 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of novel membranes for blood purification therapies based on copolymers of N-vinylpyrrolidone and n-butylmethacrylate
M. Tijinka, J. Janssenb, M. Timmera, J. Austenc, Y. Aldenhoffb, J. Koomand, L. Kooleef, J. Damoiseauxc, R. van Oerleg, Y. Henskensh and D. Stamatialis*a
aInstitute for biomedical technology and technical medicine MIRA, Biomaterials Science and Technology, University of Twente, Faculty of Science and Technology, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: d.stamatialis@utwente.nl; Fax: +31 53 4892155; Tel: +31 53 4894675
bInterface BIOmaterials BV, Burg. Lemmensstraat 364, 6163 JT Geleen, The Netherlands
cDepartment of Internal Medicine, Immunology laboratory, Maastricht University Medical Center, PO Box 5800, 6202 AZ Maastricht, The Netherlands
dDepartment of Internal Medicine, Division of Nephrology, Maastricht University Medical Center, PO Box 5800, 6202 AZ Maastricht, The Netherlands
eDepartment of Biomedical Engineering/Biomaterials Science, Maastricht University, Faculty of Health, Medicine & Life Sciences, Maastricht, The Netherlands
fUniversity of Malaya, Department of Biomedical Engineering, Kuala Lumpur, Malaysia
gDepartment of Internal Medicine, Clin. Thrombosis & Hemostasis laboratory, Maastricht University Medical Center, PO Box 5800, 6202 AZ Maastricht, The Netherlands
hDepartment of Internal Medicine, Central Diagnostic Laboratory, Maastricht University Medical Center, PO Box 5800, 6202 AZ Maastricht, The Netherlands
First published on 2nd September 2013
Abstract
Developments in membrane based blood purification therapies often come with longer treatment times and therefore longer blood–material contact, which requires long-term membrane biocompatibility. In this study, we develop for the first time membranes for blood purification using the material SlipSkin™, which is a copolymer, made from N-vinylpyrrolidone (NVP) and butylmethacrylate (BMA). Specific attention is focused on understanding the mechanism of pore formation and the tailoring of the membrane mechanical and transport properties to obtain the optimal membrane for blood purification therapies. Polymer composition, solvent type and solvent evaporation time influence membrane morphology and membranes with sieving properties of cascade filters in plasma fractionation applications are developed. The new membranes have very good blood compatibility properties; in fact compared to benchmark flat membranes currently used in the clinic, they have lower platelet adhesion while all other properties (contact activation, thrombogenicity, leukocyte adhesion, hemolysis and complement activation) are also very good and comparable to the benchmarks.
1. Introduction
In blood purification treatments, membranes with optimal separation properties as well as optimal biocompatibility are required. For example, apheresis membranes, plasma separation membranes and hemodialysis membranes have optimized molecular weight cut offs for their applications and show good biocompatibility for a few hours of application. New developments in blood purification therapies often require longer durations. For example, nocturnal dialysis (where 7.85 hour treatment instead of 3.75 h for conventional hemodialysis is applied) leads to improvements in patient survival.1,2 However, longer filtration times mean more contact with the membrane material, requiring long-term membrane biocompatibility. This holds for wearable and portable artificial kidneys too. Davenport et al. reported about a pilot study with a wearable artificial hemodialysis device, however, 2 out of 8 patients had clotting problems and the treatment was discontinued, highlighting the need for improved biocompatible membranes.3Currently, blood purification membranes made of synthetic polymers dominate the market. Often polymer blends are used in which a hydrophobic membrane material (e.g. polysulfone or polyethersulfone) is combined with a hydrophilic additive (e.g. polyvinylpyrrolidone (PVP) or polyethylene glycol (PEG)). Also hydrophilic or hydrophilized copolymers are used (poly(ethylene vinyl alcohol) or polyacrylonitrile methallylsulfonate).4 Membranes with hydrophilic patches in a hydrophobic membrane matrix are generally thought to be biocompatible.5 However, the hydrophilic modifier can elute from the dialysis membrane surface into the patients' circulation.6 Another approach is the use of surface modifications such as heparin immobilization onto polysulfone membranes7 or albumin grafting onto polyethersulfone based membranes.8 However, in most of these studies the biocompatibility of blood purification membranes is not fully investigated. Although the effect of biocompatibility of different membrane materials is hard to show in clinical trials,9 both nurses and physicians prioritize biocompatibility for future innovations on blood purification membranes above efficiency and removal capacity.10
In this study, we develop via phase inversion membranes suitable for blood purification therapies using the SlipSkin™ biomaterial which is a copolymer of N-vinylpyrrolidone (NVP) (hydrophilic building block) and n-butylmethacrylate (BMA) (hydrophobic building block). This is actually the most common method for preparation of membranes for blood purification.11 In general, due to the combination of hydrophilic and hydrophobic properties in one material, copolymers may offer favorable and often tunable biocompatibility and cytocompatibility properties. However, at the same time this combination often imposes challenges in the fabrication of porous membranes with such copolymers. As a result very few studies discuss in detail the science and technology for their optimization for specific applications. In this work, we pay specific attention to this and aim to understand the mechanism of pore formation and the tailoring of the membrane's mechanical and transport properties to obtain the optimal membrane for blood purification therapies. We use various polymer compositions in which the molar ratio of the hydrophilic and hydrophobic building blocks is varied and prepare under various process conditions (tailoring solvent type and solvent evaporation time) porous flat sheet membranes suitable for plasma fractionation. Although SlipSkin has been used before as a surface coating for several blood-contacting medical devices (catheters and guide wires) and its cytocompatibility and hemocompatibility have been documented,12–14 the biocompatibility of SlipSkin™ porous membranes has not been studied yet. In our opinion, this study is absolutely necessary since it is well known that the membrane biocompatibility can be seriously influenced by the membrane processing conditions, surface roughness, pore and surface morphology, even if the membrane forming material has good biocompatibility. Therefore here, we also thoroughly investigate the biocompatibility of the new membranes based on the ISO guidelines 10993: “Biological evaluation of medical devices, part 4, selection of tests for interactions with blood”. Following the advice of these guidelines, for external communication devices like hemodialysis equipment, we perform tests in all 5-test categories of thrombosis, coagulation, platelets, hematology and complement system. In all cases, we compare the performance of the newly developed membranes with several benchmark flat membranes.
2. Materials and methods
2.1. Materials
SlipSkin™, a copolymer of N-vinylpyrrolidone (NVP, hydrophilic) and n-butylmethacrylate (BMA, hydrophobic) (Fig. 1), was used as the membrane material and was kindly provided by INterface BIOmaterials BV (Geleen, The Netherlands). Polyethersulfone (PES) (ULTRASON, E6020P) (BASF, Arnhem, The Netherlands) and polyvinylpyrrolidone (PVP) (K90, MW ≈ 360![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) (Fluka, Sigma-Aldrich, Germany) were used as membrane material and additive, respectively. Acetone (Biosolve, Valkenswaard, The Netherlands) and extra pure N-methylpyrrolidone (NMP) (Acros organics, Geel, Belgium) were employed as solvents. Ultrapure water, prepared using a Millipore purification unit, was used as a non-solvent in the coagulation baths.
000) (Fluka, Sigma-Aldrich, Germany) were used as membrane material and additive, respectively. Acetone (Biosolve, Valkenswaard, The Netherlands) and extra pure N-methylpyrrolidone (NMP) (Acros organics, Geel, Belgium) were employed as solvents. Ultrapure water, prepared using a Millipore purification unit, was used as a non-solvent in the coagulation baths. | ||
| Fig. 1 Chemical formulae of (a) N-vinylpyrrolidone (NVP) and (b) n-butylmethacrylate (BMA). | ||
Creatinine (113.2 Da), myoglobin (17.8 kDa), bovine serum albumin (BSA) (66 kDa), γ-globulin (150 kDa) and thyroglobulin (670 kDa) were purchased from Sigma-Aldrich and dissolved in ultrapure water.
For biocompatibility tests, we selected several benchmark membranes for comparison with our SlipSkin membranes. PES/PVP blends are often used by the industry for the preparation of blood purification membranes.4,15 Commercial hemodialysis or plasma fractionation membranes are always in hollow fiber configuration, but for our hemocompatibility testing, flat sheet membranes were necessary. Therefore we prepared flat sheet (home-made) membranes, based on a PES/PVP polymer blend. Besides, commercial flat sheet PES based membranes (14650-47-D, molecular weight cut off = 50000 Da, Sartorius, Nieuwegein, The Netherlands) were selected. A flat sheet polyester membrane, which is in clinical use for blood transfusion applications, was selected as well and was taken out of blood transfusion filters SQ40SE (PALL, Portsmouth, UK). For complement activation experiments, two more benchmark materials, regenerated cellulose (RC58, Whatman, Sigma-Aldrich) and DEAE cellulose (DE81 DEAE cellulose, Whatman, Sigma-Aldrich), were used as positive control as described before.16,17 Glass coverslips were obtained from VWR (Amsterdam, The Netherlands).
The following reagents for a thrombin generation assay were purchased from Thrombinoscope BV (Maastricht, The Netherlands). FluCa reagent contains 2.5 mM fluorogenic substrate for thrombin (Z-Gly-Gly-Arg-AMC) and 87 mM calcium chloride. The MP reagent contains 24 μM phospholipids at 20:20:60 mol% PS:PE:PC. The thrombin calibrator contains a fixed amount of thrombin-α2-macroglobulin complex. For the second thrombin generation assay, a Z-Gly-Gly-Arg-AMC fluorogenic substrate was purchased from Bachem (Bubendorf, Switzerland), dimethyl sulfoxide was purchased from Acros Organics (Geel, Belgium), and calcium chloride was purchased from Merck (Darmstadt, Germany). For the platelet adhesion assay triton X100 was purchased from Acros Organics and CytoTox 96® non-radioactive cytotoxicity kit was purchased from Promega Madison (USA). Platelets were fixed with glutaraldehyde from Sigma-Aldrich (Zwijndrecht, The Netherlands).
2.2. Membrane preparation
SlipSkin™ membranes were prepared from 10 wt% SlipSkin™ in acetone or NMP polymer dopes. Different types of SlipSkin™ co-polymers were used with NVP:BMA molar ratios of 30:70, 50:50 and 70:30. Home-made PES membranes were prepared from 15 wt% PES and 7 wt% PVP in NMP.18 The solutions were mixed on a roller bank, and afterwards degassed overnight at room temperature. All membranes were prepared by phase inversion. Solutions were cast onto a glass plate using a casting knife with a slit of 300 μm for the SlipSkin™ membranes and a 150 μm slit for the homemade PES membranes. Casting was immediately followed by immersion in the coagulation bath unless otherwise stated. Coagulation baths of ultrapure water and 60 wt% NMP and 40% ultrapure water were employed for the SlipSkin™ and homemade PES membranes, respectively. In some cases, a solvent evaporation step of 30 seconds, 5 or 10 minutes was performed after which the cast films were immersed into the coagulation bath. After the membrane formation process, the membranes were rinsed and stored in ultrapure water for further use.All membranes with accompanying codes and details are described in Table 1. SlipSkin™ membranes are indicated with M, and the first number in this code indicates the molar ratio of NVP:BMA building blocks in the polymer composition. When NMP was used as solvent for SlipSkin™, it was also indicated in the code while for the other SlipSkin™ membranes, acetone was used as solvent. Furthermore, the evaporation step was indicated as ‘ev’ in the code for a solvent evaporation time of 30 seconds, while the solvent evaporation times of 5 and 10 minutes were indicated in the code as ev5 and ev10, respectively.
| Code | Manufacturer | Material | NVP:BMA | Solvent | Solvent evaporation time |
|---|---|---|---|---|---|
| M30NMP | Homemade | SlipSkin™ | 30:70 | NMP | 0 |
| M50NMP | Homemade | SlipSkin™ | 50:50 | NMP | 0 |
| M70NMP | Homemade | SlipSkin™ | 70:30 | NMP | 0 |
| M30 | Homemade | SlipSkin™ | 30:70 | Acetone | 0 |
| M50 | Homemade | SlipSkin™ | 50:50 | Acetone | 0 |
| M70 | Homemade | SlipSkin™ | 70:30 | Acetone | 0 |
| M50ev | Homemade | SlipSkin™ | 50:50 | Acetone | 30 seconds |
| M50ev5 | Homemade | SlipSkin™ | 50:50 | Acetone | 5 minutes |
| M50ev10 | Homemade | SlipSkin™ | 50:50 | Acetone | 10 minutes |
| G | VWR | Glass | — | — | — |
| PESh | Homemade | PES/PVP | — | — | — |
| PESc | Sartorius | PES | — | — | — |
| PE | PALL | Polyester | — | — | — |
| RC | Whatman | Regenerated cellulose | — | — | — |
| DC | Whatman | DEAE cellulose | — | — | — |
2.3. Membrane characterization
where J represents the clean water flux expressed in L m−2 h−1, ΔP is the transmembrane pressure difference in bar and Lp is the clean water permeance in L m−2 h−1 bar−1.

The membrane sieving coefficient (SC) was determined using an air pressurized dead end Amicon ultrafiltration cell. The following compounds were dissolved in ultrapure water with initial concentrations of 0.1 mg mL−1 creatinine, 0.25 mg mL−1 myoglobin, 1 mg mL−1 BSA, 0.5 mg mL−1 γ-globulin and 1 mg mL−1 thyroglobulin and pressurized through the membrane (active surface area: 12.6 cm2) at 0.5 bar for 30 minutes. Then the concentrations of the feed and permeate compartments were analyzed using a photospectrometer (Varian, Cary 300 Scan UV-visible spectrophotometer) at 230, 409, 278, 278, and 280 nm for creatinine, myoglobin, BSA, γ-globulin and thyroglobulin, respectively. The SC was calculated as follows:

2.4. Biocompatibility tests
Platelet poor plasma (PPP) was prepared by a centrifugation step at 2000 × g for 5 minutes and a second centrifugation step at 10000 × g for 10 minutes. PPP was frozen at −80 °C until further use. To investigate contact activation, circular membrane pieces (diameter = 6 mm) and glass coverslips for 5 repeats were put on the bottom of a flat bottom 96 well plate in ultrapure water. Also empty wells without membranes were used to incubate PPP as blank. Before addition of 120 μL PPP, the ultrapure water was aspirated. The PPP was incubated at room temperature under gentle shaking conditions for 15 minutes while no calcium ions or phospholipids were present. Then 80 μL of the incubated PPP was aspirated and transferred to round bottom 96 well plates. Also a control of plasma, which was not incubated, and a pooled normal plasma internal control were taken into account. 20 μL MP reagent was added to all wells and the plate was pre-warmed at 37 °C for 10 minutes. The calibrated automated thrombogram (CAT) assay was performed as previously described.19 Thrombin generation was started by addition of 20 μL FluCa reagent. The calibration was performed with a mixture of 80 μL plasma and 20 μL of the thrombin calibrator and 20 μL FluCa reagent. Fluorescence of the thrombin split fluorogenic substrate was read in a Fluoroskan Ascent reader (Thermo Labsystems OY, Helsinki, Finland) equipped with a 390/460 filter set. Thrombin generation curves were generated with the Thrombinoscope software (Thrombinoscope BV, Maastricht, The Netherlands). Also the data analysis was performed using this software. The lag time and peak height were calculated from each separate curve.
For the second thrombin generation test, the bottom of the wells of a 96 well plate was covered with the test membranes and glass coverslips. Fresh blood was taken in Vacuette pre-citrated tubes from 3 healthy donors (donors 4, 5, and 6). The whole blood was anticoagulated with citrate (11 mM) and platelet rich plasma (PRP) was obtained by a centrifugation step at 200g for 15 minutes. The obtained PRP was aspirated and used within 2 h. The fluorogenic substrate was added to the PRP and just before the start of the experiment, PRP was recalcified. 200 μL of this mixture was added to the wells and also empty wells were used as blank. This thrombin generation test was performed as described before.14 Fluorescence was measured at 365/460 nm filter set with a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, USA). For quantification, the time between the start of the experiment and the moment at which the thrombin concentration first exceeds the 5 nM level was used (thrombin generation time).
For qualitative platelet adhesion, PRP of two donors was used and membranes and glass coverslips were put on the bottom of a 24 well plate in ultrapure water. Before addition of 100 μL PRP (of donors 5 and 6) the ultrapure water was aspirated. The PRP was incubated for 45 minutes at 37 °C. Then the PRP was removed. The test membranes and glass coverslips were washed in phosphate buffered saline (PBS) 3 times and then transferred to a clean vial and 900 μL of 2.5% glutaraldehyde was added and incubated for >1 h at 4 °C. Afterwards, the test membranes and glass coverslips were washed in a 0.1 M phosphate buffer (pH = 7.2) and with ultrapure water. After drying, the membranes and glass coverslips were put on a SEM holder, gold sputtered and examined using SEM, as described earlier.
Platelet adhesion was quantified using the lactate dehydrogenase (LDH) assay.20 Membrane circles (diameter = 6 mm) were immersed in ultrapure water (in duplo) in a 96 well plate, and 250 μL PRP (of donors 4, 5, and 6) was added and incubated at 37 °C for 60 minutes after aspiration of ultrapure water. Then, the PRP was removed, and the membrane samples were washed three times with PBS to remove non-adherent platelets. The membrane samples were transferred into clean vials and incubated for 60 minutes at room temperature in 200 μL lysis buffer. 50 μL of the supernatant was transferred in duplo to a 96 well plate and 50 μL substrate mix was added and this mixture was incubated for 30 minutes in the dark. Then, 50 μL stop solution was added and the optical density was measured at 490 nm on the SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, USA). Glass was not used as a control here because of non-accurate estimation of adhered platelets on glass.20 Platelets were quantified by a standard curve plotted from samples containing known amounts of platelets measured with a Beckman Coulter Ac-T diff analyzer (California, USA).
For the leukocyte adhesion test, empty wells were also taken into account for the incubation of the donor blood. After the incubation, blood was aspirated and analyzed using a Sysmex XE-5000 (Sysmex Europe GmbH, Germany) for leukocyte, basophil, eosinophil, neutrophil, monocyte and lymphocyte counts and hematocrit.
For the hemolysis test, additional wells were taken into account containing 600 μL saline or 600 μL sterile ultrapure water; these wells were used as negative and positive controls, respectively.24 After the incubated step, blood was aspirated and centrifuged at 4000 rpm for 6 minutes. Light absorbance was measured at 542 nm on the supernatant to analyze the free hemoglobin concentration. The percentage of hemolysis (H) was obtained as follows:

The bottom of 24 well plates was covered with 15 mm circles of the test membranes in duplo and washed in sterile ultrapure water. Blood was taken from 3 healthy donors (donors 1, 2, and 3), collected in BD Vacutainer SSTII Advance tubes (BD Plymouth, UK) and left to clot for 30 minutes. Then serum was obtained by a centrifugation step at 2000g for 10 minutes and the collected serum was pooled together per donor. The incubation step procedure was based on ref. 21, our own experience and the amount of available serum. Aliquots were immediately frozen for the not incubated serum control. Furthermore, empty wells were taken into account for the blank incubation of serum. 800 μL serum was added per well and incubated for 1 h at 37 °C under gentle shaking. After incubation, the serum was aspirated and directly analyzed further. The classical and alternative pathways were measured by ELISA using the Wieslab Complement system classical pathway and Wieslab complement system alternative pathway kits (Euro Diagnostica, Malmö, Sweden) according to the instructions of the manufacturer.25 The internal controls with a normal and pathological pooled serum show results within the valid range confirming the validity of both ELISA tests.
3. Results and discussion
3.1. Membrane fabrication
Fig. 2a shows SlipSkin™ membranes prepared using NMP as solvent and the 30:70, 50:50 and 70:30 copolymers: M30NMP (A and D), M50NMP (B and E) and M70NMP (C and F). Although the mechanical stability of these membranes was not systematically studied, during handling we observed that the M70NMP membranes were very fragile, whereas the M50NMP and M30NMP membranes were less fragile. All these membranes are porous, and below a thin skin layer large macro voids throughout the membrane cross-section are present. The mutual affinity (or miscibility) between the solvent–non-solvent pair NMP–water is very high, leading to instantaneous demixing and macro void formation during membrane fabrication.26–29 | ||
| Fig. 2 Scanning electron microscopy images of SlipSkin™ based membranes. (a) Membranes M30NMP (A and D), M50NMP (B and E) and M70NMP (C and F), formed using polymer dopes of 10 wt% SlipSkin™ dissolved in NMP. (b) Membranes M30 (A) and M50 (B) formed using polymer dopes of 10 wt% SlipSkin™ dissolved in acetone. (c) Membranes formed from a 10 wt% 50:50 SlipSkin™ in acetone with evaporation steps of 0 seconds M50 (A, B, and C), 30 seconds M50ev (D, E, and F), 5 minutes M50ev5 (G, H, and I) and 10 minutes M50ev10 (J, K, and L) with magnifications of 2000× (A, D, G, and J) or 5000× (B, C, E, F, H, I, K, and L). Images show cross-sections (A, B, D, E, G, H, J, and K) and top surfaces (C, F, I, and L). | ||
Fig. 2b presents SEM images of membranes formed using polymer dopes of 10 wt% SlipSkin™ dissolved in acetone. The M30 (A) and M50 (B) membranes have a porous sponge like structure. M70 membranes are brittle and a representative SEM picture could not be obtained. The solvent–non solvent pair acetone–water has lower mutual affinity than NMP–water and therefore delayed onset of de-mixing takes place, leading to macro void free membranes.27 Similar effects of solvents on membrane structures made from different polymers have been described before. For example, using water as non-solvent, a dope of PMMA dissolved in NMP gave finger like macro void structures while a dope of PMMA dissolved in acetone resulted in a symmetric porous sponge like structure.28 Besides, it seems that M50 shrinks more during the phase inversion process than the M30 membrane. This may be due to the lower amount of BMA present in the M50 membrane, which is the hydrophobic building block providing strength to the membrane during solidification.
Fig. 2c shows SEM images of membranes formed from a 10 wt% 50:50 SlipSkin™ in acetone with no solvent evaporation steps (A, B, and C), or solvent evaporation for 30 seconds (D, E and F), 5 minutes (G, H, and I) and 10 minutes (J, K, and L). All membranes seem to have a porous structure. The M50ev membranes with 30 second solvent evaporation seem to have bigger pores in the cross-sectional image compared to the M50 membranes. However, the surface images of the M50ev membranes show somewhat smaller pores. Due to the solvent evaporation the membranes with 5 and 10 minute evaporation steps seem to have pores through the membrane thickness but it looks like they are closed cellular pores without good pore interconnectivity.
Macroscopically, shrinkage was observed while drying the membranes in air. Also, the thicknesses in SEM pictures are smaller than the thicknesses of the membranes measured in a wet state using the micrometer, see Table 2. As expected, thinner membranes are formed after longer solvent evaporation. However, there was no difference in thickness between the membranes obtained after the 5 and 10 minute evaporation step.
| Membrane | Evaporation step | n | Average membrane thickness (μm) ± sd |
|---|---|---|---|
| M50 | 0 seconds | 7 | 99.9 ± 9.7 |
| M50ev | 30 seconds | 8 | 63.8 ± 5.3 |
| M50ev5 | 5 minutes | 5 | 34.8 ± 3.5 |
| M50ev10 | 10 minutes | 5 | 33.5 ± 2.5 |
3.2. Membrane transport properties
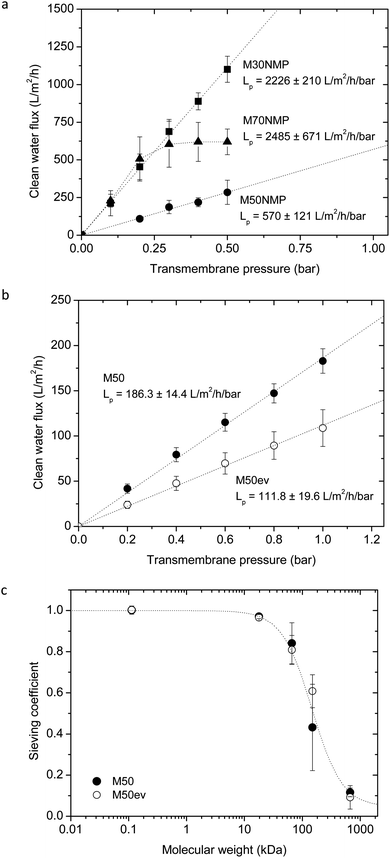 | ||
| Fig. 3 Membrane transport properties. (a) Average clean water fluxes plotted against transmembrane pressure for the membranes formed from 10 wt% SlipSkin™ dissolved in NMP:M30NMP (n = 2), M50NMP (n = 2) and M70NMP (n = 2) The error bars indicate standard deviations. (b) Average clean water flux plotted against the transmembrane pressure for membranes M50 (●) (n = 5) and M50ev (○) (n = 3) formed out of a 10 wt% 50:50 SlipSkin™ dissolved in acetone. The error bars indicate standard deviations. (c) Average sieving coefficients of molecular weight markers for the membranes M50 (●) and M50ev (○). The following markers were used: creatinine (MW 112.13 Da), myoglobin (MW 17 kDa), BSA (MW 66 kDa), gamma globulin (MW 150 kDa) and thyroglobulin (MW 670 kDa). Sieving coefficients of creatinine were measured in duplo, the other markers were measured in triplo or more. Error bars indicate standard deviations. | ||
Fig. 3b presents the clean water flux versus pressure relationship for the M50 and M50ev membranes. The M50 membranes have a clean water permeance of 186 ± 14 L−1 m−2 h−1 bar−1 and the flux–pressure relationship is linear. Although the SEM pictures show a porous structure, the M30 membranes show no water permeation after applying a pressure of 1 bar for several hours. The M70 membranes have poor mechanical stability and could not be tested for the clean water permeance. The M50ev membranes have a lower clean water permeance of 112 ± 20 L−1 m−2 h−1 bar−1 (p < 0.05) compared to the M50 membranes fabricated without a solvent evaporation step probably due to membrane densification. The flux–pressure relationship there is linear. Membranes M50ev5 and M50ev10 fabricated with 5 minute and 10 minute evaporation have no water permeability when they were tested for several hours. This is probably due to the lack of pore interconnectivity, consistent with the SEM analysis of these membranes, which mainly shows a closed cell pore structure. Since our target here is to develop ultrafiltration membranes with good mechanical properties we continued further investigation of sieving coefficients and biocompatibility only for the M50 and M50ev membranes.
For most of the commercial membranes however, there is hardly any information available in the scientific literature about biocompatibility. In the next section we investigate extensively the biocompatibility properties of our SlipSkin™ membranes M50 and M50ev and compare this to several benchmark materials.
3.3. Biocompatibility results
We performed several biocompatibility tests. The tests are selected based on the ISO guidelines. For the category of coagulation, two different thrombin generation tests were performed. In the category of thrombosis, platelet adhesion and aggregation were analyzed by SEM, while a LDH assay was performed for a quantitative analysis of platelet adhesion in the category of platelets. Leukocyte adhesion experiments and hemolysis evaluation were performed for the hematology category. For the complement category, we performed a classical and alternative pathway complement test.Fig. 4a shows typical thrombin generation curves of the positive control glass, the M50 membrane and the negative control blank (incubated PPP). The time until the rapid increase in thrombin concentration (thrombin generation time, TGT) as well as the peak height is analyzed and the peak height gives the most distinguishable and consistent results. The SlipSkin™ M50 membrane peak height is between the peak height values of the positive control glass and the negative control blank. Fig. 4b presents the average peak height of the thrombin concentration (nM) of donors 1, 2, and 3. The not incubated PPP shows similar results to the incubated plasma, the blank. The thrombin peak height of our SlipSkin™ membranes M50 and M50ev is similar to the benchmark materials and much lower than the glass (positive control) showing that the SlipSkin™ membranes have excellent properties concerning contact activation. Besides the above, we also measured thrombin generation real time, where PRP was incubated on the membrane in the presence of calcium. In this assay, the thrombin generation time was used for further analysis and was an indicator for the thrombogenicity of the material.14,37Fig. 5a shows typical results of thrombin generation curves over time for the M50 membrane and negative (blank) and positive (glass) control. Fig. 5b presents the results of the thrombin generation times and again, the SlipSkin™ membranes M50 and M50ev show very good properties, comparable to currently clinical in use PE membranes and other benchmark membranes in terms of thrombogenicity.
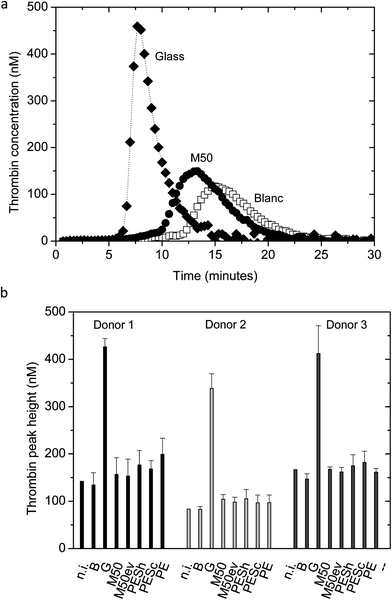 | ||
| Fig. 4 (a) Typical thrombin generation curve for blank (incubated PPP) (□), glass (♦)and a M50 membrane (●). (b) Average thrombin peak height presented for the categories: n.i. (not incubated PPP), B (blank, incubated PPP), glass, M50, M50ev, PESh, PESc and PE for donors 1, 2, and 3. Error bars indicate standard deviations. | ||
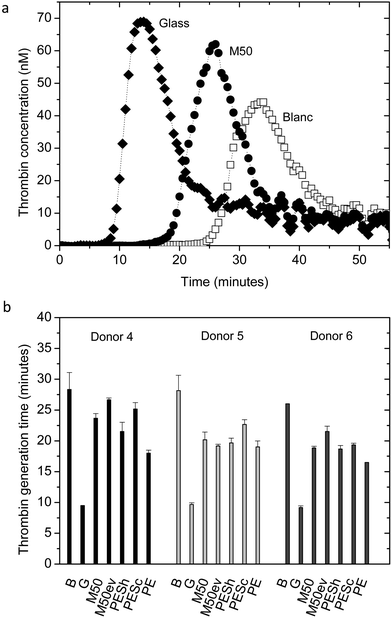 | ||
| Fig. 5 (a) Typical thrombin generation curve of glass (♦), M50 (●) and blank (incubated PRP) (□). (b) Average thrombin generation times presented for donors 4, 5, and 6, for the following materials: B (blank, incubated PRP), G (glass), M50, M50ev, ESh, PESc, PE membranes. Error bars indicate standard deviations. Thrombin generation times were measured in triplicate for each material and each donor. | ||
Here, we have performed two tests: SEM image analysis for a qualitative measure for platelet adhesion and aggregation, while quantitative results about platelet adhesion were obtained using the LDH platelet adhesion assay.20Fig. 6a shows SEM pictures of platelets, which adhered onto the tested membranes and glass. Both SlipSkin™ membranes (M50 and M50ev) (C, D, E, and F) show little platelet adhesion, no severe cluster formation and the adhered platelets are mostly round with little or no pseudopodia formation. Besides there are hardly any deposits. On glass, single platelets and small clusters are adhered with mostly long pseudopodia and small amounts of deposits (A and B). The PESh membranes (G and H) show deposits on the surface, where the platelets are mostly round with small pseudopodia but no clusters are observed. Fig. 6a (I and J) show clusters of platelets with pseudopodia on the PESc surface, and a lot of deposits on the membrane surface. On the PE blood transfusion filter, round and flat platelets are observed, mostly without pseudopodia, although some platelets have long pseudopodia (K and L). Fig. 6b presents the average adhered platelets per cm2 material. In every donor a similar trend can be observed. Only a few platelets adhere to the M50 and M50ev membranes, whereas many platelets seem to adhere to the other benchmark materials, especially to the PESc. The low platelet adhesion to the M50 and M50ev membranes could be due to the formation of a mosaic of hydrophilic and hydrophobic regions on the membrane surface leading to relatively low protein adhesion and thereby low platelet adhesion. The search for this optimal mosaic structure is one of the major reasons for investigating membrane fabrication using copolymers.
 | ||
| Fig. 6 (a) SEM pictures of adhered platelets on the materials glass (A and B), M50 (C and D), M50ev (E and F), PESh (G and H), PESc (I and J) and PE (K and L). (b) Average number of adhered platelets per cm2 material presented for the following membranes: M50, M50ev, PESh PESC, and PE, for donors 5, 6 and 7. Error bars indicate standard deviations. | ||
 | ||
| Fig. 7 Average number of leukocytes present in blood. For n.i. (not incubated blood), B (blank, incubated blood), M50, M50ev, PESh, PESc and PE membranes for donors 1, 2, and 3. Error bars indicate standard deviations. | ||
| Membrane | Donor 1 | Donor 2 | Donor 3 |
|---|---|---|---|
| M50 | 1.7 ± 2.1 | 1.7 ± 0.4 | 0.9 ± 0.4 |
| M50ev | 0.6 ± 0.5 | 1.1 ± 0.4 | 0.8 ± 0.4 |
| PESh | 1.5 ± 0.4 | 1.2 ± 0.3 | 0.8 ± 0.3 |
| PESc | 2.6 ± 2.4 | 0.9 ± 0.2 | 0.8 ± 0.5 |
| PE | 1.3 ± 0.3 | 2.0 ± 0.8 | 0.6 ± 0.3 |
Fig. 8 shows the average % complement activity of the classical (Fig. 8a) and alternative (Fig. 8b) pathways of the complement system. The not incubated serum and the blank (incubated serum) show similar results in both cases. This indicates that the incubation step itself did not lead to complement activation; therefore a decrease in complement activity in these assays would indicate a relative deficiency of any of the complement components, due to complement activation via the responsible pathway by the membrane. Overall, there was no activation of the classical pathway of the complement system by any of them, see Fig. 8a. In donors 1 and 3 all the materials show values close to 100% complement activity. In donor 2, the complement activity for DEAE cellulose seems to be lower than the rest, but this was not observed in the other donors. Activation of the classical pathway usually occurs via immune complexes rather than artificial surfaces. The alternative pathway of the complement system, on the other hand, can be activated by artificial surfaces such as membranes.
 | ||
| Fig. 8 (a) Average % classical pathway complement activity in serum. For n.i. (not incubated serum), B (blank, incubated serum), M50, M50ev, PESh, PESC, PE, RC and DC membranes. (b) Average % alternative pathway complement activity in serum. For n.i. (not incubated serum), B (blank, incubated serum), M50, M50ev, PESh, PESc, PE, RC and DC membranes. Tested in donors 1, 2, and 3. Error bars indicate standard deviations. | ||
4. Conclusions
This work presents the preparation and characterization of flat sheet SlipSkin™ membranes with excellent biocompatibility. In our tests these membranes have lower platelet adhesion and have equivalently good properties in relation to contact activation, thrombogenicity based on thrombin generation, leukocyte adhesion, hemolysis and complement activation compared to benchmark materials PESh, PESc and clinical in use PE membranes. The new membranes have a sieving curve similar to cascade filters in plasma fractionation applications. In the future we plan to develop SlipSkin™ hollow fiber membranes suitable for hemodialysis with a lower molecular weight cut-off so that albumin is retained. Besides membranes purely made from SlipSkin™, this material may be used for preparing porous coatings of existing membrane supports, so that it can offer its biocompatibility characteristics while the mechanical stability can be provided by a support membrane material. Furthermore, the very promising biocompatibility results of our membranes make it interesting to further investigate their biocompatibility under flow conditions.5. Conflict-of-interest notification
The authors Y. Aldenhoff, L. H. Koole and J. Janssen declare that they are shareholders of the company INterface BIOmaterials BV (Geleen, the Netherlands) that manufactures and commercializes the SlipSkin™ biomaterial.Acknowledgements
M. Tijink would like to acknowledge the Dutch Kidney Foundation for the financial support of this project.References
- E. Lacson Jr, J. Xu, R. S. Suri, G. Nesrallah, R. Lindsay, A. X. Garg, K. Lester, N. Ofsthun, M. Lazarus and R. M. Hakim, J. Am. Soc. Nephrol., 2012, 23, 687–695 CrossRef PubMed.
- R. P. Pauly, J. S. Gill, C. L. Rose, R. A. Asad, A. Chery, A. Pierratos and C. T. Chan, Nephrol., Dial., Transplant., 2009, 24, 2915–2919 CrossRef PubMed.
- A. Davenport, V. Gura, C. Ronco, M. Beizai, C. Ezon and E. Rambod, Lancet, 2007, 370, 2005–2010 CrossRef.
- B. Krause, M. Storr, T. Ertl, R. Buck, H. Hildwein, R. Deppisch and H. Göhl, Chem. Ing. Tech., 2003, 75, 1725–1732 CrossRef CAS.
- R. Deppisch, M. Storr, R. Buck and H. Gohl, Sep. Purif. Technol., 1998, 14, 241–254 CrossRef CAS.
- M. Matsuda, M. Sato, H. Sakata, T. Ogawa, K. I. Yamamoto, T. Yakushiji, M. Fukuda, T. Miyasaka and K. Sakai, J. Artif. Organs, 2008, 11, 148–155 CrossRef CAS PubMed.
- X. J. Huang, D. Guduru, Z. K. Xu, J. Vienken and T. Groth, Macromol. Biosci., 2011, 11, 131–140 CrossRef CAS PubMed.
- B. Fang, Q. Ling, W. Zhao, Y. Ma, P. Bai, Q. Wei, H. Li and C. Zhao, J. Membr. Sci., 2009, 329, 46–55 CrossRef CAS PubMed.
- F. Locatelli, F. Mastrangelo, B. Redaelli, C. Ronco, D. Marcelli, G. La Greca, G. Orlandini, M. Corti, G. Pontoriero, S. Di Filippo, R. Rossi, R. Farma, P. Colantonio, D. Martinelli, A. Vasile, V. Allegra, G. Mengozzi, L. Martinbianco, V. Bonomini, S. Stefoni, L. Colì, G. Cianciolo, L. De Sanctis, V. De Blasi, L. Alfonso, S. Rizzelli, P. Patruno, A. Brendolan, C. Crepaldi, M. D. Casu, S. Mureddu, R. Maiorca, L. Cristinelli, G. Setti, C. Camerini, M. Salvadori, S. Bandini, A. Rosati, G. Mancini, G. Panzetta, M. Ianche, F. Dagostino, G. Del Mastro, G. Cogoni, G. P. Accalai, M. A. Sechi, S. Alloatti, G. Paternoster, A. Gaiter, M. Manes, F. P. Schena, G. Pertosa, D. Bottalico, S. Cantafio, E. Salvo, F. D'Agostino, A. Guido, D. Limido, P. Beretta, M. R. Viganò, G. Dadone, G. Verzetti, C. Navino, M. C. Barbè, C. E. Ruva, M. Amato, S. Aterini, E. Ippolito, G. Splendiani, A. Sturniolo, S. Costanzi, S. Passalacqua, A. Scatizzi, A. Di Maggio, N. Montemurro, A. Vercellone, G. Segoloni, A. Pacitti, S. Maffei, F. Scaccia, R. Fini, G. Bandiani, G. Antiga, M. A. Nicolini, E. Asproni, F. Cadinu, N. Salvietti, D. Urti, P. Lorusso, L. Dani, C. Peona, O. Filiberti, M. C. Porcu, C. Barbieri, S. Costa, F. Nicrosini, V. Mioli, L. Bibiano and P. Carletti, Kidney Int., 1996, 50, 1293–1302 CrossRef CAS.
- Gambro AB Scientific Affairs, Dialysis Opinions 2008 – Focus on Innovation, 2008, http://www.gambro.com/Global/Globalweb/About/Research_development/Dialysis%20opinions/Dialysis%20opinion%202008.pdf Search PubMed.
- D. F. Stamatialis, B. J. Papenburg, M. Girones, S. Saiful, S. N. M. Bettahalli, S. Schmitmeier and M. Wessling, J. Membr. Sci., 2008, 308, 1–34 CrossRef CAS PubMed.
- Y. B. J. Aldenhoff, J. H. L. Hanssen, M. L. W. Knetsch and L. H. Koole, J. Vasc. Interv. Radiol., 2007, 18, 419–425 CrossRef PubMed.
- H. H. L. Hanssen, G. M. R. Wetzels, A. Benzina, F. H. Van Der Veen, T. Lindhout and L. H. Koole, J. Biomed. Mater. Res., 1999, 48, 820–828 CrossRef CAS.
- K. N. J. Stevens, O. Crespo-Biel, E. E. M. van den Bosch, A. A. Dias, M. L. W. Knetsch, Y. B. J. Aldenhoff, F. H. van der Veen, J. G. Maessen, E. E. Stobberingh and L. H. Koole, Biomaterials, 2009, 30, 3682–3690 CrossRef CAS PubMed.
- C. Zweigart, M. Neubauer, M. Storr, T. Böhler and B. Krause, in Comprehensive Membrane Science and Engineering, ed. D. Enrico and G. Lidietta, Elsevier, Oxford, 2010, pp. 351–390 Search PubMed.
- A. Erlenkötter, P. Endres, B. Nederlof, C. Hornig and J. Vienken, Artif. Organs, 2008, 32, 962–969 CrossRef PubMed.
- L. Muthusubramaniam, R. Lowe, W. H. Fissell, L. Li, R. E. Marchant, T. A. Desai and S. Roy, Ann. Biomed. Eng., 2011, 39, 1296–1305 CrossRef PubMed.
- M. S. L. Tijink, M. Wester, J. Sun, A. Saris, L. A. M. Bolhuis-Versteeg, S. Saiful, J. A. Joles, Z. Borneman, M. Wessling and D. F. Stamatialis, Acta Biomater., 2012, 8, 2279–2287 CrossRef CAS PubMed.
- H. C. Hemker, P. Giesen, R. Al Dieri, V. Regnault, E. De Smedt, R. Wagenvoord, T. Lecompte and S. Béguin, Pathophysiol. Haemostasis Thromb., 2003, 33, 4–15 CrossRef CAS PubMed.
- Y. Tamada, E. A. Kulik and Y. Ikada, Biomaterials, 1995, 16, 259–261 CrossRef CAS.
- T. Y. Liu, W. C. Lin, L. Y. Huang, S. Y. Chen and M. C. Yang, Biomaterials, 2005, 26, 1437–1444 CrossRef CAS PubMed.
- L. Uzun, H. Yavuz, B. Osman, H. Çelik and A. Denizli, Int. J. Biol. Macromol., 2010, 47, 44–49 CrossRef CAS PubMed.
- Y. Van Kooyk, P. Weder, K. Heije and C. G. Figdor, J. Cell Biol., 1994, 124, 1061–1070 CrossRef CAS.
- G. J. Dahe, R. S. Teotia, S. S. Kadam and J. R. Bellare, Biomaterials, 2011, 32, 352–365 CrossRef CAS PubMed.
- M. A. Seelen, A. Roos, J. Wieslander, T. E. Mollnes, A. G. Sjöholm, R. Wurzner, M. Loos, F. Tedesco, R. B. Sim, P. Garred, E. Alexopoulos, M. W. Turner and M. R. Daha, J. Immunol. Methods, 2005, 296, 187–198 CrossRef CAS PubMed.
- S. A. McKelvey and W. J. Koros, J. Membr. Sci., 1996, 112, 29–39 CrossRef CAS.
- M. Mulder, Basic principles of membrane technology, Kluwer Academic Publishers, Enschede, 1996 Search PubMed.
- C. Jao-Ming, W. Da-Ming, L. Fung-Ching and L. Juin-Yih, J. Membr. Sci., 1996, 109, 93–107 CrossRef.
- Z. L. Xu and F. A. Qusay, J. Membr. Sci., 2004, 233, 101–111 CrossRef CAS PubMed.
- W. R. Clark, R. J. Hamburger and M. J. Lysaght, Kidney Int., 1999, 56, 2005–2015 CrossRef CAS PubMed.
- M. J. Lysaght, Contrib. Nephrol., 1988, 61, 1–17 CAS.
- B. G. Stegmayr, Transfus. Apher. Sci., 2005, 32, 209–220 CrossRef PubMed.
- S. Nakaji and T. Yamamoto, Ther. Apheresis, 2002, 6, 267–270 CrossRef CAS.
- A. Sueoka, T. Miyahara, K. Takakura and Y. Ueda, Ther. Apheresis, 2000, 4, 65–67 CrossRef CAS.
- A. Sueoka, Ther. Apheresis, 2000, 4, 211–212 CrossRef CAS.
- M. N. Levy and R. M. Berne, Principles of physiology, Mosby, 2000 Search PubMed.
- K. N. Stevens, M. L. Knetsch, A. Sen, V. Sambhy and L. H. Koole, ACS Appl. Mater. Interfaces, 2009, 1, 2049–2054 CAS.
- S. Itoh, C. Susuki and T. Tsuji, J. Biomed. Mater. Res., Part A, 2006, 77, 294–303 CrossRef PubMed.
- M. P. Grooteman, J. C. Bos, A. J. van Houte, J. van Limbeek, M. Schoorl and M. J. Nubé, Nephrol., Dial., Transplant., 1997, 12, 492–499 CrossRef CAS PubMed.
- Y. Rousseau, N. Haeffner-Cavaillon, J.-L. Poignet, A. Meyrier and M.-P. Carreno, Cytokine, 2000, 12, 506–517 CrossRef CAS PubMed.
- U. T. Seyfert and F. Jung, Infusionstherapie und Transfusionsmedizin, 2000, 27, 317–322 Search PubMed.
- M. J. Walport, N. Engl. J. Med., 2001, 344, 1058–1066 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2013 |