Electronic and magnetic structure of C60/Fe3O4(001): a hybrid interface for organic spintronics
P. K. Johnny
Wong
*a,
Wen
Zhang
a,
Kai
Wang
a,
Gerrit
van der Laan
b,
Yongbing
Xu
cd,
Wilfred G.
van der Wiel
a and
Michel P.
de Jong
*a
aNanoElectronics Group, MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, Enschede, 7500 AE, The Netherlands. E-mail: p.k.j.wong@utwente.nl; m.p.dejong@utwente.nl; Tel: +31-53-489-6205
bDiamond Light Source, Chilton, Didcot, OX11 0DE, UK
cSpintronics and Nanodevice Laboratory, Department of Electronics, University of York, York, YO10 5DD, UK
dNanjing-York International Center of Spintronics and NanoEngineering, School of Electronics Science and Engineering, Nanjing University, Nanjing 210093, China
First published on 3rd December 2012
Abstract
We report on the electronic and magnetic characterization of the hybrid interface constituted of C60 molecules and an epitaxial Fe3O4(001) surface grown on GaAs(001). Using X-ray absorption spectroscopy (XAS) and X-ray magnetic circular dichroism (XMCD), we demonstrate that a stable C60 sub-monolayer (ML) can be retained on the Fe3O4(001) surface upon in situ annealing at 250 °C. A carbon K-edge dichroic signal of 1% with respect to the XAS C 1s → π* peak intensity has been observed, indicative of a weaker electronic interaction of C60 with Fe3O4(001) compared to the previously reported case of C60/Fe(001). Remarkably, the Fe L-edge XMCD spectrum of Fe3O4(001) reveals a reduced Fe3+/Fe2+ ratio upon C60 sub-ML adsorption. This observation has been ascribed to electron donation by the C60 molecules, as a consequence of the high work function of Fe3O4(001). Our present work underlines the significance of chemical interactions between inorganic magnetic surfaces and molecular adsorbates for tuning of the electronic and magnetic properties of the interfaces, which have a profound impact on spin-polarized charge transport in hybrid organic–inorganic spintronic devices.
1 Introduction
Organic spintronics,1–4 a novel research field aimed at combining the advantageous properties of organic semiconductors with the weak spin relaxation and dephasing mechanisms intrinsic to light-weight carbon-based materials,5,6 forms a highly promising platform for the development of next-generation electronic technologies. Although an increasing amount of experimental studies showing strong magnetoresistance (MR) effects on hybrid inorganic–organic spin valves have been reported in the literature,4,6–9 the microscopic mechanisms behind these effects and their relationship to the interfacial spin-dependent properties remain poorly understood. This is largely due to the presence of ill-defined interfaces in the devices, which hampers systematic studies of the effects of interfacial properties on spin dependent transport. The critical role played by hybrid interfaces has started to draw a great deal of attention, as elucidated by several studies.8,10–13 Our recent work13 on well-defined interfaces between C60 and bcc-Fe(001) showed that the hybridization between the π-electronic states of C60 and the 3d band of the Fe(001) surface is quite strong and produces a significant magnetic polarization of the C60-derived electronic states. This is expected to play an important role in spin injection/extraction across this interface. Recently, it has also been proposed that spin-dependent hybridization between metal states and molecular electronic states in nanometer-scale La0.7Sr0.3MnO3/tris(8-hydroxyquinoline)Al/Co (LSMO/Alq3/Co) magnetic-tunnel junctions plays a decisive role in the observation of sign reversal of the MR effects.8 The relevance and influence of such hybridization phenomena for/on spin transport across organic–inorganic interfaces remain a largely open question. Experimental evaluation of well-defined and well-characterized interfaces that allow for systematic analysis with theories is required to advance the knowledge on this issue.In this work, we present a study of the electronic and magnetic structure of the interface between C60 molecules and an ultrathin ferrimagnetic transition-metal oxide, Fe3O4(001), epitaxially grown on GaAs(001). Such interfaces constitute an important and interesting type of model interfaces for organic spintronics because of the following reasons. (1) The less reactive nature of Fe3O4 surfaces is expected to lead to weaker hybridization effects when compared to those occurring at the C60/Fe(001) interface. (2) As we have demonstrated previously, high-quality Fe3O4 films with well-defined magnetic anisotropy can be prepared in a straightforward manner by post-deposition annealing of an ultrathin epitaxial Fe film on various technologically relevant semiconductor surfaces in an oxygen-rich environment.14 (3) Fe3O4 exhibits promising properties for spintronic applications. Fe3O4 is a mixed-valence ferrimagnetic oxide with an exceptionally high Curie temperature of ∼850 K. More importantly, according to theoretical studies of its spin-polarized band structure, Fe3O4 is a half-metallic material with the majority-spin electrons exhibiting an insulating (or semiconducting) behavior, while the minority-spin electrons feature a metallic behavior. Therefore, only minority-spin electrons are present at the Fermi level, which leads to −100% spin polarization of bulk Fe3O4.15 The highest achieved surface spin polarization in practice thus far is about −80% for an Fe3O4(111) surface.16 (4) Epitaxial Fe3O4 films, which have a cubic inverse-spinel structure with a distorted fcc oxygen sublattice, offer good prospects for forming well-ordered, crystalline C60 molecular films.
2 Experimental
Experiments were carried out on beamline D1011 at MAX-Laboratory in Lund, Sweden. Thin films of C60 molecules and Fe3O4(001) were prepared in situ in the measurement chamber for X-ray absorption spectroscopy (XAS) and X-ray magnetic circular dichroism (XMCD), with a base pressure of 10−10 mbar. Different stages of the hetero-epitaxy were carefully followed and monitored by low-energy electron diffraction (LEED). The substrates used in this study were As-capped GaAs(001). A ∼0.5 μm layer of homo-epitaxial GaAs was grown on a commercial wafer to provide the smoothest possible GaAs surface. The As cap was desorbed at about 340 °C and the substrate was further annealed to 550 °C for 60 min to obtain a clean and ordered surface,17 on top of which a 3 nm Fe film was epitaxially grown at room temperature (RT) using a mini e-beam evaporator. The Fe layer was then exposed to molecular oxygen, introduced into the UHV chamber with a precision leak valve, at an O2 partial pressure of 8 × 10−5 mbar for 10 min, while the sample temperature was kept at 230 °C.14 Multilayers of C60 molecules were deposited onto the ferrite surface by evaporation from a simple custom-built Knudsen-cell. To examine the C60/Fe3O4(001) interface, the samples were annealed to 250 °C for 5 min to desorb C60 overlayers, retaining only the more strongly bound C60 molecules on the Fe3O4 surface. XAS and XMCD spectra were collected at RT in total-electron-yield (TEY) mode, in which the sample drain current is recorded as a function of the photon energy. The spectra were normalized to the incident photon flux obtained using the TEY from a gold grid. The angle of incidence of the photon beam was set to 45° relative to the sample normal. The sample was magnetically saturated along its in-plane magnetic easy axis using a pulsed magnetic field of ±600 Oe, and the XMCD was measured at remanence using 75%-circularly polarized X-rays.3 Results and discussion
The LEED patterns of a cleaned GaAs(001) substrate, exhibiting a (1 × 1) surface unit cell, and a 3 nm thick Fe(001) epitaxial film grown at the top, are displayed in Fig. 1(a) and (b), respectively. Post-oxidation of the magnetic film into Fe3O4(001) is illustrated and verified in Fig. 1(c). A 45° in-plane rotation of the magnetite lattice with respect to the GaAs(001) unit cell can be observed, as well as the emergence of half-order spots, in agreement with our previous study.14 After annealing-off the multilayer C60 film from the ferrite surface, the diffraction pattern transforms into a (1 × 1) structure, as indicated in Fig. 1(d). This may be related to strong adsorption of the C60 sub-ML on the oxide surface, as will be elaborated below. | ||
| Fig. 1 LEED patterns taken from the GaAs(001) substrate, epitaxial bcc-Fe(001), bare Fe3O4(001), and C60 adsorbed on Fe3O4(001) surfaces. (a) GaAs surface after in situ annealing; (b) after growth of a 3 nm Fe(001) at RT; (c) after post-growth oxidation of Fe(001); and (d) after in situ annealing of a multilayer of C60 molecules. | ||
Fig. 2(a) shows the C K-edge XAS spectra of an (initial) multilayer C60 film on Fe3O4(001) before and after annealing at 250 °C for 5 min. The multilayer C60 spectrum well resembles the corresponding spectrum of bulk C60.18 The first peak at 284.5 eV is due to a C 1s → π* transition to the (three-fold degenerate) lowest unoccupied molecular orbital (LUMO). The next three peaks, labeled LUMO+1, LUMO+2, and LUMO+3, respectively, are excitations to other (groups of) π* orbitals.19 Above the ionization threshold of 290 eV, the broader structures are due to σ* shape resonances. After thermal treatment, a sub-ML of C60 is retained on the Fe3O4 substrate. Using photoemission spectroscopy, the C60 coverage was estimated to be ∼0.5 ML. For C60 on a variety of metal surfaces, a stable C60 ML can be obtained by thermal desorption of the weakly bound overlayers (see e.g.ref. 13). Strong hybridization effects occur at the C60/metal interfaces, as is evidenced by the merging of the LUMO+1 and LUMO+2 resonances into a single peak.13,18 The lack of such spectral features, in combination with the low retained coverage (∼0.5 ML), indicates a weaker interfacial interaction between C60 and the Fe3O4 surface. The stability of the ∼0.5 ML C60 molecules was further examined by annealing for a longer duration (20 min) at the same temperature, which resulted in an identical XAS spectrum. This reveals that the interaction at the sub-ML C60/Fe3O4 interface is not exclusively of the van der Waals-type but a sufficiently strong interaction that keeps both materials together. Compared to the multilayer spectrum, the LUMO+1, +2 energy positions of the sub-ML C K-edge XAS seem to be slightly shifted with an additional minor shoulder emerging between 286 and 287 eV, as shown in Fig. 2(b). These spectral features again indicate a non-negligible electronic interaction at the sub-ML C60/Fe3O4 interface. Nonetheless, these signs of an interfacial hybridization in the present case are less significant than for C60/Fe(001).
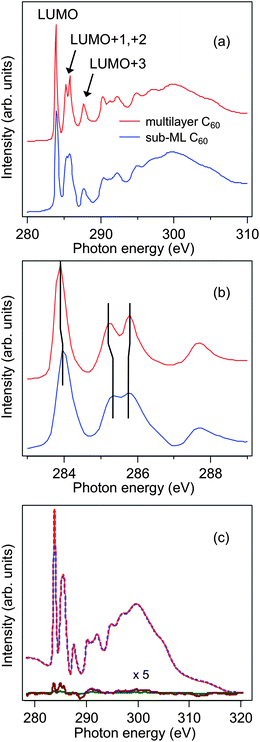 | ||
| Fig. 2 (a) C K-edge XAS spectra of a multilayer and sub-ML C60 on Fe3O4(001). (b) Lower photon energy region of XAS spectra shown in (a). The vertical lines are guides for the eyes to help identify the energy positions of the LUMO-derived peaks. (c) C K-edge XAS and XMCD spectra of a sub-ML C60/Fe3O4(001). The XMCD spectra were obtained at remanence by taking the difference between the XAS spectra recorded with parallel and antiparallel alignments of applied magnetization and photon helicity. | ||
As has been shown in a number of previous experimental studies, a sizeable interfacial magnetic moment and spin polarization can be induced in the π-conjugated states of several carbon-based systems. This is caused by the hybridization of the π-electronic states with the valence and conduction bands of ferromagnetic metal surfaces.13,20 The induced magnetic effects observed in the XMCD experiments, here quantified in the form of C K-edge XMCD intensity with respect to the C 1s → π* peak intensity, are, e.g., 3% for C60/Fe(001)13 and 5% for graphene/Ni(111).20 For the C60 sub-ML on Fe3O4(001) we can also observe a weak dichroic signal of about 1% at the C K-edge under similar experimental conditions, as depicted in Fig. 2(c). Depending on the photon energy, the interfacial magnetic polarization of the C60 molecules changes sign, which is similar to the theoretical and experimental results reported by Atodiresei et al.11 and Tran et al.,13 respectively. It is noteworthy that the electronic interaction (and orbital hybridization) of C60 with Fe(001) and Fe3O4(001) could naively be expected to differ considerably from the case of graphene on Ni(111)20 and flat, aromatic molecules on Fe/W(110),11 due to the “soccer-ball” shape of the C60 molecule. More precisely, C60 is a truncated icosahedron consisting of 12 pentagons and 20 hexagons.21 However, as pointed out by Maxwell et al.,18 the hybridization between the molecular orbitals of C60 and the metal substrate does not merely affect the C atoms that are in direct contact with the surface, due to delocalization of the electrons over the C60 molecule.
We now address the impact of C60 adsorption on the electronic and magnetic properties of the Fe3O4 surface. XAS and XMCD are especially suitable for studying the effect of the observed electronic interactions on the surface spin-dependent electronic and magnetic structure of Fe3O4, since these techniques can provide direct information on the oxidation state and local structure around the different Fe cation sites of magnetite.14,22Fig. 3(a) shows the Fe L2,3-edge XMCD spectra of Fe3O4(001) with and without the adsorbed sub-ML C60. Three different Fe cation environments can be clearly distinguished. The two negative peaks in the L3-edge at 705.8 and 707.5 eV arise from the octahedral (Oh) Fe2+ and Fe3+ cations, respectively, while the positive peak at 706.7 eV is caused by the tetrahedral (Th) Fe3+ sites. The positive and negative signs of these peaks originate from the antiferromagnetic coupling between the octahedral and tetrahedral sublattices in the inverse-spinel structure of magnetite. The line shape of the XMCD spectrum serves as a distinct fingerprint,22 indicating that the stoichiometry of our magnetite film is indeed Fe3O4 while spurious phases cannot be detected. It is also clearly discernible that the adsorption of a C60 sub-ML results in a decrease in the XMCD intensities of both Th and Oh Fe3+ cations, relative to that of the Fe2+ ions. For ease of comparison, we have normalized the measured XMCD spectra to the Oh Fe3+ peak height in Fig. 3(b), where a relative increase of Oh Fe2+ is evidenced. It has been well established that the XMCD peak-ratio Fe3+/Fe2+ of Fe3O4 reflects the balance between Fe2+ and Fe3+ ions in the ferrite.22,23 A change of this ratio usually refers to non-stoichiometry of the ferrite, which is mostly triggered by: (i) substitution of the Fe sites by other chemical elements24 or (ii) oxidation/reduction of the Fe cations in the ferrite.23,25 Here, possible oxygen vacancies created by partial desorption of oxygen in our magnetite film due to annealing in UHV appears unlikely, as the temperature being used (250 °C) is quite low23 compared to the magnetite to wüstite (FeO) transition temperature of 410 °C in vacuum.26 This account would appear even more convincing if we compare the O K-edge XAS spectra of our sample, as shown in Fig. 3(c). Clearly discernible in the spectra are the two regions, where the first sharp peak is attributed to the O 2p states hybridized with the Fe 3d states, while the second broad region corresponds to the O 2p state hybridized with the Fe 4s and 4p states.27 Park et al. previously reported the impact of annealing on the stoichiometry and electronic structure of magnetite under an experimental condition similar to ours.28 Their combined study of O K-edge XAS and Fe L-edge XMCD at various annealing temperatures suggested that even a slightly reduced magnetite film, i.e., Fe3O4−δ, would exhibit a significantly reduced intensity and splitting of the first O K-edge peak (at the lowest excitation energy) compared to that of single-phase Fe3O4. Given the lack of any of these spectral changes in our O K-edge spectra upon thermal annealing, reduction of the ferrite film via oxygen loss in our present study can be safely ruled out as a major mechanism for causing the observed changes in dichroism in Fig. 3(a) and (b). We are, however, unable to entirely eliminate any oxygen vacancies that are below the detection limit of the spectroscopic technique. Nevertheless, our results tentatively point to a correlation between the Fe3+/Fe2+ ratio modification and the above-mentioned hybridization of C60 molecules with the Fe3O4(001) surface.
 | ||
| Fig. 3 (a) Fe L2,3-edge XMCD spectra of Fe3O4(001) with and without a sub-ML C60. Inset shows the XMCD peak intensity of Oh Fe2+ of clean Fe3O4(001) as a function of applied magnetic field strength. (b) Fe L2,3-edge XMCD spectra of Fe3O4(001) normalized to the Oh Fe3+ peak height. (c) O K-edge XAS of Fe3O4(001) before and after in situ thermal treatment. | ||
To obtain additional information concerning the mechanism involved, XMCD sum rules,29 which permit quantitative analysis of the spin and orbital magnetic moments, were applied to the integrated Fe L2,3-edge XMCD spectra shown in Fig. 4. The general shape of the XAS spectra is similar to the results in the literature.23 The non-zero integral of the XMCD signal corresponds to a small orbital contribution to the total magnetic moment of the Fe3O4 layer. A two-step background was subtracted from the XAS spectra,30 and the number of Fe 3d holes was taken equal to 4.5.31,32 We also show in the inset of Fig. 3(a) the XMCD hysteresis of the clean magnetite film as a function of magnetic field along the incident X-ray direction (45° off the sample normal) measured at the fixed photon energy of the Oh Fe2+ peak. The high degree of squareness of the hysteresis with a coercivity of 300 Oe justifies the use of our remanence data to extract the magnetic moments.
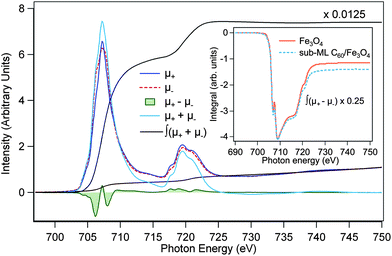 | ||
| Fig. 4 Sum-rule analysis of Fe L2,3-edge XAS and XMCD spectra of Fe3O4(001) with and without sub-ML C60 adsorption. μ+ and μ− are the 2p → 3d XAS intensities for parallel and antiparallel alignments of photon helicity and magnetization. ∫(μ+ + μ−) and ∫(μ+ − μ−) are the integrated intensities over the XAS and XMCD spectra, respectively. | ||
Table 1 summarizes the results of the sum-rule analysis, which have been corrected for the photon incident angle and degree of circular polarization. The orbital magnetic moment, which is very sensitive to hybridization effects, does not change considerably upon sub-ML C60 adsorption. The unquenched orbital moments observed here, although remaining substantially smaller than the values presented in ref. 31, are in contrast to that of a bulk Fe3O4 crystal.32 This can be generally expected for ultrathin magnetic films because of decreased crystal symmetry and reduced delocalization effects at the surface. On the other hand, the spin moment has been found to decrease from 2.95 ± 0.21 to 2.62 ± 0.18 μB/f.u. upon C60 adsorption. Here, we propose that the relative increase of the Oh Fe2+ concentration is related to electron transfer from C60 to the outer Fe atoms of the Fe3O4 underlayer. Energy-level alignment at various C60/substrate interfaces has been determined previously as a function of the substrate work function by Osikowicz et al.33 Following their model, electron transfer from C60 to a particular substrate would become possible if the substrate's work function is higher than 5.5 eV. This scenario could indeed be reached in the case of Fe3O4(001) because of its high work function (5.3–5.8 eV).34 However, one might simply expect that such an electron transfer mechanism (if present) at the C60/Fe3O4 interface would instead increase the value of the spin moment. According to Hund's rule and the high-spin (HS) electronic configuration of magnetite,35 the d-orbitals in an octahedral crystal field are split into low-energy t2g and high-energy eg states, where the states are filled to achieve maximum spin multiplicity, i.e., maximum number of unpaired electrons. The HS 3d6 (Fe2+) t32g↑e2g↑t12g↓ configuration has one unpaired electron less than the 3d5 (Fe3+) t32g↑e2g↑, which therefore explains the lower spin moment after sub-ML C60 adsorption. Similarly, the observed marginal enhancement of the orbital magnetic moment can be readily attributed to the 3d5 → 3d6 transitions, since the half-filled 3d5 shell exhibits a negligible orbital moment. It is worth noting that the observation of hybridization-induced magnetization of C60 in this present study can be linked to the electron donation mechanism from C60 to the Fe atoms of Fe3O4. Electron transfer from C60 leaves an uncompensated spin on the molecule, and hence, induces magnetism, while at the same time it affects the spin- and orbital moments at the Fe sites of the magnetite substrate.
| Fe3O4(001) | Sub-ML C60/Fe3O4(001) | |
|---|---|---|
| m spin | 2.95 ± 0.21 | 2.62 ± 0.18 |
| m orb | 0.29 ± 0.03 | 0.32 ± 0.03 |
| m orb/mspin | 0.10 | 0.12 |
It should be pointed out that since the bonding between C60 and Fe3O4 is strictly a surface/interface effect, its quantitative influence on the magnetic moments of the magnetite film is not probed exclusively, due to the significant contribution of the bulk atoms to the XAS signal measured in TEY. Only very recently has it been demonstrated that the ferrimagnetic ordering in 1 nm thick Fe3O4(111) crystals grown on a Ru(0001) substrate can be preserved at RT,36 which could, in principle, provide a suitable template for studying the abovementioned effect. Nonetheless, our results well illustrate the non-negligible electronic interactions at the C60/Fe3O4(001) hybrid interface. We suspect that surface defects, such as terrace steps with a high density of broken or dangling bonds, can be one possible origin responsible for the rather strong adsorption strength of C60 on the ferrite surface. Detailed structural characterization, e.g., by atomic resolution scanning tunneling microscopy, will be a further step forward to the understanding of this new hybrid organic–inorganic system.
4 Conclusions
The electronic and magnetic interfacial properties of in situ prepared sub-ML C60/Fe3O4(001) on GaAs(001) have been investigated by XAS and XMCD measurements at the Fe L2,3- and C K-edges. The C K-edge XAS spectra show evidence of the electronic interaction between C60 π (π*) and Fe3O4 3d states, leading to a small, but observable induced magnetic moment of C60-derived interfacial electronic states. This is weak, however, compared to the case of C60/Fe(001). The Fe L-edge XMCD suggests that the sub-ML C60 is able to reduce the outer Fe atoms of the ferrite to Fe2+ by electron donation. Our results illustrate the crucial role of the interplay between the characteristics of inorganic surfaces (electronic structure, magnetic moments, and work function) and molecular properties in tuning the electronic and magnetic structure of hybrid organic–inorganic interfaces. These effects are expected to have a profound impact on interfacial spin-polarized charge transport in organic and molecular spintronic devices. The spin polarization of injected/extracted charges depends sensitively on the magnetic- and electronic structure of the interfaces, in particular the spin polarization of the hybrid states near the Fermi energy. It has been argued in ref. 8 that the strength of the molecule/substrate interaction may even inverse the sign of the spin polarization of the injected (or extracted) charges. It would be interesting to test such phenomena using devices that comprise a series of well-characterized hybrid interfaces, such as the C60/Fe3O4 interface described here, featuring different interaction strength between the molecules and the ferromagnetic contacts.Acknowledgements
This work is funded by the European Research Council (ERC Starting Grants no. 280020 and no. 240433) and the research program of the Foundation for Fundamental Research on Matter (FOM, grant no. 10PR2808), which is part of the Netherlands Organization for Scientific Research (NWO).References
- W. J. M. Naber, S. Faez and W. G. van der Wiel, J. Phys. D: Appl. Phys., 2007, 40, R205 CrossRef CAS.
- V. A. Dediu, L. E. Hueso, I. Bergenti and C. Taliani, Nat. Mater., 2009, 8, 707 CrossRef CAS.
- V. Dediu, M. Murgia, F. C. Matacotta and C. Taliani, Solid State Commun., 2002, 122, 181 CrossRef CAS.
- Z. H. Xiong, D. Wu, Z. V. Vardeny and J. Shi, Nature, 2004, 427, 821 CrossRef CAS.
- C. B. Harris, R. L. Schlupp and H. Schuch, Phys. Rev. Lett., 1973, 30, 1019 CrossRef CAS.
- V. I. Krinichnyi, S. D. Chemerisov and Y. S. Lebedev, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 16233 CrossRef CAS.
- T. S. Santos, J. S. Lee, P. Migdal, I. C. Lekshmi, B. Satpati and J. S. Moodera, Phys. Rev. Lett., 2007, 98, 016601 CrossRef CAS.
- C. Barraud, P. Seneor, R. Mattana, S. Fusil, K. Bouzehouane, C. Deranlot, P. Graziosi, L. Hueso, I. Bergenti, V. Dediu, F. Petroff and A. Fert, Nat. Phys., 2010, 6, 615–620 CrossRef CAS.
- V. Dediu, L. E. Hueso, I. Bergenti, A. Riminucci, F. Borgatti, P. Graziosi, C. Newby, F. Casoli, M. P. de Jong, C. Taliani and Y. Zhan, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 115203 CrossRef.
- S. Javaid, M. Bowen, S. Boukari, L. Joly, J.-B. Beaufrand, X. Chen, Y. J. Dappe, F. Scheurer, J.-P. Kappler, J. Arabski, W. Wulfhekel, M. Alouani and E. Beaurepaire, Phys. Rev. Lett., 2010, 105, 077201 CrossRef CAS.
- N. Atodiresei, J. Brede, P. Lazic, V. Caciuc, G. Hoffmann, R. Wiesendanger and S. Blugel, Phys. Rev. Lett., 2010, 105, 066601 CrossRef.
- L. Schulz, L. Nuccio, M. Willis, P. Desai, P. Shakya, T. Kreouzis, V. K. Malik, C. Bernhard, F. L. Pratt, N. A. Morley, A. Suter, G. J. Nieuwenhuys, T. Prokscha, E. Morenzoni, W. P. Gillin and A. J. Drew, Nat. Mater., 2011, 10, 39 CrossRef CAS.
- T. L. A. Tran, P. K. J. Wong, M. P. de Jong, W. G. van der Wiel, Y. Q. Zhan and M. Fahlman, Appl. Phys. Lett., 2011, 98, 222505 CrossRef.
- Y. X. Lu, J. S. Claydon, Y. B. Xu, S. M. Thompson, K. Wilson and G. van der Laan, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 233304 CrossRef; P. K. J. Wong, W. Zhang, X. G. Cui, Y. B. Xu, J. Wu, Z. K. Tao, X. Li, Z. L. Xie, R. Zhang and G. van der Laan, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 035419 CrossRef; W. Zhang, J. Z. Zhang, P. K. J. Wong, Z. C. Huang, L. Sun, J. L. Liao, Y. Zhai, Y. B. Xu and G. van der Laan, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 104451 CrossRef.
- Z. Zhang and S. Satpathy, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 13319 CrossRef CAS.
- Y. S. Dedkov, U. Rüdiger and G. Güntherodt, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 064417 CrossRef.
- Y. B. Xu, E. T. M. Kernohan, D. J. Freeland, A. Ercole, M. Tselepi and J. A. C. Bland, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 890 CrossRef CAS.
- A. J. Maxwell, P. A. Brühwiler, D. Arvanitis, J. Hasselström, M. K.-J. Johansson and N. Mårtensson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 7312 CrossRef CAS.
- B. Wästberg, S. Lunell, C. Enkvist, P. A. Brühwiler, A. J. Maxwell and N. Mårtensson, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 13031 CrossRef.
- M. Wesser, Y. Rehder, K. Horn, M. Sicot, M. Fonin and A. B. Preobrajenski, Appl. Phys. Lett., 2010, 96, 012504 CrossRef.
- S. Saito and A. Oshiyama, Phys. Rev. Lett., 1991, 66, 2637 CrossRef CAS.
- P. Morrall, F. Schedin, G. S. Case, M. F. Thomas, E. Dudzik, G. van der Laan and G. Thornton, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 214408 CrossRef.
- F. Schedin, E. W. Hill, G. van der Laan and G. Thornton, J. Appl. Phys., 2004, 96, 1165 CrossRef CAS.
- R. A. D. Pattrick, G. van der Laan, C. M. B. Henderson, P. Kuiper, E. Dudzik and D. J. Vaughan, Eur. J. Mineral., 2002, 14, 1095 CrossRef CAS.
- V. S. Coker, C. I. Pearce, C. Lang, G. van der Laan, R. A. D. Pattrick, N. D. Telling, D. Schüler, E. Arenholz and J. R. Lloyd, Eur. J. Mineral., 2007, 19, 707 CrossRef CAS.
- F. Bertram, C. Deiter, K. Pflaum, M. Suendorf, C. Otte and J. Wollschlager, J. Appl. Phys., 2011, 110, 102208 CrossRef.
- H.-J. Kim, J.-H. Park and E. Vescovo, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 15284 CrossRef CAS; F. M. F. de Groot, M. Grioni, J. C. Fuggle, J. Ghijsen, G. A. Sawatzky and H. Petersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 5715 CrossRef.
- B.-G. Park, J.-Y. Kim, J.-H. Park and H. Lee, J. Korean Phys. Soc., 2009, 54, 712 CrossRef CAS.
- B. T. Thole, P. Carra, F. Sette and G. van der Laan, Phys. Rev. Lett., 1992, 68, 1943 CrossRef CAS; P. Carra, B. T. Thole, M. Altarelli and X. D. Wang, Phys. Rev. Lett., 1993, 70, 694 CrossRef.
- C. T. Chen, Y. U. Idzerda, H.-J. Lin, N. V. Smith, G. Meigs, E. Chaban, G. H. Ho, E. Pellegrin and F. Sette, Phys. Rev. Lett., 1995, 75, 152 CrossRef CAS.
- D. J. Huang, C. F. Chang, H.-T. Jeng, G. Y. Guo, H.-J. Lin, W. B. Wu, H. C. Ku, A. Fujimori, Y. Takahashi and C. T. Chen, Phys. Rev. Lett., 2004, 93, 077204 CrossRef CAS.
- E. Goering, S. Gold, M. Lafkioti and G. Schütz, Europhys. Lett., 2006, 73, 97 CrossRef CAS.
- W. Osikowicz, M. P. de Jong and W. R. Salaneck, Adv. Mater., 2007, 19, 4213 CrossRef CAS.
- M. Fonin, R. Pentcheva, Y. S. Dedkov, M. Sperlich, D. V. Vyalikh, M. Scheffler, U. Rüdiger and G. Güntherodt, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 104436 CrossRef; A. A. Fursina, R. G. S. Sofin, I. V. Shvets and D. Natelson, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245112 CrossRef.
- J. Chen, D. J. Huang, A. Tanaka, C. F. Chang, S. C. Chung, W. B. Wu and C. T. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 085107 CrossRef.
- M. Monti, B. Santos, A. Mascaraque, O. R. de la Fuente, M. A. Nino, T. O. Mentes, A. Locatelli, K. F. McCarty, J. F. Marco and J. de la Figuera, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 020404R CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |