 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu coated Ni nanowires with tuneable dimensions for biomedical applications†
Kirsten M. Pondmana, A. Wouter Maijenburgb, F. Burcu Celikkolc, Ansar A. Pathand, Uday Kishored, Bennie ten Hakena and Johan E. ten Elshof*b
aNeuro Imaging, MIRA Institute, University of Twente, Enschede, The Netherlands
bInorganic Material Science, MESA+ Institute for nanotechnology, University of Twente, Enschede, The Netherlands. E-mail: j.e.tenelshof@utwente.nl
cNanobiophysics, MIRA Institute, University of Twente, Enschede, The Netherlands
dCentre for Infection, Immunity and Disease Mechanisms, Biosciences, Brunel University, London, UK
First published on 19th September 2013
Abstract
Due to their shape anisotropy, high aspect ratio magnetic nanoparticles offer many advantages in biomedical applications. For biocompatibility, it is essential to have full control over the dimensions and surface chemistry of the particles. The aim of this study was to synthesize biocompatible nanowires with tuneable dimensions. This was achieved by electrodeposition of Ni in polycarbonate membranes. To ensure biocompatibility, a continuous gold coating was deposited onto the Ni wires by a newly developed electroless deposition method. The coating was analysed using electron microscopy and X-ray diffraction. Magnetic properties, anisotropy and Au film thickness were studied using vibrating sample magnetometry. After biofunctionalization, no significant cytotoxic effects were found in studies involving a diverse range of primary and tumour cells exposed to increasing concentrations of nanowires for up to 7 days. These nanowires may thus be used for in vivo applications such as magnetic drug delivery.
Introduction
Magnetic nanoparticles offer attractive possibilities in biomedicine, e.g. in magnetic separation, therapeutic drug, gene and radionuclide delivery, and hyperthermia, and as contrast agents in magnetic resonance imaging.1 To date, the majority of magnetic nanoparticles used are spherical, consisting of one or multiple magnetic cores with a biocompatible coating and active ligands. Within biomedicine, magnetic nanowires are receiving growing attention.2–6 Due to their elongated shape and anisotropic physical properties, these particles interact very differently with tissues, cells and biomolecules.3,7–9 Due to their increased surface to volume ratio, more drugs and ligands can be bound for drug delivery or immunoassays. While the small diameter of nanowires makes it possible for the particles to pass through the narrow capillaries, the large remanent magnetization increases the range and effectiveness of magnetic interactions at a distance from external magnets. This allows not only magnetic drug targeting at locations deeper inside the body4 and cell guidance,10 but also increases the effectiveness of cell separation significantly.6,9,11,12 Nanowires, with lengths matching the cell diameter, have previously been shown to outperform magnetic beads for cell separation, highlighting the importance of the dimensions of the nanowires.9,11 Whereas spherical particles only respond to magnetic flux gradients, high aspect ratio particles allow the application of torque with a relatively weak external field to perform dynamic targeted cell therapy.13 The torque can also be used to align the nanowires, and after their uptake by cells, it allows for the spatial organization of cells.3,12Most research on elongated nanoparticles was performed with carbon nanotubes. This research showed that elongated particles are capable of entering cells by penetration, allowing for direct access to the cytosol without the need for endosome escape strategies.7,14,15
Several chemical methods to synthesize elongated nanoparticles can be found in the literature (Fig. 1).16
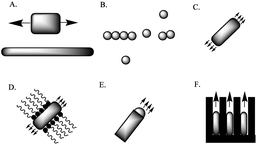 | ||
| Fig. 1 (A) Size reduction of a 1D-microstructure; (B) (self)assembly of spherical nanostructures; (C) spontaneous synthesis by dictation by the anisotropic crystallographic structure of a solid; (D) kinetic control by a capping reagent; (E) confinement by a liquid droplet in a vapor–liquid–solid procedure; (F) direction through the use of a template. | ||
Templated electrodeposition has several advantages in this application, including ease of fabrication, reproducibility, and low cost. Nanowires made by this method are always continuous since any breakage would halt the growth of the nanowires. Depending on the template and deposition time used, nanowires can be made with nanometer to micrometer dimensions, spanning several relevant biological length scales. Various magnetic metals such as iron (Fe), cobalt (Co) and nickel (Ni), and their alloys have been used to form nanowires inside the pores of silica, anodized alumina, or polycarbonate membranes.17–20
Having full control over the dimensions of nanowires is essential for biomedical applications. Previous research on asbestos and carbon nanotubes has suggested that cytotoxicity is highly dependent on aspect ratio. Long nanowires have been associated with inflammation, fibrosis and malignant mesothelioma and are likely to generate reactive oxygen species that can cause DNA damage and cell death.21 Unfortunately, most proposed nanowires8,9,22 are larger than cells, leaving the wires to protrude from the cells, thereby reducing the viability of the cells. Particles longer than 5 μm cannot be considered as “safe” in terms of uptake and clearance by macrophages and possibly induce inflammation and carcinogenic effects.23 A length below 5 μm will also make it possible for the nanowires to flow through the curvy capillaries allowing for local drug delivery.
In addition to dimensional restrictions, the materials and surface chemistry of the particles should be biocompatible and stable during use. Fe is often considered the most advantageous option, but due to the ease of oxidation of Fe, Ni is often preferred as a magnetic phase for nanowires. Although Ni is a naturally occurring transition material and an essential nutrient,24 there are indications that Ni may be a carcinogen and induce apoptosis.25–27 Studies on the toxicity of Ni wires are divided; some studies indicate no toxicity of Ni nanowires at low concentrations, and the need for long term exposure to act as carcinogens.4,28 But at higher concentration, even short incubation times have been shown to be lethal to cells.8 Some authors indicate the benefits of induced apoptosis by Ni nanowires, e.g. reactive oxygen species mediated apoptosis caused by Ni nanowires in human pancreatic adenocarcinoma cells.29 Unfortunately, these apoptotic effects are not specific to carcinoma cells and would therefore also occur in other healthy bystander cells. Shielding of the potentially toxic Ni from contact with the body was therefore considered essential in this study. The coating is also important to protect the Ni from oxidation. For this purpose, various polymers, proteins and phospholipids have been proposed.22,30,31 These coatings are biocompatible but cannot shield the particles from oxygen. Therefore, a solid Au shell is considered most advantageous as Au is known for its excellent biocompatibility and it cannot be removed by factors within the body.32 Furthermore, Au provides a platform for functionalization, such as conjugation of biomolecules, and targeting moieties and drugs.33,34
This work aims at the development of a magnetic nanowire for application in magnetic drug delivery and similar biomedical applications. The proposed electrodeposited Ni nanowire has a tuneable length below 5 μm with narrow size distribution. To increase the biocompatibility, the nanowires were coated with an Au shell. To limit toxic effects, the use of cyanide, which is commonly used in Au coatings,32 was avoided. Therefore, a new coating procedure was developed, inspired by a method to synthesize Au nanotubes by electroless deposition.35 The nanowires were characterized using a variety of techniques in order to determine their dimensions, magnetic properties, crystallographic structure and biocompatibility.
Materials and methods
Materials
Commercially available polycarbonate track-etched membranes (PCTE, Whatman, UK) with a nominal (quoted by the supplier) pore size of 50 nm were used as template membranes. Nickel(II) sulfate hexahydrate (NiSO4·6H2O), boric acid (H3BO3), ammonia (NH4OH, 37%), and glutathione (GSH) were obtained from Sigma-Aldrich and were used without further purification. Thiolated polyethylene glycol (mPEG-SH, 2 kDa) was purchased from Rapp Polymere (Tübingen, Germany). Dichloromethane (CH2Cl2) was used as obtained from Thermo Fisher Scientific (Waltham, USA). Trifluoroacetic acid, 11-aminoundecanoic acid, sodium sulfite (Na2SO3), sodium carbonate (NaHCO3), silver nitrate (AgNO3), and formaldehyde were purchased from Acros (Geel, Belgium). Tin(II) chloride (SnCl2) was purchased from Alfa Aesar (Ward Hill, USA). Oromerse-B was purchased from Technic Inc. (Cranston, RI, USA). Materials for cell culture: Dulbecco's modified Eagle medium (DMEM) and fetal bovine serum (heat inactivated) were purchased from PAA laboratories (Pasching, Austria), penicillin–streptomycin, trypan blue and phosphate buffered saline (PBS) from Invitrogen (Carlsbad, CA, USA), and CellTiter-Blue (CTB) from Promega Corp (Madison, WI).Nanowire synthesis
Ni nanowires were synthesized by templated electrodeposition in a conventional three-electrode setup. First, an Au layer was sputtered onto a PCTE membrane with a pore size of 50 nm using a Perkin-Elmer 2400 sputtering system under Ar. The Au layer (also called the back electrode) was used as the working electrode in the electrochemical deposition process. Prior to deposition, the backside of the back electrode was isolated using a glass plate. A small Pt mesh was used as the counter electrode and Ag/AgCl in 3 M KCl (Metrohm Autolab) was used as the reference electrode. All reported potentials are with respect to the reference electrode. The electrodes were connected to a potentiostat (Autolab PGSTAT 128N from Metrohm Autolab, Netherlands). For electrodeposition of nanowires, the electrodes were placed inside a plating solution containing NiSO4·6H2O (1.4 M) and H3BO3 (0.73 M), and a voltage of −1.0 V versus reference was applied for 6 min.After deposition, the membranes were dissolved in dichloromethane and washed at least 3 times with dichloromethane and 3 times with MilliQ water by centrifugation.
Electroless Au coating of Ni nanowires
Au coating was achieved using 5 subsequent steps adapted from ref. 31 as depicted schematically in Fig. 2.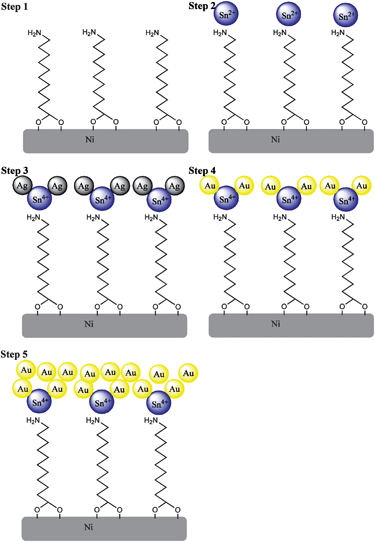 | ||
| Fig. 2 Scheme showing stepwise Au coating on Ni nanowires. (1) Amination, (2) Sn2+ deposition, (3) Ag deposition, (4) replacement of Ag by Au and (5) growth of the Au layer. | ||
Clean Ni nanowires were dispersed by sonication in an aqueous solution of 10 mM 11-aminoundecanoic acid for 2 h, to form an aminated monolayer (step 1 in Fig. 2). The nanowires were then washed at least 3 times with H2O.
A fresh aqueous solution containing 54 mM SnCl2 and 70 mM trifluoroacetic acid was added to the nanowires for 45 min. Sn(II) ions interacted with the electronegative amine groups forming a layer of electrostatically bound Sn2+ on the surface (step 2 in Fig. 2). After incubation, the nanowires were washed repeatedly with H2O.
Sn2+ was oxidized by Ag+via Sn2+ + 2Ag+ → Sn4+ + 2Ag0, by adding an aqueous solution containing 29 mM AgNO3 and 100 mM NH4OH (step 3 in Fig. 2). After repeated washing with H2O, the Ag phase can be replaced by Au.
A solution of 1 g Na2SO3, 0.6 g NaHCO3, 6 mL Na3Au(SO3)2 (Oromerse B) and 480 mL H2O was prepared. The nanowires were placed in 10 mL aliquots of this solution and sonicated at 3.6 °C for 30 min.
To allow for the formation of a closed layer of Au on the Ni nanowires, 25 μL mL−1 formaldehyde was added to the solution. This destabilized the Au+–sulfite complex, and induced the galvanic replacement reaction Ag0 + Au+ → Ag+ + Au0 (steps 4 and 5 in Fig. 2), and further electroless growth of the Au layer. The nanowires were sonicated for 12 h at 3.6 °C, and washed at least 5 times with H2O to remove free ions. Nanowires prepared using this method are named Au–Ni nanowires.
Biofunctionalization
The biofunctionalization of the Au–Ni nanowires was accomplished by incubating 100 μg mL−1 Ni or Au–Ni nanowires with 9 mM glutathione and 1 mM mPEG-SH in PBS for 24 h at 4 °C. Excess surfactant was removed by washing with PBS. The functionalization was analysed by Fourier transform infrared spectroscopy (FTIR, Thermo Scientific smart orbit Nicolet 6700 ATR-IR spectrometer) on KBr pellets with 0.5% dried and crushed Au–Ni nanowires.Characterization
Analysis of the nanowires was performed with a Zeiss HR-LEO 1550 FEG Scanning Electron Microscope (SEM) operating at 2.0 kV. X-ray diffraction (XRD) was performed on samples randomly deposited on a glass plate with a Bruker D2 Phaser with a Cu Kα X-ray source and a wavelength of 1.54 Å.Magnetic characterization was performed using a vibrating sample magnetometer (Model 10 Mark II VSM, Microsense). Samples were dried in a glass container before measurement; a blank measurement was subtracted from the sample data. Dispersed samples were measured in a glass container closed with Parafilm, and a blank containing the liquid in which the dispersion was made was subtracted from the data.
Cell culture
A number of tumour as well as non-tumour cell lines were used to assess the effect of nanoparticles on cell viability. These included: THP1 (a monocytic cell line derived from acute monocytic leukemia); Jurkat cell line (a T cell line derived from acute T cell leukemia); U937 (a monocytic cell line derived from histiocytic lymphoma); Raji (a B cell line derived from Burkitt's lymphoma); GBM (a primary cell line derived from glioblastoma multiforme); HeLa (a human cervical carcinoma epithelial cell line); A431 (an epithelial cell line derived from epidermoid carcinoma); RAW 264.7 (a murine macrophage cell line) and A549 (an epithelial cell line derived from lung carcinoma). The non-adherent cell lines THP1, Jurkat, U937, and Raji cell lines were grown in complete RPMI containing 10% heat inactivated FCS, 2 mM L-glutamine, 100 U mL−1 of penicillin and 100 μg mL−1 of streptomycin and 1 mM of sodium pyruvate, whereas adherent cell lines A431, A549 and GBM, and RAW 264.7 and HeLa were grown in complete DMEM containing the above mentioned supplements.Human monocytes and lymphocytes, and PBMC were isolated from blood samples from 2 healthy subjects using Ficol-Paque plus (GE Healthcare) gradient according to the manufacturer's instructions. PBMC obtained from the interface of plasma and Ficoll-Paque were washed twice with RPMI and suspended in complete RPMI. 5 × 106 PBMC in complete RPMI were plated out in each well of a 6 well plate (SPL Life Sciences), and pre-coated with human AB serum overnight. After 2 h of incubation at 37 °C in 5% CO2, the non-adherent PBMC (containing T, B and NK cells) were gently removed from adherent cells (monocytes).
Confocal imaging
A Zeiss (Göttingen, Germany) LSM 510 confocal laser scanning microscope (CLSM) was used to image uptake of Au–Ni nanowires by cells in bright field. 104 cells per well were incubated in a 96-well plate. Cells were allowed to adhere for 2 h after which Au–Ni nanowires were added with a nanowire concentration of approximately 10 nanowires per cell. This concentration was estimated by assuming that the nanowires have the density of bulk Ni and Au and average dimensions as estimated from SEM images. Control samples contained no nanowires. Images were obtained after 24 h incubation at 37 °C in a 5% CO2 atmosphere.Concentration dependent short term cell viability on phagocytotic and non-phagocytotic cells
Adherent cells, RAW 264.7 and HeLa, were seeded on 96-well culture plates at 5 × 103 cells per well in 100 μL of cell medium. Cells were allowed to adhere to the plate for 24 h at 37 °C in a 5% CO2 atmosphere. Incubation of Ni and Au–Ni nanowires was done by adding 100 μL of Au–Ni nanowire dispersion in cell medium to the cells on the 96-well culture plates, for 48 h at 37 °C in a 5% CO2 atmosphere. The final concentrations of the Au–Ni nanowires were 0, 1, 10, 100 and 1000 nanowires per cell. All experiments were performed in triplicate.After 48 h the cell medium was carefully removed and the cells were washed with PBS, thereafter 100 μL of cell medium was added with 10 μL of CellTiter-Blue and incubated for 4 h. The fluorescence was measured on a spectrophotometer (Victor, Perkin Elmer) with an excitation wavelength of 560 nm and an emission wavelength of 590 nm. The fluorescence values were normalized by the controls and expressed as percent viability. Validity of the assay was assessed by cell counting and visual inspection of the cells.
Long term cell viability following exposure to Au–Ni nanowires
The long term cytotoxicity of Au–Ni nanowires was investigated by measuring viable and dead cells. Non-adherent cells were seeded at 105 cells per well and adherent cells at 2 × 104 cells per well and were allowed to adhere for 2 h at 37 °C in 5% CO2. Au–Ni nanowires in PBS were added to duplicate wells at a concentration of 100 nanowires per cell. Duplicate control wells were treated with an equal volume of PBS. The cells were then incubated at 37 °C in 5% CO2 for 12 h, 1, 2, 5 and 7 days. At each time point, the cells were suspended (using trypsin–EDTA to detach adherent cells). 20 μL of the cell suspensions were then mixed with an equal volume of trypan blue (0.4%). At least 100 cells from each duplicate culture were observed for the morphology and staining of cells using a haemocytometer and an inverted microscope (Leica). Blue stained cells were considered dead. Adherent and non-adherent PBMC were cultured at 5 × 105 cells per well and the ratio of cells to nanowires was similar to other cell lines. Approximately 200 cells from each culture were observed under the microscope using trypan blue at each time point as mentioned above.Results and discussion
Ni nanowires with an average diameter of 150 nm and a length of 2.6 ± 0.3 μm (SEM, n = 100) were obtained by electrodeposition of Ni in the pores of a PCTE membrane for 6 min at −1.0 V versus an Ag/AgCl reference electrode. The nanowires were released into suspension by dissolving the membrane and several washing steps with dichloromethane and water. An Au coating was deposited by electroless deposition onto the Ni nanowires and the morphology of the Au-coated Ni nanowires was investigated by HR-SEM as shown in Fig. 3. | ||
| Fig. 3 HR-SEM images of Ni nanowires and Au–Ni nanowires. (a) Original Ni nanowires, (b) Oromerse-B Ni nanowires, (c) Sn–Ag–Au–Ni nanowires, and (d) 11-aminoundecanoic acid–Sn–Ag–Au nanowires, hereafter indicated as Au–Ni nanowires. | ||
Employing only the Au deposition reaction with Oromerse B (steps 4 and 5 in Fig. 2) did not result in Au nucleation on the Ni nanowire surface as can be seen in Fig. 3b. Therefore, an activation step was introduced in which Sn nuclei were deposited, followed by galvanic replacement by Ag, and finally growth of Au on the Ag nuclei. Unfortunately, the Au coating was still discontinuous (Fig. 3c), most likely due to an insufficient concentration of Sn nuclei on the Ni surface. An amination step, using 11-aminoundecanoic acid, was introduced to provide more homogeneous Sn deposition, allowing a continuous Au coating to form over the entire nanowire surface (Fig. 3d), with an approximate thickness of 40 nm as estimated from SEM. Longer Au deposition will result in the formation of a thicker Au layer.
The phase and grain size of the Ni nanowires and Au coating were investigated by XRD (Fig. 4). Ni nanowires showed diffraction peaks corresponding to the (111) and (200) planes of cubic Ni. The peak at 2θ 38° can be assigned to the Au (111) plane of the back electrode. After the complete Au coating procedure diffraction peaks were detected, which can be assigned to Au (110), (111), (200), (220) and (311). The Ni (111) and Au (200) peaks overlap, therefore it is not possible to distinguish between them. The broader peaks in the Au spectrum compared to the Ni spectrum indicate a smaller crystallite size for the Au layer. Using the Debye–Scherrer equation τ = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, the crystallite sizes of Ni and Au can be estimated. Here, τ is the mean size of the crystalline domain, K the shape factor, β the full width at half maximum, θ the Bragg angle and λ the X-ray wavelength. The estimated crystallite sizes are 54 nm for Ni and 20 nm for Au. By comparing the crystallite sizes from XRD with SEM imaging, we can conclude that the crystalline domain size in the central Ni nanowire is about 1/3 of its diameter, and it is surrounded by an Au layer with an equivalent thickness of about 2 grains.
cosθ, the crystallite sizes of Ni and Au can be estimated. Here, τ is the mean size of the crystalline domain, K the shape factor, β the full width at half maximum, θ the Bragg angle and λ the X-ray wavelength. The estimated crystallite sizes are 54 nm for Ni and 20 nm for Au. By comparing the crystallite sizes from XRD with SEM imaging, we can conclude that the crystalline domain size in the central Ni nanowire is about 1/3 of its diameter, and it is surrounded by an Au layer with an equivalent thickness of about 2 grains.
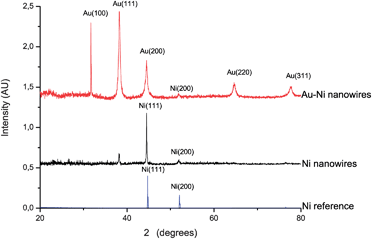 | ||
| Fig. 4 XRD patterns of randomly oriented Ni nanowires and Au–Ni nanowires and a pure Ni reference sample. | ||
Fig. 5 shows the room temperature magnetization curves for uncoated Ni nanowires inside the template with magnetic field (H) applied in two directions, i.e. parallel and perpendicular to the wire direction. The squareness ratio Mr/Ms and coercivity are known to increase significantly with aspect ratio.36 The obtained hysteresis loops show magnetic anisotropy where the magnetic easy direction is along the axis, as can be seen from the squareness ratio Mr/Ms = 0.48 in the parallel direction. From VSM measurements on randomly orientated nanowires as depicted in Fig. 8 the saturation magnetization of the Ni nanowires was found to be 34 emu g−1, which is significantly less than the value for bulk Ni (55.4 emu g−1). This can be explained by the surface anisotropy which is significant for a wire with small diameter. After Au coating of the Ni nanowires the saturation magnetization decreased to 10 emu g−1 (Fig. 6). From this value, the diameter and length of the Ni nanowire and the densities of Ni (8.9 × 106 g m−3) and Au (19.3 × 106 g m−3), the shell thickness can be calculated. Neglecting the influence of the other coating layers the Au layer thickness is approximately 34 nm, which is in good agreement with the thickness estimated from HR-SEM images.
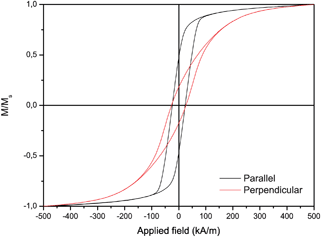 | ||
| Fig. 5 Magnetic hysteresis curves of Ni nanowires measured inside the membrane under an external field parallel and perpendicular to the nanowire long axis. Parallel coercivity value of 302 Oe and Mr/Ms = 0.48 and perpendicular coercivity of 378 Oe and Mr/Ms = 0.17. | ||
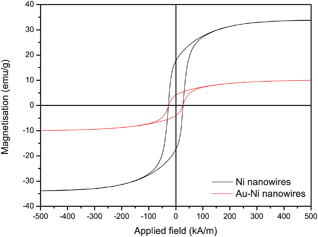 | ||
| Fig. 6 Hysteresis curves of Ni nanowires and Au–Ni nanowires in random orientation under an external field. Ms = 34 emu g−1 for uncoated and Ms = 10 emu g−1 for Au coated Ni nanowires, suggesting a coating thickness of 34 nm on a 2.6 μm long Ni nanowire with 150 nm diameter. | ||
After biofunctionalisation using SH-PEG and glutathione, a suspension of Au–Ni nanowires was obtained (Fig. 7). The nanowires could be easily collected using a permanent magnet and dispersed by vortexing. Due to the possibility to move freely in the liquid, the magnetization curves of suspended nanowires showed superparamagnetic behaviour, with no remnant magnetisation (Fig. 8). Attachment of GSH and mPEG-SH was examined by FTIR spectroscopy (Fig. 9). GSH attachment can be identified by two bands around 1600 cm−1 for both cysteine and glutamic acid-carbonyl. The anchoring is via the thiol group as the band at 2500 cm−1 for SH stretching is not visible on the Au–Ni nanowires. Binding of mPEG-SH can be seen by a strong band at 1380 cm−1 showing C–H stretching, and the band at 1080 cm−1 shows C–O–C stretching from mPEG-SH.
 | ||
| Fig. 7 (a) Suspension of Au–Ni nanowires stabilised with GSH and mPEG-SH. (b and c) The Au–Ni nanowires can be collected using a permanent magnet. | ||
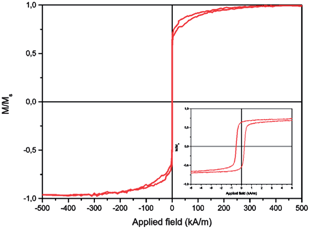 | ||
| Fig. 8 Magnetisation loop M vs. applied field H for suspended Au–Ni nanowires showing superparamagnetic behaviour. The inset shows that the coercivity of the Au–Ni nanowires is approximately 1 kA m−1 for dispersed samples. | ||
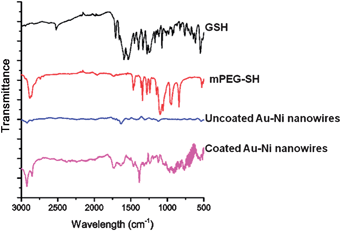 | ||
| Fig. 9 FTIR spectra of GSH, mPEG-SH, and uncoated Au–Ni nanowires, and the attachment of GSH and mPEG-SH to the Au–Ni nanowires. | ||
The uptake of Au–Ni nanowires was studied by confocal microscopy (Fig. 10). 3D imaging (not shown here) ascertained that the particles were inside the cells and not simply adhered to the membrane. After 24 h of incubation, RAW 264.7 cells had taken up many nanowires and clusters of nanowires. As these cells are macrophages, the uptake pathway is most likely phagocytosis. Confocal microscopy analysis showed that the nanowires were completely taken up by these cells without signs of frustrated phagocytosis. This indicates that macrophages will be able to remove the particles from the bloodstream. Jurkat cells are not capable of phagocytosis and only take up particles by receptor mediated endocytosis. As expected, these cells did not take up any nanowires, and appeared as healthy as control untreated cells (see ESI†).
 | ||
| Fig. 10 Interaction of Au–Ni nanowires with (a) RAW 264.7 cells and (b) Jurkat cells. | ||
The effects of Au–Ni nanowires on cell viability were evaluated using a CTB assay. Au–Ni nanowires did not show a decrease in cell viability of both HeLa and RAW cells over 48 h (Fig. 11). In a study between phagocytic and non-phagocytic cells, a slightly higher cytotoxicity was observed for phagocytic (RAW 264.7) cells compared to non-phagocytic (HeLa) cells, which might indicate the importance of uptake on cytotoxicity. It should be noted that the high concentration of 1000 nanowires per cell is much higher than targeted in potential applications in biomedicine, and is only used here to investigate the biocompatibility of the nanowires. In RAW cells Ni nanowires showed increased cytotoxicity compared to the Au coated Ni nanowires, indicating the necessity of the Au coating. The used concentration of nanowires is much higher than the lethal concentration of Ni nanowires with a length of 20 μm found by Byrne et al.,8 which indicates and illustrates the high biocompatibility of the much shorter Ni and Au–Ni nanowires.
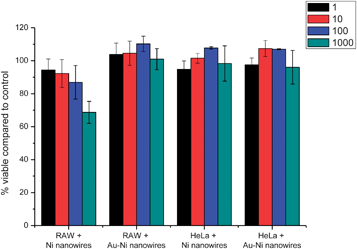 | ||
| Fig. 11 Short term viability compared to untreated control of RAW and HeLa cells assessed by CTB assay after 48 h incubation with Ni and Au–Ni nanowires. The bars indicate standard deviation. | ||
A range of cells – primary and cancerous, phagocytic and non-phagocytic, adherent and non-adherent – were used in order to systematically investigate cytotoxicity over a long term (Table 1). All 9 different cell types used in this study did not show significant cytotoxicity of the Au–Ni nanowires for periods up to 7 days.
| Cell type | 12 h | 24 h | 48 h | 5 days | 7 days | |
|---|---|---|---|---|---|---|
| U937 (non-adherent) | Control | 97.6 ± 1.0 | 96.3 ± 0.6 | 96.9 ± 0.4 | 98.1 ± 0.4 | 97.2 ± 0.1 |
| Au–Ni wires | 96.5 ± 0.7 | 96.5 ± 0.7 | 96.7 ± 0.6 | 98.7 ± 1.4 | 98.2 ± 1.2 | |
| THP1 (non-adherent) | Control | 99.7 ± 0.5 | 95.7 ± 0.1 | 98.0 ± 1.5 | 99.0 ± 0.3 | 98.5 ± 0.4 |
| Au–Ni wires | 98.5 ± 0.5 | 93.7 ± 0.1 | 95.5 ± 1.8 | 99.0 ± 0.4 | 99.3 ± 0.3 | |
| A549 (adherent) | Control | 95.3 ± 1.4 | 98.1 ± 0.6 | 92.9 ± 0.3 | 98.5 ± 1.4 | 97.0 ± 0.9 |
| Au–Ni wires | 98.8 ± 0.6 | 98.6 ± 0.2 | 94.2 ± 1.4 | 98.5 ± 1.3 | 96.6 ± 0.7 | |
| Raji (non-adherent) | Control | 97.7 ± 0.3 | 98.2 ± 0.4 | 98.7 ± 0.1 | 99.5 ± 0.1 | 96.6 ± 0.6 |
| Au–Ni wires | 97.9 ± 0.1 | 97.7 ± 0.4 | 98.6 ± 0.7 | 98.4 ± 0.4 | 95.4 ± 2.9 | |
| GBM (adherent) | Control | 98.7 ± 0.1 | 97.8 ± 3.1 | 99.1 ± 0.5 | 96.0 ± 0.7 | 96.3 ± 2.5 |
| Au–Ni wires | 98.9 ± 0.2 | 98.4 ± 0.0 | 98.7 ± 0.7 | 96.1 ± 0.7 | 97.1 ± 0.9 | |
| Jurkat (non-adherent) | Control | 98.4 ± 0.3 | 96.9 ± 0.6 | 97.8 ± 0.7 | 94.6 ± 0.8 | 93.6 ± 1.7 |
| Au–Ni wires | 97.8 ± 0.5 | 97.9 ± 0.2 | 98.1 ± 1.0 | 94.6 ± 0.1 | 92.0 ± 1.0 | |
| PBMC (adherent) | Control | 99.5 ± 0.2 | 98.4 ± 0.6 | 99.5 ± 0.4 | 98.4 ± 1.0 | 98.4 ± 0.5 |
| Au–Ni wires | 99.8 ± 0.2 | 99.3 ± 0.5 | 99.5 ± 0.4 | 98.4 ± 0.9 | 98.5 ± 0.4 | |
| PBMC (non-adherent) | Control | 99.5 ± 0.4 | 97.8 ± 0.9 | 99.8 ± 0.3 | 96.3 ± 1.3 | 98.2 ± 0.9 |
| Au–Ni wires | 99.6 ± 0.4 | 98.5 ± 0.8 | 99.7 ± 0.2 | 96.1 ± 1.4 | 97.7 ± 0.6 | |
| A431 (adherent) | Control | 98.7 ± 0.1 | 98.6 ± 0.1 | 96.2 ± 1.9 | 96.1 ± 3.8 | 96.1 ± 3.8 |
| Au–Ni wires | 98.9 ± 0.2 | 98.2 ± 1.1 | 95.6 ± 0.2 | 95.2 ± 1.7 | 95.2 ± 1.7 |
Conclusions
Biocompatible nanowires with tuneable dimensions were prepared by templated electrodeposition of Ni in PCTE membranes. The dimensions of the nanowires can be fully tuned by adapting the deposition time and template dimensions. An Au coating procedure was developed to ensure biocompatibility by complete shielding of the Ni phase from the biological environment. The newly developed procedure involves amination of the Ni surface, activation by Sn and Ag, followed by electroless Au deposition. A coating thickness of approximately 40 nm was achieved by 12 h electroless Au deposition. SEM imaging shows a smooth and continuous layer of Au on the Ni nanowires. Elemental analysis confirmed the formation of Au on Ni. Magnetic measurements were used to reveal the shape anisotropy and ferromagnetic properties of the Au–Ni nanowires. Stable suspensions of nanowires were obtained by biofunctionalisation using GSH and mPEG-SH. Cell studies indicate that the nanowires can be completely engulfed by cells without signs of frustrated phagocytosis. No significant cytotoxic effects and morphological alterations were found when nanowires were exposed to a variety of cell lines, indicating the potential use of the nanowires in biomedical applications, such as magnetic drug delivery.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the Chemical Sciences division of the Netherlands Organization for Scientific Research (NWO-CW) is gratefully acknowledged. The authors thank R. Besselink (Inorganic Materials Science) for support and suggestions, M. Sobik (Neuro Imaging) for data acquisition and interpretation and M.M.S.M. Wösten (University Utrecht, Veterinary Faculty Division infection biology) and Robert Sim (Oxford University, Department of Pharmacology) for advice on cell viability tests.References
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- Q. Hu, Y. Q. Liu, N. Li, C. Cheng, S. G. Xu, N. Wang, W. Qin and B. Z. Tang, Nano, 2013, 8, 1350029 CrossRef.
- A. Hultgren, M. Tanase, C. S. Chen, G. J. Meyer and D. H. Reich, J. Appl. Phys., 2003, 93, 7554–7556 CrossRef CAS.
- A. Prina-Mello, Z. Diao and J. M. Coey, J. Nanobiotechnol., 2006, 4, 9 CrossRef PubMed.
- H. T. Huang, T. R. Ger, Y. H. Lin and Z. H. Wei, Lab Chip, 2013, 13, 3098–3104 RSC.
- A. Sharma, Y. C. Zhu, S. Thor, F. Zhou, B. Stadler and A. Hubel, IEEE Trans. Magn., 2013, 49, 453–456 CrossRef.
- M. Safi, M. H. Yan, M. A. Guedeau-Boudeville, H. Conjeaud, V. Garnier-Thibaud, N. Boggetto, A. Baeza-Squiban, F. Niedergang, D. Averbeck and J. F. Berret, ACS Nano, 2011, 5, 5354–5364 CrossRef CAS PubMed.
- F. Byrne, A. Prina-Mello, A. Whelan, B. M. Mohamed, A. Davies, Y. K. Gun'ko, J. M. D. Coey and Y. Volkov, J. Magn. Magn. Mater., 2009, 321, 1341–1345 CrossRef CAS PubMed.
- A. Hultgren, M. Tanase, C. S. Chen and D. H. Reich, IEEE Trans. Magn., 2004, 40, 2988–2990 CrossRef.
- F. Johansson, M. Jonsson, K. Alm and M. Kanje, Exp. Cell Res., 2010, 316, 688–694 CrossRef CAS PubMed.
- A. Hultgren, M. Tanase, E. J. Felton, K. Bhadriraju, A. K. Salem, C. S. Chen and D. H. Reich, Biotechnol. Prog., 2005, 21, 509–515 CrossRef CAS PubMed.
- M. Tanase, E. J. Felton, D. S. Gray, A. Hultgren, C. S. Chen and D. H. Reich, Lab Chip, 2005, 5, 598–605 RSC.
- A. O. Fung, V. Kapadia, E. Pierstorff, D. Ho and Y. Chen, J. Phys. Chem. C, 2008, 112, 15085–15088 CAS.
- M. M. Song, W. J. Song, H. Bi, J. Wang, W. L. Wu, J. Sun and M. Yu, Biomaterials, 2010, 31, 1509–1517 CrossRef CAS PubMed.
- X. H. Shi, A. von dem Bussche, R. H. Hurt, A. B. Kane and H. J. Gao, Nat. Nanotechnol., 2011, 6, 714–719 CrossRef CAS PubMed.
- Y. N. Xia, P. D. Yang, Y. G. Sun, Y. Y. Wu, B. Mayers, B. Gates, Y. D. Yin, F. Kim and Y. Q. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- H. Schlorb, V. Haehnel, M. S. Khatri, A. Srivastav, A. Kumar, L. Schultz and S. Fahler, Phys. Status Solidi B, 2010, 247, 2364–2379 CrossRef.
- M. G. Maas, E. J. B. Rodijk, A. W. Maijenburg, D. H. A. Blank and J. E. ten Elshof, J. Mater. Res., 2011, 26, 2261–2267 CrossRef CAS.
- A. George, A. W. Maijenburg, M. G. Maas, D. H. A. Blank and J. E. ten Elshof, ACS Appl. Mater. Interfaces, 2011, 3, 3666–3672 CAS.
- A. W. Maijenburg, E. J. B. Rodijk, M. G. Maas, M. Enculescu, D. H. A. Blank and J. E. ten Elshof, Small, 2011, 7, 2709–2713 CrossRef CAS PubMed.
- V. C. Sanchez, J. R. Pietruska, N. R. Miselis, R. H. Hurt and A. B. Kane, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 511–529 CrossRef CAS PubMed.
- L. A. Bauer, D. H. Reich and G. J. Meyer, Langmuir, 2003, 19, 7043–7048 CrossRef CAS.
- P. H. Hoet, I. Bruske-Hohlfeld and O. V. Salata, J. Nanobiotechnol., 2004, 2, 12 CrossRef PubMed.
- F. H. Nielsen, Nutr. Today, 1993, 28, 14–19 CrossRef PubMed.
- J. J. Pan, Q. S. Chang, X. Wang, Y. Son, Z. Zhang, G. Chen, J. Luo, Y. Y. Bi, F. Chen and X. L. Shi, Chem. Res. Toxicol., 2010, 23, 568–577 CrossRef CAS PubMed.
- J. Zhao, L. Bowman, X. Zhang, X. Shi, B. Jiang, V. Castranova and M. Ding, J. Nanobiotechnol., 2009, 7, 2 CrossRef PubMed.
- M. Costa, Fresenius' J. Anal. Chem., 1998, 361, 381–385 CrossRef CAS.
- N. Gao, H. J. Wang and E. H. Yang, Nanotechnology, 2010, 21, 105–107 Search PubMed.
- M. Z. Hossain and M. G. Kleve, Int. J. Nanomed., 2011, 6, 1475–1485 CrossRef CAS PubMed.
- D. Magnin, V. Callegari, S. Matefi-Tempfli, M. Matefi-Tempfli, K. Glinel, A. M. Jonas and S. Demoustier-Champagne, Biomacromolecules, 2008, 9, 2517–2522 CrossRef CAS PubMed.
- N. S. Birenbaum, B. T. Lai, C. S. Chen, D. H. Reich and G. J. Meyer, Langmuir, 2003, 19, 9580–9582 CrossRef CAS.
- I. T. Jeon, M. K. Cho, J. W. Cho, B. H. An, J. H. Wu, R. Kringel, D. S. Choi and Y. K. Kim, J. Mater. Chem., 2011, 21, 12089–12095 RSC.
- G. Han, P. Ghosh and V. M. Rotello, Nanomedicine, 2007, 2, 113–123 CrossRef CAS PubMed.
- R. Venkatesan, A. Pichaimani, K. Hari, P. K. Balasubramanian, J. Kulandaivel and K. Premkumar, J. Mater. Chem. B, 2013, 1, 1010–1018 RSC.
- P. Kohli, J. E. Wharton, O. Braide and C. R. Martin, J. Nanosci. Nanotechnol., 2004, 4, 605–610 CrossRef CAS PubMed.
- B. Das, K. Mandal, P. Sen and S. K. Bandopadhyay, J. Appl. Phys., 2008, 103, 013908-1-5 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Control cell microscopy images of RAW 264.7 and Jurkat cells. See DOI: 10.1039/c3tb20808g |
| This journal is © The Royal Society of Chemistry 2013 |