Thiol modification of silicon-substituted hydroxyapatite nanocrystals facilitates fluorescent labelling and visualisation of cellular internalisation†
Received
1st June 2013
, Accepted 19th July 2013
First published on 22nd July 2013
Abstract
Calcium phosphates are used widely as orthopaedic implants and in nanocrystalline form to enable the transfer of genetic material into cells. Despite widespread use, little is known about their fate after they have crossed the cell membrane. Here we present a method of surface modification of silicon-substituted hydroxyapatite (SiHA) through a silane group, which enables the engraftment of a fluorescent dye to facilitate real-time biological tracking. Surface modification of the nanocrystal surface was undertaken using (3-mercaptopropyl)trimethoxysilane (MPTS), which presented a thiol for the further attachment of a fluorophore. Successful modification of the surface was demonstrated using zeta potential measurements and fluorescence microscopy and the number of thiol groups at the surface was quantified using Ellman's reagent. In vitro experiments using the fluorescently modified particles enabled the discrimination of the calcium phosphate particulate from other biological debris following internalisation by a population of MC3T3 (pre-osteoblast) cells and the particles were shown to maintain fluorescence for 24 hours without quenching. The successful modification of the surface of SiHA with thiol groups offers the tantalising possibility of the intracellular growth factor delivery.
1 Introduction
Calcium phosphate ceramics such as hydroxyapatite (HA) have been used widely for the restoration of function in diseased and damaged hard tissue. In addition, they have found application in a diverse selection of sectors as food additives, adsorbents in chromatography columns and even as substrates to enable absorption of pollutants from wastewater.1,2 Within the biomedical sector, calcium phosphate salts have been used principally because of their similarity to the mineral component of bone and also since their dissolution products are non-toxic. Relatively recent work has seen calcium phosphate salts used for the delivery of biological materials into cells in the form of peptides, polymers and DNA sequences.3–8 Calcium phosphate salts have a critical safety advantage over other vectors such as viruses in that they pose no risk of pathogenicity due to mutation. Although it is known that calcium phosphate–DNA complexes cause no apparent cytotoxicity, the fate of the particles upon internalisation is not yet known. The difficulty in tracking the particles can be related to the visual similarity to granulation within the cells.
The large crystal lattice of the apatites means that their structure may incorporate numerous substitutions, which can be used to tailor material chemistry to induce a particular biological reaction.9–12 One main focus of research into the development of new calcium phosphate based materials has been the substitution of silicon into the hydroxyapatite lattice (SiHA) to enable additional biomolecule attachment and controlled release in vivo. Silicon substitution is of particular interest since silicon is well established to be an important factor in the production of new bone matrix and functions to assist in the production of collagen by osteoblasts.12–15 Calcium phosphate composites have been used widely as implant materials due to the low toxicity of their ionic components and the intimate bond that they are able to form with a wide range of hard and soft tissues. There have been proposed to be two sites in crystalline HA that may be exploited for functionalisation; a hydroxyl group and the phosphate group.16–18 Previous studies have shown that the surface hydroxyl (OH) groups of HA may react with organic isocyanate groups and this has been exploited for the attachment of polymers such as poly(ethylene glycol) (PEG), poly(methyl methacrylate) (PMMA), poly(n-butyl methacrylate) (PBMA), poly(2-hydroxyethyl methacrylate) (poly(HEMA)) to the crystal surfaces.19,20 In addition, it has been found that substitution by alkylphosphonates causes calcium phosphate monolith structures to become mesoporous with high specific surface area,21 which is of course crucial for biological applications where cell attachment, proliferation, bioresorbability, and tissue/interface regeneration rely on materials with specific surface areas approaching that of native biomineral. Of these reagents containing hydroxyl groups, phosphoric acid based reactants are favoured because the P–OH groups facilitate the adsorption of proteins, as demonstrated for the model protein, bovine serum albumin.22 However, the limited availability and reactivity of OH groups can result in low numbers of biomolecules being grafted on to the HA surface.7,23 The Ca2+ ions in calcium phosphates, including HA, have been exploited for the ionic bonding of various functional groups. For example, Lee et al.7 have reported the thiol modification of HA using 3-mercaptopropionic acid and Ganesan et al.8 attached porphyrins to the HA surface as a potential drug carrier. Silicon substitution has drawn much attention as a route to surface modification24 with organosilanes, particularly those containing amino or thiol terminal groups. For example, previous work has demonstrated that osteoblast adhesion to HA is increased when functionalised with 3-aminopropyltriethoxysilane.25 Although much attention has been given to the use of (3-mercaptopropyl)trimethoxysilane (MPTS) for the surface modification of silicon oxide,26–28 there is scarcely any research on the surface modification of calcium phosphates with MPTS.
Here we report a new method for the surface modification of SiHA nanoparticles that enables the engraftment of fluorescent markers onto the surface of the nano particulate. The method involves the functionalisation of SiHA nano particles with the silane MPTS via covalent bonding, which in turn presents a thiol functional group from the particle surface. The influence of the modification method on the physicochemical properties of the material were determined using X-ray diffraction (XRD), X-ray fluorescence (XRF), Zeta potential (ZP) measurement, and Fourier Transform Infra-red Spectroscopy (FTIR). The surface modification of the SiHA particles was evaluated by the attachment of the commercially available thiol reactive probe fluorescein-5-maleimide (Fig. 1) and demonstrated by live cell confocal fluorescence imaging of the particles after internalisation by MC3T3 cells.
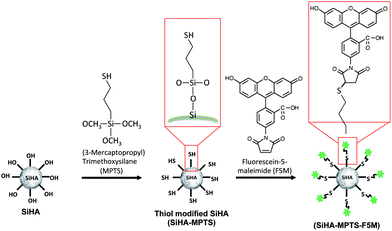 |
| | Fig. 1 Schematic diagram of the thiol functionalisation process of SiHA using MTPS and the subsequent labelling with fluorescein-5-maleimide (F5M). | |
2 Experimental
2.1 HA/SiHA synthesis, functionalisation and dye labelling
HA and SiHA synthesis: HA was synthesised by a wet chemical precipitation method. A 250 mL solution of (NH4)2HPO4 (239 mM) and a 350 mL solution of (CaNO3)2·4H2O (264 mM) were prepared with Millipore water, which was pre-boiled under reflux for 2 hours to remove CO2 and cooled to room temperature. The (CaNO3)2·4H2O was transferred to a closed glass reaction vessel during magnetic stirring at 200 rpm and nitrogen bubbling. The pH of the solution was adjusted to 10 by the addition of NH4OH. The (NH4)2HPO4 solution was added to the reaction vessel drop wise by burette, while maintaining the pH of the reaction at 10 by addition of NH4OH, then left to age overnight. The resulting HA precipitate was washed 5 times with Millipore water by centrifugation at 4000 rpm for 10 minutes and resuspended in water for later use at a concentration of approximately 10 mg mL−1. SiHA was synthesised using the method outlined above, but with a 250 mL aqueous solution of Si(OCOCH3)4 (41 mM) and (NH4)2HPO4 (203 mM) added drop wise to a 350 mL aqueous solution of (CaNO3)2·4H2O (290 mM). Attachment of thiol functional groups via MPTS: 230 mg of HA/SiHA in double distilled water was centrifuged at 4000 rpm for 10 min and the supernatant removed. The particles were then resuspended in 16.4 mL of absolute ethanol filtered with a 0.22 μm pore size syringe filter before adding 100 μL of 3-mercaptopropyltrimethoxysilane (MPTS). The samples were mixed using a ThermoMixer (ThermoMixer Comfort, Eppendorf UK Ltd., UK) at 37 °C for 3 hours at a mixing speed of 1000 rpm and then washed 5 times with double distilled water by centrifuging at 4000 rpm for 10 minutes. Dye labelling with Fluorescein-5-Maleimide (F5M): A stock solution of 5.2 mg Fluorescein-5-Maleimide (Life Technologies Ltd, UK) in 1 mL PBS (without Mg2+ and Ca2+). 1 mL of MPTS functionalised SiHA/HA (SIHA/HA-MPTS) (approximately 6 mg solid material in water) was centrifuged at 4000 rpm for 10 min. The supernatant was removed and the pellet resuspended in 500 μL of PBS before adding 470 μL of the fluorescein-5-maleimide stock solution and mixed on a ThermoMixer for 2 h at 37 °C. Finally, the SiHA-MPTS-F5M particles were washed twice with absolute ethanol and five times with double distilled water (both filtered beforehand using a 0.2 μm pore size filter). As a control, SiHA particles, without thiol modification, were mixed with fluorescein-5-maleimide using the above method.
2.2 XRD
X-ray diffraction was used to determine the crystalline composition of the samples. HA/SiHA in solution was centrifuged at 4000 rpm for 10 min. The pellet was removed, dried in an oven at 65 °C overnight and ground into a fine powder. XRD patterns of the powder samples were obtained with a X-ray diffractometer (D5000, Bruker Corp. USA.) using the Cu Kα line. Data were collected from 2θ = 5 to 60° with a 0.02° step-size and a step time of 0.5 s/°. These as-precipitated samples are referred to as HA and SiHA herein. To allow a better comparison between HA/SiHA and the ICDD reference patterns, a proportion of the precipitated materials were sintered at 650 °C prior to XRD analysis. Sintered samples are referred to as HA-650 and SiHA-650 herein.
2.3 TEM
HA and SiHA samples were diluted from 10 mg mL−1 to 400 μg mL−1 and a 5 μL drop placed on mesh-400 copper TEM grid (Agar Scientific). Samples for TEM were imaged using a JEOL JEM 1200EX microscope with a beam energy of 80 kV.
2.4 XRF
HA and SiHA solution was dried in an incubator set to 85 °C. The elemental composition of each sample was also determined using an X-ray fluorescence spectrometer (S8 TIGER, Bruker Corp., U.S.A). Powder forms of the samples were prepared as described above. 500 mg of the sample powder was mixed with 2.5 grams of wax and pressed into a pellet.
2.5 FTIR
HA, SiHA, HA-MPTS and SiHA-MPTS powders were prepared as previously described and heated overnight at 85 °C to remove moisture prior to analysis. 2 mg of sample powder was mixed with 198 mg of KBr (1%w/w), milled and then pressed into a pellet. FTIR spectra were acquired using a ThermoScientific Nicolette 380 FTIR instrument (ThermoScientific, UK) and represent an average of 64 runs corrected with a background measurement of a 200 mg pure KBr pellet.
2.6 Zeta-potential measurements
HA, SiHA, HA-MPTS and SiHA-MPTS particle dispersions were mixed with 10 mM KCl solution at a concentration of 0.05 mg mL−1. The pH of the sample solutions was adjusted by the addition of 100 mM HCl and 100 mM KOH at 25 °C and left overnight to equilibrate. Zeta potential profiles as a function of cell z-position were collected using a Beckman Coulter Delsa Nano C. At each of the 9 z-positions 10 accumulations were performed repeated over three runs before repeating the whole process with a fresh sample three times. The pH of each sample was checked and recorded immediately before analysis, corrected (if necessary) and recorded again after analysis.
2.7 Quantitating thiol groups of the silane functionalised HA/SiHA
Thiol presentation on the particle surface was quantified based on an assay using 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB), also known as Ellman's Reagent, that binds to free –SH groups to form the yellow-coloured product 2-nitro-5-thiobenzoic acid (TNB). The absorbance of the assay solution is proportional to the concentration of free –SH groups in the sample. First, a ‘Reaction Buffer’ was made consisting of 100 mM Na(PO)4 and 1 mM EDTA in deionised water set to pH 8.0 by drop wise addition of Na(OH). 50 μL of Ellman's reagent solution (4 mg Ellman's reagent in 1 mL Reaction Buffer) was added to 2.50 mL of Reaction Buffer in a centrifuge tube with a separate tube for each sample plus an additional tube for a control sample. 250 μL of the functionalised HA/SiHA particle dispersion was then added to the tube, mixed using vortex mixer and incubated at room temperature for 15 minutes to form an ‘assay solution’. For the control, an addition 250 μL of reaction buffer was added to the tube instead of HA/SiHA particle solution. After incubation, 1 mL of the assay solution was transferred to a clean cuvette and the absorbance measured at 412 nm using a spectrophotometer (Cecil CE7500, Buck Scientific, US) zeroed on the control sample. The relationship between molar absorptivity, E (M−1 cm−1), and concentration of TNB, c (moles per litre), can be defined as follows:| | 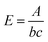 | (1) |
where A = measured absorbance and b = path length of the cuvette in centimetres. The concentration of TNB (and hence concentration of free –SH groups) in the solution in the cuvette was then calculated by solving eqn (1) for c and substituting b = 1 cm and E = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 150 M−1 cm−1. The number of moles of –SH groups in the assay solution, massay, was then calculated using eqn (2):
150 M−1 cm−1. The number of moles of –SH groups in the assay solution, massay, was then calculated using eqn (2):| | 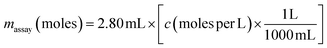 | (2) |
where the factor ‘2.80 mL’ represents the total volume of the assay solution when 250 μL of sample and 50 μL of Ellman's reagent is added to the 2.5 mL of reaction buffer. Given that the –SH groups were contributed solely by the 250 μL fraction of the assay solution from the addition of the particle solution, the final molar concentration of free –SH groups in the original particle solutions, Csample was determined to be:| | 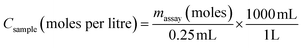 | (3) |
2.8 DNA binding to HA, SiHA and SiHA-MPTS
100 μg of particles in solution was mixed with 10, 20, 50, 80 and 100 μg of DNA (31149, Sigma-Aldrich, Dorset, UK) and incubated for 15 minutes at 37 °C. The particle-DNA solutions were then centrifuged and the absorption of the supernatant at 260 nm was measured using a Varian Cary 5000 Spectrophotometer (Agilent Technologies UK Limited, Stockport, UK). The absorbance of the sample was proportional to the amount of DNA not bound to the particle as determined through a calibration curve.
2.9 Live/dead assay
MC3T3 cells seeded on to glass coverslips with 2 mL of supplemented media containing 0.6 μg mL−1 of SiHA-MPTS-F5M particles. After 24 hours, 7 μL of Calcein AM and 25 μL of Propidium Iodide solutions in supplemented media to a total volume of 2 mL was added to the cells and incubated at 37 °C for 15 minutes prior to imaging with an epi-fluorescence microscope.
2.10 Uptake of HA/SiHA by MC3T3 osteoblast precursor cells
MC3T3 (passage 10) osteoblast precursor cells were cultured in DMEM (D6545, Sigma-Aldrich, Dorset, UK) supplemented with (v/v) 10% Fetal Bovine Serum (A15-105 Mycoplex, PAA, Yeovil, UK), 2.4% Hepes Buffer (H0887, Sigma-Aldrich, Dorset, UK), 2.4% L-Glutamine (G7513, Sigma-Aldrich, Dorset, UK) and 1% Penicillin/Streptomycin (P4333, Sigma-Aldrich, Dorset, UK). The cells were seeded at a density of 3 × 104 cells per quadrant in a 4-segmented live cell imaging dish (code: 627870, Greiner-Bio One Ltd., Gloucester, UK) and incubated overnight at 37 °C in the supplemented media. HA and SiHA in supplemented media was prepared at concentrations of 0 μg mL−1 and 300 μg mL−1, mixed using a vortex mixer and sonicated for 5 min before being kept in a water bath at 37 °C until required. After 24 h, the media was removed from each well and replaced with 1 mL of supplemented media before adding 1 mL HA or SiHA-media solutions to each quadrant while gently agitating the dish to ensure even distribution of the particles across the surface. The media was removed 24 hours after exposure to the particles, the cells in each quadrant washed three times with 1 mL PBS and replaced with 1 mL per quadrant of cell imaging media.
Fluorescein stained samples were imaged with a Zeiss LSM 710 ConfoCor 3 confocal system (Carl Zeiss Ltd, U.K.) attached to a Zeiss Axio Observer.Z1 inverted microscope and equipped with a Zeiss EC Plan-Neofluar ×63 NA = 1.40 oil objective lens, 488 nm laser diode, a 458 nm/488 nm beam splitter and a 34-channel spectral detector, which was used to divert fluorescence between 500 nm and 650 nm to a photomultiplier tube detector. Bright field images were obtained simultaneously with the fluorescence images by detecting the transmitted excitation laser light with a second photomultiplier tube detector.
3 Results and discussion
3.1 Structure and composition analysis
X-ray diffraction patterns of as-precipitated HA and SiHA along with HA and SiHA sintered at 650 °C (HA-650 and SiHA-650 respectively) were obtained and are shown in Fig. 2. The broad diffraction peaks of the as-precipitated HA and SiHA samples suggested that they were of low crystallinity and composed of sub-micron sized crystals, which was expected from a room temperature precipitation method without thermal treatment.17,29,30 Peaks were identified at around 26°, 32°, 40° and between 47 and 53° and appeared to align well with the most intense peaks from reference data for HA (ICDD PDF card no. 00-009-0432). However, it was evident that the broad HA peak at 32° in Fig. 2 was an envelope of the three most intense peaks in the reference pattern data and thus imposing a limit on quality of the match between the measured data and the HA reference. Two shoulder peaks were identified within the main peak of SiHA, but were still poorly resolved. Furthermore, it is known that the XRD patterns of various apatites could resemble those of as-precipitated HA and therefore an assessment of the chemical/structural changes in sintered samples is required in order to confirm the phase composition and structure of the original as-precipitated samples. Fig. 2 shows the XRD pattern of HA sintered at 650 °C for 2 hours. Dominant peaks were identified at 25.83°, 31.81°, 32.20°, 32.88° and were attributed to the (002), (211), (112) and (300) planes of HA respectively. Sharper and more intense peaks were observed in sintered HA (HA 650) compared to as-prepared HA, indicating an increase in the crystallinity of the sample. In contrast, sintering did not increase the crystallinity of SiHA since no change in the sharpness of the diffraction peaks and little or no change in the peak intensity was observed when compared to as-precipitated SiHA. These observations are consistent with other works that have explored the effect of sintering temperature on Si doped HA where the lack of change (or decrease in cases of Si doping above 2–3%wt) in crystallinity has been attributed to Si incorporation into the HA crystal lattice.13,31,32 Silicon substitution did not appear to change the angular position or relative intensity of the peaks when compared to HA and HA-650. Secondary phases consisting of α-TCP and β-TCP were not observed in the diffraction patterns of the HA or SiHA samples when compared to ICDD reference patterns 00-009-0348 and 00-009-0169 respectively. TCP phases were not detected in HA even after sintering at 900 °C, but SiHA decomposed into a mixture of HA and α-TCP (data not shown). The TEM images of HA and SiHA shown in Fig. 3 displayed the needle like morphology commonly reported with wet chemical precipitation methods.7,33,34 Additional phases such as α-TCP, β-TCP and CaCO3, which normally have a plate-like morphology, were not observed in either the HA or SiHA samples. The SiHA crystals appeared smaller and their edges were not as clearly defined compared with HA which may indicate that the SiHA sample was of lower crystallinity compared to HA. This observation corresponds well with the broad peaks observed for SiHA in the XRD patterns.
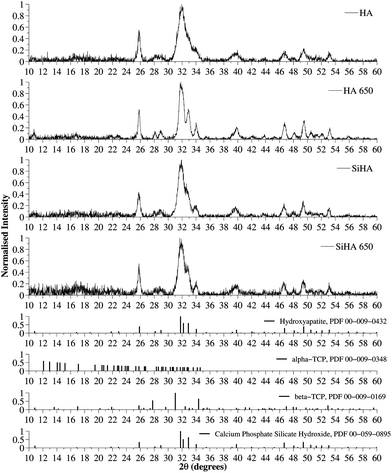 |
| | Fig. 2 XRD patterns of as precipitated HA/SiHA and HA/SiHA sintered at 650 degrees (HA 650/SiHA 650) and JCPDS card number 00-009-0432 (Hydroxyapatite). Intensity values of all samples were normalised to the most intense diffraction peak of the HA sample. The broad diffraction peaks suggested the samples were made of nanosized crystals and no other phases of calcium phosphate were detected. | |
 |
| | Fig. 3 TEM images of as-precipitated (a) HA and (b) SiHA. Both samples showed the well reported needle-like morphology, but the crystal edges are not as well defined compared to those of HA, indicating reduced crystallinity. | |
XRF was used to determine the %wt of Ca and P oxides and corresponding mole ratios of the elements are presented in Table 1. The Ca:P ratio of the HA sample was 1.64 and compares well with the theoretical Ca:P ratio of HA of 1.67 to within experimental error using this technique. Ca:P ratio of HA-MPTS (1.95) was higher than expected and may have been due to CO32− ions substituting the PO43− ions in the crystal structure, thus reducing the phosphorous content of the sample and hence increasing the Ca:P ratio.
Table 1 Ca:P ratios determined from XRF data and zeta potential data for HA, HA-MPTS, SiHA and SiHA-MPTS
| Sample |
Ca:P ratio |
Zeta potential (mV) at pH 7.4 |
| HA |
1.64 |
−1.97 |
| HA-MPTS |
1.95 |
−1.99 |
| SiHA |
1.97 |
−1.66 |
| SiHA-MPTS |
1.94 |
−11.66 |
Fig. 4 shows the FTIR spectra of HA, SiHA along with thiol-terminated silane functionalised HA and SiHA (HA-MPTS and SiHA-MPTS respectively). A summary of the peaks identified from the spectra, along with the chemical bond and mode the peak was assigned to, is presented in Table 2. The characteristic peaks of hydroxyapatite were identified in all of the samples including the OH modes (3569 cm−1 and 632 cm−1), the P–O stretching modes (1091, 1044–1032 and 962 cm−1), the O–P–O bending modes (602, 575–561 cm−1) and the O–P–O stretching modes (473, 463–460 cm−1) from the PO43− groups. The OH stretching mode at 3569 cm−1 was partially obscured by the water band between 2500 and 3800 cm−1 as the samples were not sintered prior to FTIR analysis. Evidence of some B-type carbonate substitution was found in both HA and SiHA by the presence of peaks located at 1454 cm−1, 1423 cm−1 and 873 cm−1. A peak assigned to the Si–O vibrational mode of the SiO44− ions was detected in the SiHA-MPTS sample at around 875 cm−1. Although the corresponding peak in the SiHA sample was not as well resolved, another SiO44− vibrational mode at 470 cm−1 SiO44− was identified. An additional source of Si–O–Si bonds could have come from the binding of MPTS via its O atom to the SiHA surface. This may explain why the peak was most prominent in SiHA-MPTS compared to HA-MPTS as the presence of SiO44− on the surface increased the number of potential binding sites for MPTS. The difficulty in detected all peaks relating to silicon substitution in both the SiHA and SiHA-MPTS sample could have been due these vibrational modes overlapping with the phosphate modes, which been noted to happen in the following cases: (i) 470 cm−1 (Si–O–Si band) and 472 cm−1 (the O–P–O bending modes of the phosphate groups) and (ii) 945 cm−1 (Si–O symmetric stretching mode) and the weak 962 cm−1 (P–O symmetric stretching mode). The translational mode of water associated to HA generally exists between 300 cm−1 and 600 cm−1, which would also obscure the peaks within this range related to the presence of SiO44−.
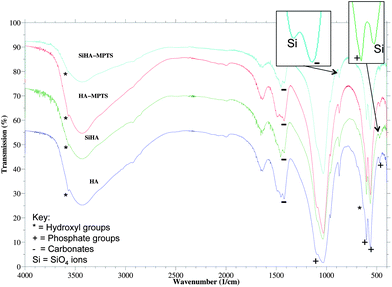 |
| | Fig. 4 FTIR spectra of as-precipitated HA and SiHA along with thiol functionalised HA and SiHA (HA-MPTS and SiHA-MPTS respectively). | |
Table 2 FTIR absorption peaks identified in HA, HA-MPTS, SiHA and SiHA-MPTS along with references to other works which support the peak assignments
| Peak (cm−1) |
Assignment |
|
|
Absorption peaks for hydroxyapatite:
|
| 3569 |
Stretching mode of OH group |
17, 31, 36 and 37
|
| 1454 |
ν
4 or ν3 bending mode of CO32− groups in A- and B-type carbonated HA |
17, 36 and 38
|
| 1423 |
B-type CO32− stretching mode |
36
|
| 1091, 1044–1032 |
Triply degenerated asymmetric stretching mode of P–O bond of the PO4 groups |
17, 31, 36, 39 and 40
|
| 962 |
Symmetric stretching mode of the P–O bonds |
17, 31, 36, 39 and 40
|
| 873 |
ν
4 or ν3 bending mode of CO32− groups in carbonated HA |
36
|
| 632 |
Vibrational mode of OH group |
31 and 36
|
| 602, 575–561 |
Triply degenerated O–P–O bending modes |
36
|
| 473, 463–460 |
Double degenerated O–P–O bending modes of the PO4 groups |
31 and 36
|
| |
|
Absorption peaks for silicates in SiHA:
|
| 880 |
Si–O vibrational mode of SiO44− |
39
|
| 875 |
Si–O vibrational mode of SiO44− |
31 and 38
|
| 470 |
Si–O–Si vibrational mode of SiO44− |
38
|
| |
|
Absorption peaks for MPTS:
|
| 1100 |
Si–O–C stretching from MPTS |
41
|
Evidence of surface modification was obtained from zeta potential measurements of HA, SiHA, HA-MPTS and SiHA-MPTS at pH 7.4, shown in Table 1. The zeta potential of HA and SiHA was measured to be −1.97 mV and −1.66 mV respectively. Most notably, the zeta potential of SiHA SiHA-MPTS was measured to be −11.66 mV and the change in the zeta potential suggested that this sample exhibited a different surface chemistry to both SiHA and HA. The result compares well with the work of Shyue et al. which also reported a decrease in the seat potential due to the presence of thiols.35 HA-MPTS did not show the same change in zeta-potential as SiHA-MPTS, which may further suggests that either none or significantly less thiol groups were attached to the HA surface in comparison to SiHA-MPTS. The presence of thiol groups was confirmed and quantified using an Ellman's reagent assay. For SiHA-MPTS, the thiol content was estimated to be 1.60 × 10−5 moles per mg of solid material. DNA binding efficiency experiments showed that 100 μg of SiHA-MPTS particles could completely bind up to 10 μg DNA whereas only 85% and 65% of this DNA mass was bound to 100 μg of unfunctionalised SiHA and HA respectively. At higher DNA masses of 50–100 μg, 20% of the DNA was bound to SiHA-MPTS while only 1–10% of the DNA was bound to SiHA and HA.
3.2 Demonstration of application: live cell imaging of SiHA internalisation by MC3T3 cells using a thiol reactive fluorescent probe
Using confocal fluorescence microscopy, unmodified SiHA treated with fluorescein-5-maleimide showed very weak fluorescence (Fig. 5b) in the regions of the image where particles could be seen in bright field (Fig. 5a and c), which demonstrated the effectiveness of the washing procedure in removing non-specifically bound dye molecules. The fluorescence from SiHA modified with MPTS (SiHA-MPTS) shown in Fig. 5e was brighter than the fluorescence from unmodified SiHA. Furthermore, the intensity of the fluorescence was even across the vast majority of the structures shown in Fig. 5d and f. The increased fluorescence intensity was attributed to the proportion of dye molecules bound to the thiols of the SiHA-MPTS since unbound molecules were expected to be removed during the washing procedure.
 |
| | Fig. 5 Bright field, confocal fluorescence and composite images of SiHA exposed to fluorescein-5-maleimide (SiHA-F5M) (a–c) and MPTS functionalised SiHA particles conjugated to fluorescein-5-maleimide dye (SiHA-MPTS-F5M) (d–f); specific binding of fluorescein-5-maleimide to the thiol group of SiHA-MPTS was demonstrated by the even fluorescence across the particulates (d–f) and very weak non-specific fluorescence in the samples not treated with MPTS (a–c). | |
The images in Fig. 6 are combined confocal fluorescence and brightfield images of MC3T3 cells after a 24 hour exposure to thiol-terminated silane functionalised SiHA particles conjugated to fluorescein-5-maleimide dye (SiHA-MPTS-F5M). Fluorescence could not be clearly seen in the low magnification image (Fig. 6a), which would most likely have been due to the low particle concentration and low numerical aperture of the objective lens used to acquire the image. However, some fluorescence was detected in multiple cells at a higher magnification (Fig. 6b). SiHA-MPTS-F5M was not found to be cytotoxic at a concentration of 0.6 μg mL−1 over 24 hours after performing a live-dead assay (Fig. S1†). In Fig. 6(c–e), the SiHA-MPTS-F5M particles appeared to form small aggregates, approximately 500–1000 nm in diameter, which aligned along the cell membrane. These structures could not be removed from the cell membrane despite repeated washing in PBS, indicating a strong affinity for the cell membrane. When focusing on an image plane through the middle of the cell (Fig. 6f–j), bright fluorescence was detected in new locations within the boundary of the cell as shown in the composite image. Furthermore, fluorescence from the few particles on the coverslip could no longer be detected at this new focal plane, demonstrating that any detected fluorescence was not an integration of fluorescence from particles at the bottom of or underneath the cell. Fluorescence spectroscopy of the sample during imaging revealed an emission profile matching that of the dye with an emission peak between 519 and 529 nm (Fig. S2†). The internalised structures measured approximately 400–500 nm in diameter and appeared to localise at various points within the cytoplasm of the MC3T3 cell, but could not determine whether this was the result of either (i) individual particles being internalised and then concentrated within the cell, or (ii) the aggregates being internalised as a whole. However, the localisation and strong intensity of the fluorescence did indicate that the fluorescent labels were still attached to the particle surface and photoactive after 24 hours in culture media and post internalisation. Internalised particles were observed again while focusing up towards the top of the cell.
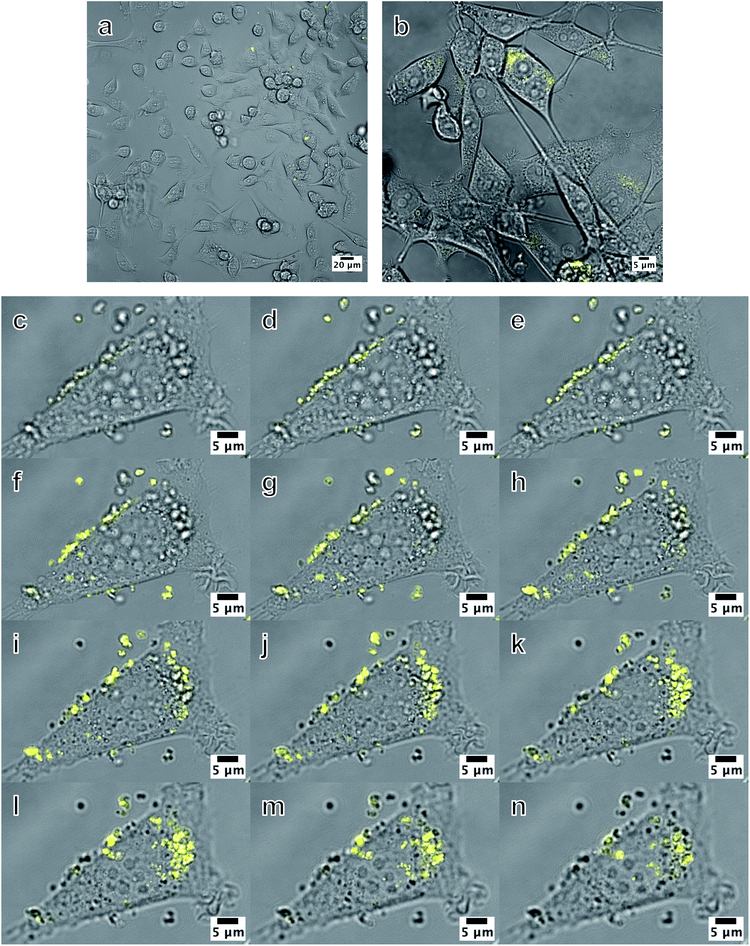 |
| | Fig. 6 Combined bright field and fluorescence images of MC3T3 cells exposed to 0.6 μg mL−1 SiHA-MPTS-F5M particles. (a) is a low magnification image showing a population of cells and (b) is a higher magnification image where fluorescence was just visible. Images (c)–(n) are slices from a confocal z-stack taken at: the coverslip/bottom of the cell plane (c–e), through the middle of the cell (f–j) and at the top of the cell (k–n). Bright localised fluorescence was observed within the cell and enabled internalised SiHA to be visually discriminated from the other cellular material of similar morphology and optical contrast as shown in the bright field images. The entire z-stack is presented as a movie in Multimedia ESI with the filename: ESI SiHA uptake Fig. 6. Movie.† | |
4 Conclusions
In this study, we have prepared nano crystalline SiHA using a wet chemical precipitation technique at room temperature without additional thermal treatment. Presentation of thiol functional groups on the surface of SiHA via silane functionalisation was achieved without perceptible change in the chemical composition of the HA or SiHA itself as determined by XRF. Zeta potential measurements showed a significant change in the surface chemistry of the SiHA particle surface, but not that of HA subjected to the functionalisation process. The presence of SiO44− in SiHA and SiHA-MPTS was identified by two different Si–O vibrational modes together with the characteristic reduction in the intensity of the OH peak in the FTIR spectra. The presence of MPTS could not be confirmed by FTIR alone as the Si–O–C stretching mode could not be reliably resolved from the phosphate peaks. However, thiols were detected using Ellman's reagent and showed that the using the functionalisation method on SiHA produced approximately 10−5 moles of thiols per milligram of solid material above the base line measurements for HA and HA-MPTS alone. Thiol group modification was visually demonstrated by the detection of fluorescence from fluorescein-5-maleimide specifically bound to the thiol groups of the modified SiHA surface despite extensive washing of the samples, suggesting that the thiol groups were covalently bonded to the SiHA surface via a Si–O–Si surface network. This was further demonstrated in the confocal fluorescence and bright field images of SiHA-MPTS-F5M internalised by MC3T3, where the bright fluorescence allowed the particles to be discriminated from other cellular material with similar morphology. Furthermore, the fluorescent labels remained attached to the particle and photoactive at for least 24 hours after internalisation. This method of functionalisation could allow time course tracking of internalisation of calcium phosphates by various bone cells in order to understand their localisation and fate during bone formation and resorption.
Acknowledgements
We would like to thank the EPSRC for funding this project (EPSRC ref: EP/F50053X/1), Advantage West Midlands for the provision of the zeta-sizer, Birmingham Advanced Light Microscopy (BALM) facility for access to the confocal microscope, Birmingham Science City for the provision of the XRD and XRF instruments and Dr Alan Smith (School of Applied Sciences, University of Huddersfield, Queensgate, Huddersfield) for the access to an alternative zeta-sizer. We would also like to acknowledge Parastoo Jamshidi (School of Chemical Engineering, University of Birmingham, Birmingham), for her assistance with XRD and XRF measurements; Dr Javier Salado (Inorganic Chemistry, Zientzia eta Teknologia Fakultatea, UPV/EHU, 644 Postakutxa, 48080, Bilbo); Laura Rowley (PSIBS, University of Birmingham, Birmingham.) for the loan of the heating mantle and their involvement in technical discussions; and Dr. James Bowen (School of Chemical Engineering, University of Birmingham, Birmingham), for his technical support.
References
- E. Landi, L. Orlandi, G. Spagna, A. Tampieri and N. Zaffaroni, Key Eng. Mater., 2000, 192, 901–904 Search PubMed.
- A. Nzihou and P. Sharrock, Waste Biomass Valoriz., 2010, 1, 163–174 CrossRef CAS.
- V. Sokolova, A. Kovtun, O. Prymak, W. Meyer-Zaika, E. A. Kubareva, E. A. Romanova, T. S. Oretskaya, R. Heumann and M. Epple, J. Mater. Chem., 2006, 17, 721–727 RSC.
- C. Ribeiro, C. Barrias and M. Barbosa, Biomaterials, 2004, 25, 4363–4373 CrossRef CAS.
- S. J. Kalita, A. Bhardwaj and H. A. Bhatt, Mater. Sci. Eng., C, 2007, 27, 441–449 CrossRef CAS.
- J. Klesing, S. Chernousova and M. Epple, J. Mater. Chem., 2012, 22, 199–204 RSC.
- S. C. Lee, H. W. Choi, H. J. Lee, K. J. Kim, J. H. Chang, S. Y. Kim, J. Choi, K.-S. Oh and Y.-K. Jeong, J. Mater. Chem., 2007, 17, 174–180 RSC.
- K. Ganesan, A. Kovtun, S. Neumann, R. Heumann and M. Epple, J. Mater. Chem., 2008, 18, 3655–3661 RSC.
- T. White, C. Ferraris, J. Kim and S. Madhavi, Rev. Mineral. Geochem., 2005, 57, 307–401 CrossRef CAS.
- N. Rameshbabu, T. Sampath Kumar, T. Prabhakar, V. Sastry, K. Murty and K. Prasad Rao, J. Biomed. Mater. Res., Part A, 2007, 80A, 581–591 CrossRef CAS.
- A. Bigi, E. Boanini, C. Capuccini and M. Gazzano, Inorg. Chim. Acta, 2007, 360, 1009–1016 CrossRef CAS.
- E. S. Thian, J. Huang, S. M. Best, Z. H. Barber, R. A. Brooks, N. Rushton and W. Bonfield, Biomaterials, 2006, 27, 2692–2698 CrossRef CAS.
- A. M. Pietak, J. W. Reid, M. J. Stott and M. Sayer, Biomaterials, 2007, 28, 4023–4032 CrossRef CAS.
- M. Shie, H. Chang and S. Ding, Int. Endod. J., 2012, 45, 337–345 CrossRef CAS.
- M. Vallet-Regí and D. Arcos, J. Mater. Chem., 2005, 15, 1509–1516 RSC.
- H. J. Lee, S. E. Kim, H. W. Choi, C. W. Kim, K. J. Kim and S. C. Lee, Eur. Polym. J., 2007, 43, 1602–1608 CrossRef CAS.
- I. Gibson, S. Best and W. Bonfield, J. Biomed. Mater. Res., 1999, 44, 422–428 CrossRef CAS.
- D. Arcos, J. Rodrıguez-Carvajal and M. Vallet-Regí, Solid State Sci., 2004, 6, 987–994 CrossRef CAS.
- J. Engstrand, A. López, H. Engqvist and C. Persson, Biomed. Mater., 2012, 7, 035013 CrossRef.
- B. M. Barth, R. Sharma, E. İ. Altınoǧlu, T. T. Morgan, S. S. Shanmugavelandy, J. M. Kaiser, C. McGovern, G. L. Matters, J. P. Smith and M. Kester,
et al.
, ACS Nano, 2010, 4, 1279–1287 CrossRef CAS.
- A. Bahdod, S. El Asri, A. Saoiabi, T. Coradin and A. Laghzizil, Water Res., 2009, 43, 313–318 CrossRef CAS.
- K. Kandori, S. Oda, M. Fukusumi and Y. Morisada, Colloids Surf., B, 2009, 73, 140–145 CrossRef CAS.
- X. Qiu, Z. Hong, J. Hu, L. Chen, X. Chen and X. Jing, Biomacromolecules, 2005, 6, 1193–1199 CrossRef CAS.
- K. Duan and R. Wang, J. Mater. Chem., 2006, 16, 2309–2321 RSC.
- M. Nelson, G. Balasundaram and T. J. Webster, Int. J. Nanomed., 2006, 1, 339 CAS.
- N. D. Meeks, S. Rankin and D. Bhattacharyya, Ind. Eng. Chem. Res., 2010, 49, 4687–4693 CrossRef CAS.
- A. Wani, E. Muthuswamy, G. H. L. Savithra, G. Mao, S. Brock and D. Oupickỳ, Pharm. Res., 2012, 1–12 Search PubMed.
- S. B. Hartono, S. Z. Qiao, J. Liu, K. Jack, B. P. Ladewig, Z. Hao and G. Q. M. Lu, J. Phys. Chem. C, 2010, 114, 8353–8362 CAS.
- I. Mobasherpour, M. S. Heshajin, A. Kazemzadeh and M. Zakeri, J. Alloys Compd., 2007, 430, 330–333 CrossRef CAS.
- M. H. Santos, M. d. Oliveira, L. P. d. F. Souza, H. S. Mansur and W. L. Vasconcelos, Mater. Res., 2004, 7, 625–630 CrossRef CAS.
- Z. Qiu, G. Li, Y. Zhang, J. Liu, W. Hu, J. Ma and S. Zhang, Biomed. Mater., 2012, 7, 045009 CrossRef CAS.
- E. Thian, J. Huang, M. Vickers, S. Best, Z. Barber and W. Bonfield, J. Mater. Sci., 2006, 41, 709–717 CrossRef CAS.
- E. Bouyer, F. Gitzhofer and M. Boulos, J. Mater. Sci.: Mater. Med., 2000, 11, 523–531 CrossRef CAS.
- V. Uskoković and D. P. Uskoković, J. Biomed. Mater. Res., Part B, 2011, 96, 152–191 CrossRef.
- J.-J. Shyue, M. R. De Guire, T. Nakanishi, Y. Masuda, K. Koumoto and C. N. Sukenik, Langmuir, 2004, 20, 8693–8698 CrossRef CAS.
- S. Koutsopoulos, J. Biomed. Mater. Res., 2002, 62, 600–612 CrossRef CAS.
- S. Sprio, A. Tampieri, E. Landi, M. Sandri, S. Martorana, G. Celotti and G. Logroscino, Mater. Sci. Eng., C, 2008, 28, 179–187 CrossRef CAS.
- A. Bianco, I. Cacciotti, M. Lombardi and L. Montanaro, Mater. Res. Bull., 2009, 44, 345–354 CrossRef CAS.
- T. Tian, D. Jiang, J. Zhang and Q. Lin, Mater. Sci. Eng., C, 2008, 28, 57–63 CrossRef CAS.
- J. Vandiver, D. Dean, N. Patel, C. Botelho, S. Best, J. D. Santos, M. A. Lopes, W. Bonfield and C. Ortiz, J. Biomed. Mater. Res., Part A, 2006, 78A, 352–363 CrossRef CAS.
- C. Wu, T. Xu and W. Yang, J. Membr. Sci., 2003, 224, 117–125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Movie of confocal z-stack used in Fig. 6, S1: epi-fluorescence images from the live/dead assay of SiHA-MPTS-F5M, and Fig. S2: fluorescence spectrum of SiHA-MPTS-F5M inside MC3T3 cells. See DOI: 10.1039/c3tb20775g |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a