 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimensionally and compositionally controlled growth of calcium phosphate nanowires for bone tissue regeneration†
Philip James Thomas Reardonab, Albertus Denny Handokoa, Lin Lib, Jie Huangb and Junwang Tang*a
aDepartment of Chemical Engineering, UCL, London, WC1E 7JE, UK. E-mail: Junwang.tang@ucl.ac.uk
bDepartment of Mechanical Engineering, UCL, London, WC1E 7JE, UK. E-mail: Jie.huang@ucl.ac.uk
First published on 10th October 2013
Abstract
Nanostructured biomaterials with controlled morphology and composition are of high interest for bone tissue regeneration. As resorbable and biocompatible materials for bone tissue engineering, calcium phosphate nanowires and nanoneedles with different aspect ratios and compositions have been first synthesized without the use of any toxic surfactants via an energy efficient microwave assisted process. Correlation between solvent composition, mixing methodology and reagent stoichiometric ratios was investigated with the aim of producing orientated growth and varied biphasic composition, resulting in dimensionally controlled growth of materials containing varying hydroxyapatite (HA)/monetite quantities. It was observed that the HA/monetite content and dimensionality could be manipulated by changing the initial ethanol (EtOH) volume in the H2O/EtOH solvent mixture. Three dimensional particles with minute amounts of HA were produced when a H2O/EtOH volumetric ratio of 20/80 was used. Conversely, high aspect ratio (ca. 54) nanowires containing ca. 38 wt% HA were obtained with a 60/40 H2O/EtOH volumetric ratio. Importantly, the quantity of HA in the high aspect ratio nanowires/needles was controlled by varying the stoichiometric ratio of the reactants, demonstrating that one-dimensional materials with close to 100% HA can be achieved when the Ca/P ratio is increased to 1.67. Additionally, significant correlation between the extent of orientated growth of the materials and the point of EtOH addition during the mixing method was observed. The findings highlight that solvent composition, reactant stoichiometric ratio and mixing procedure can be used in tandem to tailor the morphology and composition of calcium phosphate materials, which are of very high importance in developing excellent materials suitable for bone tissue regeneration.
Introduction
An ever aging and expanding population have led to increasing cases of bone disease and fractures. The new generation of nanostructured biomaterials with controlled morphology and composition are of great interest in the development of bone tissue engineering, which is of high importance in meeting this increasing demand on the modern healthcare system.Dimensionality is an important factor in controlling nanomaterial properties. Specifically, one-dimensional materials have attracted intense interest due to their unique physical and chemical properties, including enhanced mechanical and biological functionality, which endow them with great potential for biomedical technologies. Generally, selectivity of nano/microparticle morphology is achieved through template-directed growth methods, using surfactants, ligands or solid membrane templates. The conventional solvothermal technique, for example, offers an effective route for the production of morphology controlled calcium phosphate materials in the presence of templates or surfactants.1–3 However, there is concern that using templating materials during conventional solvothermal synthesis may result in unwanted organic residues being incorporated into the final products.1,4,5 Furthermore, these processes require lengthy reaction times of many hours.1–5
Calcium phosphates are a group of inorganic materials that are of special interest as biomaterials due to their similar composition to that of mammalian bone tissue. Their unique biocompatibility has led them to be used for many biomedical applications, most notably bone tissue engineering and therapeutic delivery.6,7 The bioresorbability of calcium phosphate biomaterials is a key parameter for productive in situ tissue regeneration. Dicalcium phosphate anhydrous (DCPA), or monetite, is a bioresorbable member of this group of bioactive materials, thus possessing a higher aqueous solubility and a greater in vivo degradation rate when compared to other commonly used calcium phosphate phases.8 The potential of monetite as a biomaterial for bone tissue regeneration has recently been highlighted by a series of in vitro, animal and human studies.8,9 Hydroxyapatite (HA) is a highly bioactive calcium phosphate biomaterial possessing very low solubility that has been widely reported as a bone graft substitute and as an implant coating. Combining the more stable phase of HA with the more soluble phase monetite produces a biphasic calcium phosphate ceramic, a class of biomaterials that has received much attention recently due to the potential for control over bioresorbability afforded by these compounds,10 thus making a biphasic material of HA and monetite an ideal candidate for bone tissue regeneration.
Wire-like calcium phosphate materials are attractive for mechanical reinforcement in fabricating biocomposites due to their excellent mechanical properties.11 In polymer composites the diameter and aspect ratio of the calcium phosphate nanoparticles will determine the strengthening effect, with high aspect ratio nanoparticles being the ideal filler material.12,13 Moreover, osteoblast cells are sensitive to surface composition, energy and roughness.3 Therefore, there have been many reports on attempts to control the size, shape and composition of calcium phosphate nanomaterials.6,14 Recently, the synthesis of HA nanowires using hydrothermal processing without the use of morphology controlling agents was demonstrated by Costa et al.12 however, their system used a relatively lengthy sol–gel-hydrothermal combination methodology and had less control over the material phase. Microwave heating is characterised by rapid, volumetric and differential heating, allowing for better reaction control that has been shown to give altered morphologies,15 increased yields, greater purity, and considerably reduced synthesis times.16,17 The employment of microwave dielectric heating to the hydrothermal/solvothermal method is an emerging technology that offers many advantages for the synthesis of morphology and size controlled calcium phosphate materials in the absence of template or surfactant materials, as we reported recently for the synthesis of porous monetite nanoplates and nanorods.15
In this study, a facile microwave-assisted chemical method using a mixed solvent system was employed to prepare both phase and morphology-controlled single and biphasic calcium phosphates without the use of additional surfactants, in effect producing samples that can be directly utilised in vivo without pretreatment. The underlying mechanism was discussed based on the material characterisations.
Materials and methods
Synthesis
Material preparation followed a two step process. Initially two aqueous solutions of the same volume of Ca(NO3)2·4H2O (Sigma) and H3PO4 (Sigma) were prepared. Then, at room temperature and standard atmospheric pressure, the H3PO4 solution was added drop-wise to the Ca(NO3)2·4H2O solution, after which a predetermined volume of ethanol (EtOH) was added to the mixture. The total volume of the reaction solution was fixed at 60 ml, but the volumetric ratio between the EtOH and water was adjusted, as given in Table 1. This clear reaction mixture was then transferred to a Kevlar reinforced sealed PTFE container and irradiated in a microwave solvothermal process to 200 °C using a predetermined temperature ramp and time programme. The microwave accelerated reaction system used (MARS, CEM) automatically adjusts the output power in order to maintain a steady temperature and pressure profile. A ramping time of 25 minutes and a hold time of 60 minutes were used in all cases. The product suspension was then transferred to a centrifuge whereby the solid product was separated, washed several times with water and EtOH, and then dried in an oven at 60 °C overnight.| Sample code | Volume (cm−3) | Conditions | |||
|---|---|---|---|---|---|
| Water | EtOH | Ethanol added | Ca/P ratio | Notes | |
| W20 × 1080 | 12 | 48 | After mixing | 1.4 | |
| W40E60 | 24 | 36 | After mixing | 1.4 | |
| W60E40 | 36 | 24 | After mixing | 1.4 | |
| W80E20 | 48 | 12 | After mixing | 1.4 | |
| W80E20-B | 48 | 12 | Before mixing | 1.4 | |
| W80E20-1.67 | 48 | 12 | After mixing | 1.67 | Higher Ca/P ratio |
| W100E0 | 60 | 0 | After mixing | 1.4 | No solid Product |
Characterisation
The X-ray diffraction (XRD) patterns of samples were obtained using a Bruker D4 Advance Powder X-ray diffractometer with a CuKα (λ = 0.1541784 nm) radiation source. Diffraction patterns were collected from 10° to 60°, with a step size of 0.02° and 4 seconds acquisition time per step. XRD data analysis and compilation were carried out using the Bruker® TOPAS 3 platform. Due to the complex structure of monetite, an alternative two step refinement strategy was devised based on the whole powder pattern decomposition (WPPD) method,18 followed by a simplified Rietveld method,19 to account for preferred orientation on the (002) and to quantify the fraction of each phase. Lattice parameters obtained from previous WPPD refinement were taken as starting points for subsequent Rietveld refinement. Atomic parameters of monetite and hydroxyapatite were taken from ref. 20 and 21 respectively and were not refined. The modified Thompson-Cox-Hastings pseudo-Voigt function was used to model the peak shape throughout the refinements. Material size and shape were observed using a JEOL JSM-7401F field emission-scanning electron microscope operating at 3–10 kV. Materials were observed in powder form, and due to the low conductivity of the inorganic samples, they were coated with a thin layer of Au nanoparticles to improve the image quality.Results
Effect of H2O/EtOH volumetric ratio
For the synthesis of calcium phosphate nanowires and nanoneedles, a dual solvent system of EtOH and water was utilised. The initial search-match comparison of the XRD patterns of materials produced using varied EtOH/H2O ratios against a standard monetite reference pattern (JCPDS 70-1425) reveals the presence of crystalline monetite in all cases (Fig. 1), with the major peaks at 2θ = 13.1, 26.4, 30.2, and 40.0° corresponding to the monetite (001), (002), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 20), and (003) reflections, respectively. Importantly, materials W20E80, W40E60, W60E40, and W80E20 have a relative XRD intensity ratio between the peaks corresponding to the (002) and (20) (selected as neutral reference) diffraction planes of around 0.9, 3.8, 12.6 and 139, respectively, compared to 1.1 for the standard pattern. This trend of more profound (002) preferred orientation as the H2O/EtOH ratios increase is supported by the refinement results (see ESI, Table S1†), and suggests a preferential growth orientation with an increased expression of {00l}. This preferred orientation observation is also corroborated from the electron micrographs of the samples (Fig. 2), which show a transition from three dimensional to one dimensional growth with an increase in the H2O/EtOH ratio, leading to the formation of nanowires (W80E20) or nanoneedles (W60E40).
20), and (003) reflections, respectively. Importantly, materials W20E80, W40E60, W60E40, and W80E20 have a relative XRD intensity ratio between the peaks corresponding to the (002) and (20) (selected as neutral reference) diffraction planes of around 0.9, 3.8, 12.6 and 139, respectively, compared to 1.1 for the standard pattern. This trend of more profound (002) preferred orientation as the H2O/EtOH ratios increase is supported by the refinement results (see ESI, Table S1†), and suggests a preferential growth orientation with an increased expression of {00l}. This preferred orientation observation is also corroborated from the electron micrographs of the samples (Fig. 2), which show a transition from three dimensional to one dimensional growth with an increase in the H2O/EtOH ratio, leading to the formation of nanowires (W80E20) or nanoneedles (W60E40).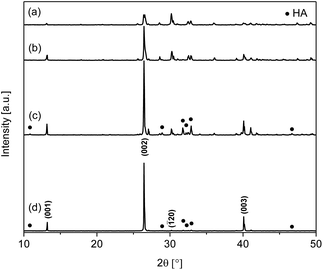 | ||
| Fig. 1 XRD patterns of materials synthesised using different H2O/EtOH volumetric ratios: (a) W20E80, (b) W40E60, (c) W60E40, and (d) W80E20. | ||
 | ||
| Fig. 2 SEM micrographs of materials produced with different H2O/EtOH volumetric ratios: (a) W20E80, (b) W40E60, (c) W60E40, and (d) W80E20. | ||
Table 2 shows the phase composition of the materials obtained by XRD data refinement when the volumetric ratio of solvents H2O/EtOH is varied. W20E80 contains almost entirely monetite with 2.4 wt% HA. In contrast, W60E40 and W80E20 show a significant amount of HA (JCPDS 09-432) in conjunction with monetite, calculated to be 37.6 and 28.0%, respectively (Table 2). It is apparent that the amount of monetite is reduced with an increase in H2O volume in the solvent composition. These results demonstrate the potential for control of calcium phosphate material composition using EtOH as a co-solvent.
SEM micrographs further reveal the changes in material morphology using different H2O/EtOH solvent ratios (Fig. 2). It can be seen that W20E80 (Fig. 2a) is a mix of various polyhedra of dimensions ca. 811 nm ± 317 nm × 593 ± 181 nm, resulting from three dimensional growth. Material W40E60 displays a plate and cube-like morphology with larger dimensions ca. 10.2 μm ± 5.4 μm × 4.4 μm ± 2.3 μm (Fig. 2b) representing a mixture of two and three dimensional growth. The wire/needle-like morphology displayed by-products W60E40 and W80E20 (Fig. 2c and d) implies that a highly directionally selective growth regime was realised with reduced EtOH volume in the solvent composition. W60E40 (Fig. 2c) has a wire or needle-like morphology of dimensions ca. 4.4 ± 2.4 μm × 231 ± 142 nm (aspect ratio ca. 19); W80E20 has a nanowire morphology of dimensions ca. 4.9 ± 1.5 μm × 83 ± 26 nm (aspect ratio ca. 54). The higher aspect ratio for the nanowire material W80E20 compared to W60E40 correlates with a reduction in three dimensional growth towards increasing one dimensionality with a reduction in EtOH volume. It is therefore observed that a reduction in EtOH volume facilitates an increase in one dimensional preferred growth orientation and reduced multidimensional particle growth.
Effect of changing the point of EtOH addition in the mixing procedure
To ascertain the effect of the presence of EtOH during the solution mixing process, the nanowire experiment (W80E20) was repeated with variation to the mixing procedure. In this different methodology instead of adding the EtOH after mixing of the H3PO4 and Ca(NO3)2·4H2O aqueous solution, EtOH was equally added to the individual aqueous precursor solutions before mixing (this material was assigned the code W80E20-B). The XRD pattern (Fig. 3) of the W80E20-B material shows the presence of both HA and monetite phases. The proportion of monetite in W80E20-B was calculated to be close (72%) to that for W80E20 (the low relative intensity of peaks corresponding to HA is due to the large degree of growth orientation with an increased expression of {00l} for monetite in W80E20), further implying that EtOH favours the production of monetite in comparison to HA. Interestingly, a 46% reduction in (002) preferred orientation was observed for W80E20-B compared to W80E20 (see ESI, Table S1†) suggesting a relative reduction in the expression of {001} (Fig. 3b). This observation is in agreement with the SEM study of this material (Fig. 4). SEM micrographs of W80E20 and W80E20-B materials show that W80E20-B is also the product of highly 1D growth displaying a needle/wire morphology (Fig. 4b) and dimensions ca. 5.4 ± 2.5 μm × 252 ± 21 nm. However, the increase in particle width and reduced aspect ratio (ca. 22) for W80E20-B (Fig. 4b) compared to W80E20 (aspect ratio of ca. 54, see Fig. 4a) suggests an increase in 3D growth when EtOH is added before reagent solution mixing.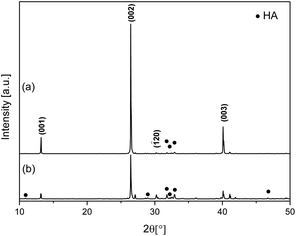 | ||
| Fig. 3 XRD Patterns of calcium phosphate materials with EtOH added to the reaction mixture: (a) after mixing of aqueous reagent solutions (W80E20) and (b) before mixing of reagent solutions (W80E20-B). | ||
 | ||
| Fig. 4 SEM micrographs of calcium phosphate materials with EtOH added to the reaction mixture: (a) after mixing of aqueous reagent solutions (W80E20) and (b) before mixing of reagent solutions (W80E20-B). | ||
Effect of Ca/P molar ratio
One dimensional calcium phosphate materials were also synthesised using different Ca/P molar ratios in the precursor mixture, with the composition of the reaction solvent consistent with that of W80E20. The XRD patterns of materials obtained with Ca/P ratios of 1.4 (W80E20) and 1.67 (this material was assigned the code W80E20–1.67) are shown in Fig. 5. A ratio of 1.4 was selected as it lies midway between the theoretical Ca/P molar ratio of monetite and that of HA in order to target a biphasic mixture, and 1.67 as it is the stoichiometric ratio for pure HA. Both products obtained with different Ca/P ratios contained crystalline monetite (JCPDS 70-1425) and HA (JCPDS 9-432) phases (Fig. 5a and b). However, the composition of W80E20-1.67 was calculated to contain a significantly larger proportion of HA (91.5%) compared to W80E20 (28.0%). This result fits with what would be expected for these materials due to HA having a higher Ca/P molar ratio of 1.67 compared to 1 for monetite, and indicates that the composition of these biphasic 1D calcium phosphate materials can be controlled by varying the Ca/P ratio of the starting materials. SEM micrographs of the products obtained by varying the Ca/P stoichiometric ratios of the starting materials (Fig. 6) show that this parameter also has an effect on the morphology of the resultant material. The resultant product obtained by using a Ca/P ratio of 1.67 had a wire/needle morphology but a smaller aspect ratio (ca. 30) with dimensions of ca. 4.6 ± 2.4 μm × 154 ± 93 nm compared to when a Ca/P ratio of 1.4 was used (aspect ratio ca. 54), correlating with a small reduction (9%) in (002) preferred orientation (see ESI, Table S1†).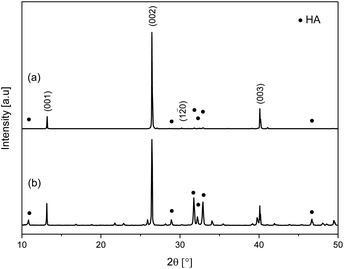 | ||
| Fig. 5 XRD patterns of products obtained using Ca/P ratios of (a) 1.4 (W80E20) and (b) 1.67 (W80E20-1.67). | ||
 | ||
| Fig. 6 SEM micrographs of products obtained using Ca/P ratios of (a) 1.4 and (b) 1.67. | ||
Discussion
The XRD data showed that the formation of HA increased with an increase in H2O volume fraction, correlating with HA being the most stable phase in H2O. However, no solid product was formed when 100% H2O was used (W100E0), implying that under purely aqueous conditions both monetite and HA have a very high solubility; this is expected given the relatively low reaction pH used (ca. 2.5). The formation of HA was observed only in the presence of ethanol, which corroborates reports that dilution with EtOH can decrease the solubility of HA and induce the formation of apatites in mixed H2O/EtOH systems.22 Conversely, at high fractions of ethanol, monetite was mainly observed accompanied by a decreasing HA formation rate, resulting in the formation of a pure crystalline monetite phase when EtOH was solely used (reported by us previously).15 The increased rate of monetite formation when large EtOH volumes are used could be explained by the reduced solubility of this phase in non-aqueous conditions.The size and aspect ratio of the materials produced were also highly dependent on the quantity of EtOH used as a co-solvent, Ca/P stoichiometric ratio of reactants, and point of addition of EtOH in the reagent mixing procedure. Generally the shape of inorganic nanoparticles is controlled by the presence of polymers, surfactants or chelating agents. In the present system rapid energy input and high-pressure conditions fostered through microwave heating produced crystalline nuclei that grew to form calcium phosphate nanoneedles and nanowires when high H2O/EtOH solvent volumetric ratios were used. Peng et al. introduced a shape evolution growth mechanism for CdSe nanocrystals, which showed that diffusion-controlled 1D growth can occur under conditions of a higher chemical potential of monomer (precursor species) in the solution to that of the unique surface of the nanoparticles.23,24 A recent study by Costa et al. reported one dimensional hydrothermal growth of HA nanowires, in which a similar diffusion-controlled mechanism was used to describe the shape evolution process of HA crystals with varying chemical potential of the solution.12 These arguments suggest that a high chemical-potential environment and an anisotropic crystal structure are key to orientated growth.
In our system, the observed morphology evolution could also be explained by this theory (Scheme 1). Here, the strong EtOH volume relation with the length and aspect ratio of the products is due to the sensitive influence of the EtOH/H2O ratio under microwave heating conditions on the concentration of the solute species ([Ca2+] and [HPO42−]) in the crystal growth phase. The concentration of solute species remaining in solution after nucleation is governed by the number of nuclei formed, with a relatively small number of nuclei desirable for elongated growth. Lerner et al. suggested that ethanol can cause weakening of the bonds in the water structure and disrupt the interaction of water molecules with calcium cations, lowering the energy barrier to deaquation and hence facilitating nucleation of the most stable calcium phosphate species.25 Also, the interaction between EtOH and microwave radiation (dielectric loss) is stronger than that between water and microwave radiation; the addition of EtOH is thus known to increase the dielectric loss (heating effect) of an aqueous medium and to induce the fast formation of calcium phosphate nuclei.
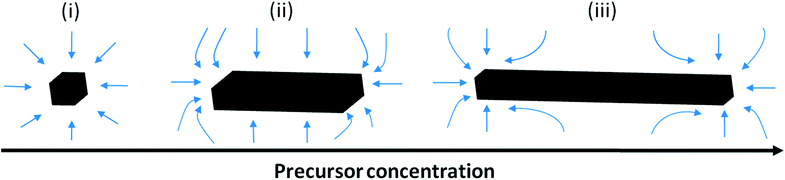 | ||
| Scheme 1 Illustration of the diffusion-controlled crystal growth process, which is governed by the precursor concentration in the bulk resulting in either 1D or 3D growth of calcium phosphate. Small blue arrows represent precursor flux. | ||
Therefore, it is proposed that this increase in nucleation rate due to EtOH addition can explain the 3D growth that is more obvious when larger EtOH volume fractions are used (W20E80 and W40E60), as the large number of nuclei produced will lead to a low precursor (solute) concentration and chemical potential in the growth stage, leading to three dimensional growth (Scheme 1(i)). Inversely, when small EtOH fractions are used (W60E40 and W80E20), the nucleation rate will be lower, leading to a reduction in the number of nuclei formed, and an associated increase in solute concentration and chemical potential in the growth stage, leading to one dimensional growth (Scheme 1(iii)).
The results suggest that both ethanol and microwave play a critical role in controlling the morphology of calcium phosphate materials. Therefore, it is hypothesised that if the point of ethanol addition is changed from after mixing of the precursor solutions to before mixing of the precursor solutions (material W80E20-B), this deaquation process would be enhanced; hence when nucleation takes place as the mixture is treated under microwave assisted solvothermal conditions, the nucleation rate will be slightly higher, thus causing a reduction in chemical potential of the growth environment leading to increased 3D growth, and a reduction in the product aspect ratio (Scheme 1(ii)). In the case where a higher Ca/P stoichiometric ratio of reagents is used, the increased presence of excess free Ca2+ would shift the chemical equilibrium leading to increased nucleation. This could explain why the nanofibres had a reduced aspect ratio when an elevated Ca/P ratio was used (Scheme 1(ii)).
This mechanism suggests that 1D or 3D growth stages may be captured by maintaining the chemical potential in the correct range (Scheme 1) through addition of different volumes of co-solvent, changing the mixing procedure, or varying the stoichiometric ratio of the precursor species, which is in agreement with the previous observations by Peng et al.23
It is believed that 1D growth can occur only if the chemical potential of monomers (precursor species) in the solution is much higher than the highest chemical potential of the atoms on the surface of the nanoparticles.23,24 Costa et al. reported the 1D growth of HA nanowires due to the chemical potential of a (precursor) amorphous calcium phosphate (ACP) solution having significantly greater chemical potential than that of the (001) associated facet (which is reported as possessing greater interfacial free energy than the other faces), which in turn caused migration of precursor to this face and hence growth perpendicular to it along the c-axis.12 In the crystal structure of monetite, the lattice contains distorted Ca-PO4 chains aligned parallel to the b-axis forming a sheet of atoms that are roughly in the (001) plane,26 which are similar to those parallel to the c-axis in HA.27 A preferential growth model of the calcium phosphate nanowires/nanorods along the [001] direction for monetite is in good agreement with the abnormally strong intensity of the monetite {001} associated peaks observed in the XRD patterns of these materials (Fig. 1). This structural feature would make the {001} very sensitive to surrounding growth conditions.
In some cases the autogenous pressure and the energy input by a hydrothermal process was found to activate the reactions and induce anisotropic growth of nanorods.28 Microwave assisted solvothermal methodology is associated with rapid and uniform increases in temperature and pressure, which has been shown to accelerate crystal growth in comparison to conventional heating.29 In addition, microwave assisted synthesis conditions will accelerate Brownian motion, giving faster effective circulation of solute in solution.
Therefore, it can be concluded that the anisotropic growth of calcium phosphate nanowires and nanoneedles in this case (without surfactants) is governed by the intrinsic chemistry of specific faces, the local solution details, and the mode of foreign energy activation (microwave assisted solvothermal heating). Based on the controllable synthesis of calcium phosphate fibres, work on films of orientated calcium phosphate fibres is underway.
Conclusions
Calcium phosphate 1D and 3D materials with varying compositions have been successfully synthesised using a rapid and facile microwave assisted method without the use of any surfactants. Calcium phosphate materials were morphologically controlled by changing the relative quantities of EtOH used as a co-solvent. Nanowire calcium phosphate materials synthesised using a H2O/EtOH volumetric ratio of 80/20 exhibited the highest aspect ratio of ca. 54 and had a biphasic composition of HA and monetite (72.0% monetite, 28.0% HA). The aspect ratio of the nanowires/nanoneedles can also be fine tuned by either changing the Ca/P stoichiometric ratio of the reactants or by changing the point of EtOH addition in the reactant mixing procedure. Varying the Ca/P reactant stoichiometry to a higher value of 1.67 appeared to increase the relative quantity of HA (up to 91.5%) in these 1D biphasic calcium phosphate materials, implying that the composition of the nanowires/nanoneedles can be controlled using this variable. Therefore, this approach could enable the production of morphology and compositionally controlled calcium phosphate 1D and 3D materials, with the potential for tuneable mechanical and degradation properties when used in composite materials for bone tissue engineering.Acknowledgements
The authors acknowledge the financial support of the EPSRC.Notes and references
- J. D. Chen, Y. J. Wang, K. Wei, S. H. Zhang and X. T. Shi, Biomaterials, 2007, 28, 2275–2280 CrossRef CAS PubMed.
- M. Cao, Y. Wang, C. Guo, Y. Qi and C. Hu, Langmuir, 2004, 20, 4784–4786 CrossRef CAS.
- K. Wei, C. Lai and Y. Wang, J. Mater. Sci., 2007, 42, 5340–5346 CrossRef CAS.
- K. Wei, C. Lai and Y. Wang, J. Macromol. Sci., Part A: Pure Appl.Chem., 2006, 43, 1531–1540 CrossRef CAS.
- C. Zhang, J. Yang, Z. Quan, P. Yang, C. Li, Z. Hou and J. Lin, Cryst. Growth Des., 2009, 9, 2725–2733 CAS.
- S. V. Dorozhkin, Acta Biomater., 2010, 6, 715 CrossRef CAS PubMed.
- V. Uskoković and D. P. Uskoković, J. Biomed. Mater. Res., Part B, 2011, 96, 152–191 CrossRef PubMed.
- F. Tamimi, J. Torres, D. Bassett, J. Barralet and E. L. Cabarcos, Biomaterials, 2010, 31, 2762–2769 CrossRef CAS PubMed.
- U. Gbureck, T. Hölzel, U. Klammert, K. Würzler, F. A. Müller and J. E. Barralet, Adv. Funct. Mater., 2007, 17, 3940–3945 CrossRef CAS.
- G. Daculsi, Biomaterials, 1998, 19, 1473–1478 CrossRef CAS.
- K. Lin, X. Liu, J. Chang and Y. Zhu, Nanoscale, 2011, 3, 3052–3055 RSC.
- D. O. Costa, S. J. Dixon and A. S. Rizkalla, ACS Appl. Mater. Interfaces, 2012, 4, 1490–1499 CAS.
- T.-Y. Liu, S.-Y. Chen and D.-M. Liu, J. Biomed. Mater. Res., Part B, 2004, 71, 116–122 Search PubMed.
- W. Suchanek, J. Mater. Res., 1998, 13, 94 CrossRef CAS.
- P. J. T. Reardon, J. Huang and J. Tang, Adv. Healthcare Mater., 2013, 2, 682–686 CrossRef CAS PubMed.
- S. Caddick and R. Fitzmaurice, Tetrahedron, 2009, 65, 3325–3355 CrossRef CAS PubMed.
- M. Baghbanzadeh, L. Carbone, P. D. Cozzoli and C. O. Kappe, Angew. Chem., Int. Ed., 2011, 50, 11312–11359 CrossRef CAS PubMed.
- H. Toraya, Rigaku J., 1989, 6, 28–34 Search PubMed.
- H. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- B. Dickens, J. S. Bowen and W. E. Brown, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 797–806 CrossRef CAS.
- A. S. Posner, A. Perloff and A. F. Diorio, Acta Crystallogr., 1958, 11, 308–309 CrossRef CAS.
- M. Larsen, A. Thorsen and S. Jensen, Calcif. Tissue Int., 1985, 37, 189–193 CrossRef CAS.
- X. Peng, Adv. Mater., 2003, 15, 459 CrossRef CAS.
- Z. A. Peng and X. G. Peng, J. Am. Chem. Soc., 2001, 123, 1389–1395 CrossRef CAS.
- E. Lerner, R. Azoury and S. Sarig, J. Cryst. Growth, 1989, 97, 725–730 CrossRef CAS.
- Z. Zou, X. Liu, L. Chen, K. Lin and J. Chang, J. Mater. Chem., 2012, 22, 22637–22641 RSC.
- W. E. Brown, J. P. Smith, J. R. Lehr and A. W. Frazier, Nature, 1962, 196, 1050–1055 CrossRef CAS.
- S.-H. Yu, Y.-S. Wu, J. Yang, Z.-H. Han, Y. Xie, Y.-T. Qian and X.-M. Liu, Chem. Mater., 1998, 10, 2309–2312 CrossRef CAS.
- S. H. Jhung, T. Jin, Y. K. Hwang and J.-S. Chang, Chem.–Eur. J., 2007, 13, 4410–4417 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Refinement details of all samples, including cell parameters and preferred orientation intensity correction factor. See DOI: 10.1039/c3tb21073a |
| This journal is © The Royal Society of Chemistry 2013 |