. Anhydrous tetrahydrofuran (50 ml) was then added, followed by degassed water (5 ml). The reaction mixture was then heated to 65 °C under Ar for 72 h. After this time, the reaction mixture was cooled to room temperature and diluted with dichloromethane (100 ml). The organic layer was washed with water (75 ml) and this aqueous wash extracted with further dichloromethane (2 × 40 ml). The organic layers were combined and washed sequentially with water (2 × 100 ml) and brine (150 ml), before being dried (MgSO4), filtered and concentrated under vacuum. Silica gel column chromatography, eluting with first 15% dichloromethane in hexane, then 1:1 dichloromethane/hexane, afforded the title compoundmesomeso-QFBOD as a bright orange powder (324 mg, 70%); Tg = 53.12 °C; δH (400.13 MHz, CDCl3) 0.60–0.90 (m, 40H), 0.97–1.27 (m, 86H), 1.39 (s, 6H), 2.01–2.14 (m, 16H), 2.33 (q, J 7.6, 4H), 2.57 (s, 6H), 7.27–7.39 (m, 5H), 7.64–7.76 (m, 13H), 7.79–7.87 (m, 7H); δC (100.61 MHz, CDCl3) 12.3, 12.7, 14.3, 14.8, 17.3, 22.8, 24.05, 24.14, 24.2, 29.4, 29.6, 30.3, 32.0, 40.6, 40.8, 55.4, 55.56, 55.58, 55.9, 119.9, 120.1, 120.17, 120.21, 120.5, 120.6, 121.6, 121.7, 123.2, 126.3, 126.4, 126.5, 127.0, 127.2, 127.3, 131.2, 133.0, 134.6, 138.6, 139.7, 140.1, 140.2, 140.3, 140.5, 140.6, 140.67, 140.70, 140.79, 140.85, 141.0, 141.2, 141.5, 141.9, 151.2, 151.71, 151.74, 152.0, 152.1, 152.2, 153.9; m/z (MALDI) 1858 (100); anal. calculated for C133H183BF2N2: C, 85.94; H, 9.92; N, 1.51; found: C, 85.95; H, 10.18; N, 1.41%.
beta-TFBOD
Mono-iodo BODIPY 18 (50 mg, 0.13 mmol, 1.0 eq.), terfluorene boronic acid 12 (252 mg, 0.21 mmol, 1.6 eq.), tris-tert-butylphosphonium tetrafluoroborate (8 mg, 0.026 mmol, 0.2 eq.) and tris(dibenzylideneacetone)dipalladium(0) (12 mg, 0.013 mmol, 0.1 eq.) were charged to a reaction flask, evaporated, then purged with Ar. This was repeated twice to ensure an oxygen free environment. Anhydrous tetrahydrofuran (20 ml) was then added, followed by potassium phosphate (55 mg, 0.26 mmol, 2.0 eq.) and degassed water (2 ml). The reaction mixture was then heated to 65 °C under Ar for 72 h. After this time, the reaction mixture was cooled to room temperature and quenched with water (100 ml). The reaction mixture extracted with dichloromethane (3 × 70 ml) and the organic layers combined, then washed with water (2 × 150 ml) and brine (150 ml), before being dried (MgSO4), filtered and concentrated under vacuum. Silica gel column chromatography, eluting with 20% dichloromethane in hexane, afforded a bright orange residue that was dissolved in the minimum volume of dichloromethane and precipitated by addition of excess cold methanol. The resulting bright orange powder was identified as the title compoundbeta-TFBOD (36 mg, 19%); Tg = 42.63 °C; δH (500.13 MHz, CDCl3) 0.60–0.89 (m, 30H), 1.05–1.26 (m, 60H), 1.99–2.14 (m, 12H), 2.41 (s, 3H), 2.48 (s, 3H), 2.56 (s, 3H), 2.58 (s, 3H), 2.70 (s, 3H), 6.11 (s, 1H), 7.21–7.22 (m, 2H), 7.32–7.40 (m, 3H), 7.65–7.71 (m, 8H), 7.76 (d, J 7.5, 1H), 7.80–7.85 (m, 5H); δC (125.76 MHz, CDCl3) 13.3, 14.1, 14.5, 15.5, 16.8, 17.5, 22.6, 23.8, 23.9, 24.0, 29.2, 29.3, 30.0, 31.8, 40.4, 55.2, 55.25, 55.35, 119.6, 119.7, 119.88, 119.95, 121.4, 121.5, 122.9, 125.1, 126.0, 126.2, 126.8, 127.0, 129.1, 132.0, 132.36, 132.43, 134.2, 137.0, 139.9, 139.99, 140.02, 140.1, 140.3, 140.4, 140.5, 140.55, 140.61, 140.8, 140.9, 141.5, 151.0, 151.2, 151.5, 151.7, 151.8, 152.4, 153.8; m/z (MALDI) 1428 (100); anal. calculated for C101H137BF2N2: C, 84.95; H, 9.67; N, 1.96; found: C, 84.58; H, 9.89; N, 2.19%.beta-QFBOD
Mono-iodo BODIPY 18 (39 mg, 0.1 mmol, 1.0 eq.), quaterfluorene boronic acid 13 (288 mg, 0.18 mmol, 1.8 eq.), tris-tert-butylphosphonium tetrafluoroborate (6 mg, 0.02 mmol, 0.2 eq.) and tris(dibenzylideneacetone)dipalladium(0) (9 mg, 0.01 mmol, 0.1 eq.) were charged to a reaction flask, evaporated, then purged with Ar. This was repeated twice to ensure an oxygen free environment. Anhydrous tetrahydrofuran (10 ml) was then added, followed by potassium phosphate (48 mg, 0.225 mmol, 2.25 eq.) and degassed water (1 ml). The reaction mixture was then heated to 65 °C under Ar for 72 h. After this time, the reaction mixture was cooled to room temperature and diluted with dichloromethane (80 ml). The organic layer was washed with water (2 × 100 ml) and brine (100 ml), before being dried (MgSO4), filtered and concentrated under vacuum. Silica gel column chromatography, eluting with 20–30% dichloromethane in hexane, afforded a bright orange residue that was dissolved in the minimum volume of dichloromethane and precipitated by addition of excess cold methanol. The resulting bright orange powder was identified as the title compoundbeta-QFBOD (40 mg, 23%); Tg = 46.63 °C; δH (500.13 MHz, CDCl3); 0.63–0.90 (m, 40H), 1.05–1.26 (m, 80H), 2.00–2.15 (m, 16H), 2.41 (s, 3H), 2.48 (s, 3H), 2.56 (s, 3H), 2.59 (s, 3H), 2.70 (s, 3H), 6.11 (s, 1H), 7.21–7.22 (m, 2H), 7.32–7.40 (m, 3H), 7.65–7.72 (m, 12H), 7.76 (d, J 7.0, 1H), 7.80–7.85 (m, 7H); δC (125.76 MHz, CDCl3) 13.3, 14.1, 14.5, 15.5, 16.8, 17.5, 22.6, 23.8, 23.9, 24.0, 29.2, 29.3, 30.0, 31.8, 40.4, 55.2, 55.3, 55.4, 119.6, 119.7, 120.0, 121.4, 121.5, 122.9, 125.1, 126.0, 126.2, 126.8, 127.0, 129.1, 132.0, 132.4, 134.2, 137.0, 139.9, 139.99, 140.03, 140.3, 140.4, 140.50, 140.54, 140.8, 140.9, 141.4, 151.0, 151.2, 151.5, 151.7, 151.8, 152.4, 153.8; m/z (MALDI) 1816 (100); anal. calculated for C130H177BF2N2: C, 85.95; H, 9.82; N, 1.54; found: C, 85.09; H, 9.91; N, 1.55%.Acknowledgements
We thank the EPSRC and the University of Strathclyde for funding and the EPSRC Mass Spectrometry Centre, Swansea, for accurate mass measurements.Notes and references
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS.
- R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS.
- Q. Zheng, G. Xu and P. N. Prasad, Chem.–Eur. J., 2008, 14, 5812 CrossRef CAS.
- Y. Ikawa, S. Moriyama and H. Furuta, Anal. Biochem., 2008, 378, 166 CrossRef CAS.
- L. Bonardi, H. Kanaan, F. Camerel, P. Jolinat, P. Retailleau and R. Ziessel, Adv. Funct. Mater., 2008, 18, 401 CrossRef CAS.
- M. M. Sartin, F. Camerel, R. Ziessel and A. J. Bard, J. Phys. Chem. C, 2008, 112, 10833 CAS.
- D. T. Chase, B. S. Young and M. M. Haley, J. Org. Chem., 2011, 76, 4043 CrossRef CAS.
- B. Turfan and E. U. Akkaya, Org. Lett., 2002, 4, 2857 CrossRef CAS.
- A. Coskun, M. D. Yilmaz and E. U. Akkaya, Org. Lett., 2007, 9, 607 CrossRef CAS.
- S. C. Dodani, Q. He and C. J. Chang, J. Am. Chem. Soc., 2009, 131, 18020 CrossRef CAS.
- T. Kowada, S. Yamaguchi, H. Fujinaga and K. Ohe, Tetrahedron, 2011, 67, 3105 CrossRef CAS.
- N. Boens, W. Qin, M. Baruah, W. M. De Borggraeve, A. Filarowski, N. Smisdom, M. Ameloot, L. Crovetto, E. M. Talavera and J. M. Alvarez-Pez, Chem.–Eur. J., 2011, 17, 10924 CrossRef CAS.
- A. L. Kanibolotsky, I. F. Perepichka and P. J. Skabara, Chem. Soc. Rev., 2010, 39, 2695 RSC.
- A. L. Kanibolotsky, R. Berridge, P. J. Skabara, I. F. Perepichka, D. D. C. Bradley and M. Koeberg, J. Am. Chem. Soc., 2004, 126, 13695 CrossRef CAS.
- K. M. Omer, A. L. Kanibolotsky, P. J. Skabara, I. F. Perepichka and A. J. Bard, J. Phys. Chem. B, 2007, 111, 6612 CrossRef CAS.
- G. Tsiminis, Y. Wang, P. E. Shaw, A. L. Kanibolotsky, I. F. Perepichka, M. D. Dawson, P. J. Skabara, G. A. Turnbull and I. D. W. Samuel, Appl. Phys. Lett., 2009, 94, 243304 CrossRef.
- A. J. C. Kuehne, D. Elfström, A. R. Mackintosh, A. L. Kanibolotsky, B. Guilhabert, E. Gu, I. F. Perepichka, P. J. Skabara, M. D. Dawson and R. A. Pethrick, Adv. Mater., 2009, 21, 781 CrossRef CAS.
- Y. Wang, G. Tsiminis, Y. Yang, A. Ruseckas, A. L. Kanibolotsky, I. F. Perepichka, P. J. Skabara, G. A. Turnbull and I. D. W. Samuel, Synth. Met., 2010, 160, 1397 CrossRef CAS.
- A. Hernandez-Santana, A. R. Mackintosh, B. Guilhabert, A. L. Kanibolotsky, M. D. Dawson, P. J. Skabara and D. Graham, J. Mater. Chem., 2011, 21, 14209 RSC.
- A. L. Kanibolotsky, F. Vilela, J. C. Forgie, S. E. T. Elmasly, P. J. Skabara, K. Zhang, B. Tieke, J. McGurk, C. R. Belton, P. N. Stavrinou and D. D. C. Bradley, Adv. Mater., 2011, 23, 2093 CrossRef CAS.
- B. Li, J. Li, Y. Fu and Z. Bo, J. Am. Chem. Soc., 2004, 126, 3430 CrossRef CAS.
- S. p. Diring, F. Puntoriero, F. Nastasi, S. Campagna and R. Ziessel, J. Am. Chem. Soc., 2009, 131, 6108 CrossRef CAS.
- A. Nagai, J. Miyake, K. Kokado, Y. Nagata and Y. Chujo, J. Am. Chem. Soc., 2008, 130, 15276 CrossRef CAS.
- G. Meng, S. Velayudham, A. Smith, R. Luck and H. Liu, Macromolecules, 2009, 42, 1995 CrossRef CAS.
- V. R. Donuru, G. K. Vegesna, S. Velayudham, S. Green and H. Liu, Chem. Mater., 2009, 21, 2130 CrossRef CAS.
- C. Thivierge, A. Loudet and K. Burgess, Macromolecules, 2011, 44, 4012 CrossRef CAS.
- G. Heliotis, G. Itskos, R. Murray, M. D. Dawson, I. M. Watson and D. D. C. Bradley, Adv. Mater., 2006, 18, 334 CrossRef CAS.
- G. Heliotis, P. N. Stavrinou, D. D. C. Bradley, E. Gu, C. Griffin, C. W. Jeon and M. D. Dawson, Appl. Phys. Lett., 2005, 87, 103505 CrossRef.
- E. Gu, H. X. Zhang, H. D. Sun, M. D. Dawson, A. R. Mackintosh, A. J. C. Kuehne, R. A. Pethrick, C. Belton and D. D. C. Bradley, Appl. Phys. Lett., 2007, 90, 031116 CrossRef.
- A. B. Nepomnyashchii, M. Bröring, J. Ahrens and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 8633 CrossRef CAS.
- S. Niu, G. Ulrich, P. Retailleau and R. Ziessel, Org. Lett., 2011, 13, 4996 CrossRef CAS.
- C.-W. Wan, A. Burghart, J. Chen, F. Bergström, L. B. Å. Johansson, M. F. Wolford, T. G. Kim, M. R. Topp, R. M. Hochstrasser and K. Burgess, Chem.–Eur. J., 2003, 9, 4430 CrossRef CAS.
- Y. Chen, J. Zhao, H. Guo and L. Xie, J. Org. Chem., 2012, 77, 2192 CrossRef CAS.
- A. B. Nepomnyashchii, S. Cho, P. J. Rossky and A. J. Bard, J. Am. Chem. Soc., 2010, 132, 17550 CrossRef CAS.
- C. Chi and G. Wegner, Macromol. Rapid Commun., 2005, 26, 1532 CrossRef CAS.
- R. Ziessel, C. Goze, G. Ulrich, M. Césario, P. Retailleau, A. Harriman and J. P. Rostron, Chem.–Eur. J., 2005, 11, 7366 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3tc00706e |
| This journal is © The Royal Society of Chemistry 2013 |