 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-responsive physical gels from poly(meth)acrylic acid-containing crosslinked particles: the relationship between structure and mechanical properties†
Silvia S.
Halacheva
*a,
Tony J.
Freemont
b and
Brian R.
Saunders
*a
aUniversity of Manchester, Materials Science Centre, Grosvenor Street, Manchester, M1 7HS, UK. E-mail: silviya.halacheva@manchester.ac.uk; Brian.Saunders@manchester.ac.uk; Tel: +44 (0)161 306 5944
bDivision of Regenerative Medicine, School of Medicine, Stopford Building, Oxford Road, Manchester, M13 9PT, UK
First published on 21st June 2013
Abstract
Gels that feature high internal porosity and have both high elasticity and ductility have potential to provide immediate load support and enable subsequent tissue regeneration of damaged soft tissue if combined with cells. Herein, we report results from a recent investigation of novel poly(methyl methacrylate-co-methacrylic acid), (PMMA-MAA) and poly(ethyl acrylate-co-methacrylic acid), (PEA-MAA) biodegradable, pH-sensitive particle gels which are with high porosity, elasticity and ductility. These gels formed at physiological pH range and are potentially injectable. The particles were prepared using solvent evaporation. They were functionalized by crosslinking the MAA groups of the particles via bis-amide formation with either cystamine (CYS) or 3,3′-dithiodipropionic acid dihydrazide (DTP) which simultaneously incorporated reversibility due to the presence of disulphide bonds within the crosslinker. The crosslinked particles were observed by dynamic light scattering to swell appreciably in size upon increasing the pH. Concentrated dispersions formed elastic and ductile physical gels within the physiological pH range. A key finding of this study was that for crosslinked particles of similar composition the formation of considerably more elastic and ductile gels was observed from the most lightly crosslinked particles. Furthermore, compared to the PMMA-MAA/CYS and PEA-MAA/CYS gels, those formed from DTP-crosslinked particles had higher elasticity, thicker pore walls and improved interconnectivity. For the PMMA-MAA/DTP gels an elastic modulus value as high as 100 kPa and a yield strain greater than 100% were observed for a gel containing only 5 wt% of particles. The improved mechanical properties of these new gel-forming dispersions imply that they now have good potential for future application as injectable gels for regenerative medicine.
1 Introduction
The use of injectable polymer/cell dispersions to repair tissue defects could constitute a superior minimally-invasive alternative to current approaches which involve surgery. As well as significantly improving patient comfort and recovery time, the injectable therapy would be simple and eliminate post-treatment complications.1,2 Recently, dispersions of star-shaped poly((S)-lactic acid)-based particles, prepared by emulsification of the polymer in glycerol, were successfully utilized as an injectable delivery system for hyaline cartilage regeneration in rabbits.3 However, the dispersions did not form physical gels and therefore could not provide immediate load support. The key essential properties of materials intended for soft tissue repair are biocompatibility, biodegradability, interconnected porosity, mechanical strength, durability and ease of preparation. Furthermore, for potential load-bearing applications they are required to transform to a high elasticity gel immediately upon application of appropriate conditions.4 A range of biocompatible hydrogels have been extensively explored for biomaterial applications.5–12 In particular, stimuli-responsive degradable hydrogels have recently attracted a great deal of attention in drug delivery, film coating, tissue engineering and in fabrication of stimuli-responsive sensors due to their ability to undergo reversible sol–gel transition in response to external chemical, physical or biochemical triggers such as temperature and pH.13–15The porosity of a hydrogel can be easily tuned by varying the structure of the polymer, the crosslinker and the extent of inter-chain crosslinks in the hydrogel's matrix. Elucidation of the structure–properties relationship can potentially enable control over the loading and release behaviour.16,17 In order to facilitate their removal from the body on demand a range of crosslinkers with biodegradable groups, such as disulphides, have been investigated.7,18–20 The disulphide linkage can be readily cleaved to the corresponding thiol(s) in the presence of reducing agents such as glutathione, which occurs naturally in the body.21 Disulphide-functionalized hydrogels have been widely explored for the preparation of stimuli-responsive gelators, reversibly crosslinked polyplexes and polymer capsules.22–24 To achieve long-lasting, injectable hydrogels that can incorporate cells into their 3D structure Lee et al.25 developed a hyaluronic acid, (HA)–Pluronic composite with tissue adhesive properties. Shu et al.26 prepared thiol modified HA using carbodiimide-mediated hydrazide chemistry. The HA hydrogels were then formed under physiological conditions by air oxidation of thiol groups to disulphides. Ghosh et al.27 crosslinked the thiol-modified HA with poly(ethylene glycol) diacrylate, (PEGDA) to form HA-based hydrogels. Their mechanical properties were evaluated as a function of the concentration and the molecular weight of the crosslinker. Synthetic physical hydrogels that incorporate biocompatible hydrophilic poly(ethylene oxide), poly(glycidol) segments along with biodegradable domains such as poly(lactic acid), poly(caprolactone) and poly(glycolic acid) or biocompatible poly(propylene glycol) have been also explored.28–32 Chitosan and alginate have emerged as promising polysaccharide-based biomaterials in the synthesis of natural 3D stimuli-responsive hydrogel scaffolds.33,34 Unfortunately, conventional hydrogels possess several disadvantages which have restricted their use in tissue engineering and drug delivery applications. Some of the disadvantages of these materials include low solubility at physiological pH, higher swelling capacity which leads to rapid dissolution and premature release of drugs, potential variability of the natural materials, depending on the source, as well as the necessity to ensure purity prior to implantation to avoid activating an immune response. Furthermore, these scaffolds often have a limited range of mechanical properties and may need to be optimized for stem cell culture.35 Increasing the elasticity and ductility remains a challenge for load-bearing applications.6,36
To generate durable microgels with interconnected micrometre-scale porosity and increased elasticity, Liu et al.37 prepared permanently crosslinked poly(MMA-co-MAA/EGD) (where EGD is ethylene glycol dimethacrylate) microgels by emulsion polymerization. Bird et al.38 prepared vinyl-functionalized pH-responsive hollow poly(MMA-co-MAA) particles which were covalently interlinked to give injectable pH-responsive gels. However, the poly(MMA-co-MAA/EGD) and vinyl-functionalized poly(MMA-co-MAA) particle-based gels did not contain any labile groups, such as disulphides, and therefore were stable under physiological conditions.
Recently, a simple technique for preparing hollow, biodegradable polymer-based particles that are both pH-responsive and redox-sensitive has been developed.21,39 The method used is based on a solvent evaporation approach and does not require the use of colloid templates, layer-by-layer assembly, small-molecule surfactants or highly hazardous materials.40–46 The particles were based on amphiphilic PMMA-MAA or PEA-MAA copolymers and were formed via copolymer migration to the dichloromethane–water interface. An advantage of this system for potential application is that MMA is used in bone cement and MAA is a hydrolysis product of MMA. In that work up to 37% of the available carboxylic acid groups in MAA were crosslinked via bis-amide formation with CYS, which simultaneously imparted elasticity and reversibility to the crosslinking procedure due to the disulphide bonds, which can be readily broken upon treatment with a reducing agent. The crosslinked hollow particles swelled in the physiological pH range and formed physical gels from their concentrated dispersions. The gels had micrometer-scale interconnected porosity and were readily disassembled upon addition of glutathione. Unfortunately, the elastic modulus values of those gels were low and did not exceed 4 kPa. Those gels were too weak to support a moderate biomechanical load.
MMA-containing biomaterials are frequently exceptional in mechanical strength and show various biological benefits in clinical applications.47 We have continued our investigation of poly(meth)acrylate based-biomaterials for use as tissue scaffold and report herein on the synthesis, characterisation and mechanical properties of new types of high porosity, high elasticity PMMA-MAA and PEA-MAA gels, which are potentially suitable for future use in minimally-invasive tissue repair. EA is used here to demonstrate the generality of our approach. A physiologically compatible strategy for preparation of the PMMA-MAA and PEA-MAA particle-based gels was employed, utilizing DTP or CYS as crosslinkers. DTP has been used previously for the preparation of reversibly crosslinked hyaluronan hydrogels, which demonstrated good potential for in situ encapsulation.26,27,48,49 The strategy for the preparation of PMMA-MAA/DTP- and PEA-MAA/DTP-based gels involves incorporation of the DTP crosslinker into the particle shells using carbodiimide-mediated amide formation, followed by formation of the physical gels under physiological conditions. As the disulphide linkage of the DTP can be readily cleaved to the corresponding thiol in the presence of reducing agents, the formation of PMMA-MAA/DTP- and PEA-MAA/DTP-based gels offers an alternative redox route (cf. CYS) for biodegradation. Herein, the mechanical properties and porosity of the CYS- or DTP-containing particle gels are explored and compared in terms of the particles' compositions, structures and pKa values. A variety of complementary characterisation methods have been employed, including dynamic light scattering (DLS), potentiometric titration, scanning electron microscopy, optical microscopy and dynamic rheology.
2 Experimental section
Materials
Tetrahydrofuran (Aldrich, anhydrous, inhibitor-free, ≥99.9%), dichloromethane (Aldrich, HPLC grade, ≥99.8%), methanol (Aldrich, HPLC grade, ≥99.9%), 2,2′-azobis(2-methylpropionitrile) (AIBN, Aldrich, 98%), methyl methacrylate (MMA, Aldrich, ≥98.5%), methacrylic acid (MAA, Aldrich, 99%), ethyl acrylate (EA, Fluka, ≥99.5%), cystamine dihydrochloride (CYS, Aldrich, 96%), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, Aldrich, ≥99%), N-hydroxysuccinimide (NHS, Aldrich, 98%), 3,3′-dithiodipropionic acid (Aldrich, 99%), hydrazine monohydrate (Fluka, purum, ≥98%), concentrated sulphuric acid (Aldrich, ACS reagent, 95–98%), poly(vinyl pyrrolidone) (PVP, Aldrich, average molecular weight 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), were used as received from the chemical supplier. Milli-Q water was used throughout, unless otherwise stated. Reactions requiring anhydrous conditions were performed under a positive pressure of nitrogen.
000 g mol−1), were used as received from the chemical supplier. Milli-Q water was used throughout, unless otherwise stated. Reactions requiring anhydrous conditions were performed under a positive pressure of nitrogen.
Synthesis of copolymers
All free-radical polymerisations were carried out in anhydrous THF, under a nitrogen atmosphere, using AIBN as an initiator. The copolymer abbreviations used here identify the percentage MAA content. For example, PMMA-29MAA contains 29 mol% MAA and 71 mol% MMA units. The following synthesis of PMMA-29MAA is representative of the procedure employed for the other copolymers. A three-neck flask, fitted with a condenser and nitrogen inlet, was charged with AIBN (0.25 g) and anhydrous THF (85 ml). The stirred solution was nitrogen purged at 55 °C for 30 min. A mixture of MMA (6.06 g, 0.7 equiv.), MAA (2.24 g, 0.3 equiv.) and AIBN (0.025 g) was dissolved in anhydrous THF (17 ml) and added to the solution at a uniform rate over a period of 90 min. After the feed the reaction was allowed to proceed at 70 °C overnight (18 h approx.). After cooling to room temperature, the reaction mixture was concentrated under reduced pressure to a volume of approximately 100 ml and then poured into 1000 ml of cold water. The precipitated polymer was removed by filtration, rinsed thoroughly with water and air-dried. Residual water was removed by freeze-drying. The molar ratio of MMA to MAA in the co-polymer was determined by 1H NMR spectroscopic analysis. The other co-polymer compositions were obtained in an analogous manner from appropriate mixtures of either MMA or EA, with MAA.Synthesis of 3,3′-dithiodipropionic acid dihydrazide
DTP was synthesized according to a previously reported method.50 Into a 250 ml flask were placed 3,3′-dithiodipropionic acid (5.00 g, 23.8 mmol) and methanol (50 ml). Three drops of concentrated sulphuric acid were added to the resulting solution and the mixture was refluxed for two hours. The reaction mixture was then cooled to room temperature and the methanol was evaporated under reduced pressure. The residue was then diluted with diethyl ether (60 ml), transferred to a separating funnel, washed sequentially with saturated NaHCO3 solution (30 ml) and water (30 ml). The organic phase was dried (MgSO4) and evaporated under reduced pressure to afford crude dimethyl 3,3′-dithiodipropionate as a transparent, pale amber oil (5.21 g, 92%), which was used without further purification.50 Dimethyl 3,3′-dithiodipropionate (5.00 g) was dissolved in methanol (100 ml) at room temperature. Hydrazine monohydrate (6.30 g, 6.0 equiv.) was then added and the reaction mixture stirred overnight (18 h approx.) at room temperature. The resulting suspension was filtered and the white solid washed sequentially with methanol (50 ml), water (10 ml) and methanol (25 ml), before being air-dried to constant weight. The desired product, DTP, was obtained as a white solid (3.10 g, 62%).51 The 1H and 13C NMR spectra of DTP in DMSO-d6 are presented in Fig. S1, ESI.†Particle preparation
A solvent evaporation method was used for the preparation of the PMMA-MAA and PEA-MAA particles. The following procedure is representative for the preparation of the non-crosslinked PMMA-MAA and PEA-MAA particles. Firstly, 2.0 g of PMMA-MAA was dissolved in 44 ml of a mixed CH2Cl2–MeOH (84:16, v/v) solvent. This was then added, at a uniform rate of 10 ml min−1, into 120 ml of water, containing 1 wt% PVP, sheared at 10000 rpm (whilst cooled to 0 °C), using a Silverson LR4 high speed mixer. The emulsification was continued for 30 seconds after addition of the polymer solution. The emulsion was then allowed to stir gently overnight to enable removal of the CH2Cl2. The product was purified by repeated centrifugation and redispersion in water in order to remove excess PVP. The resultant suspension was then filtered through a 50 μm filter and stored at 2–4 °C.
Preparation of crosslinked particles
The following procedure for the preparation of PMMA-20MAA/9CYS is representative of the general procedure employed: 4.25 ml of the concentrated PMMA-29MAA particle stock dispersion (8 wt%, 1 mmol COOH groups) was diluted with 18.4 ml of 0.1 M pH 6.0 phosphate buffer to afford a final concentration of 1.5 wt%. The pH of the buffer utilized depended upon the pKa of the particles. (The relationship between the pH at which crosslinking was performed and the particle pKa is discussed later.) To the resulting stirred suspension, NHS (0.140 g, 1.22 equiv.) was added followed by EDC hydrochloride (0.153 g, 0.80 equiv.). Stirring was continued for a further fifteen minutes before CYS (0.095 g, 0.42 equiv.) was added. The reaction mixture was then stirred at room temperature for 24 hours and the crosslinked particles were isolated by centrifugation, followed by three cycles of redispersion in water and centrifugation. The DTP crosslinked particles were prepared in an analogous manner by substituting DTP for CYS. The crosslinked particle abbreviation used here identifies the mol% non-functionalized MAA groups and the mol% CYS or DTP incorporated into the particles. For example PMMA-20MAA/9CYS contained 20 mol% MAA and 9 mol% CYS.Analysis
Proton magnetic resonance spectra (1H NMR) were recorded on Bruker Avance 400 and 500 MHz spectrometers. Chemical shifts (δH) are quoted in parts per million and are referenced to the residual solvent peak. GPC analyses were performed on a Shodex R101 Refractive index detector. Samples were dissolved in anhydrous, inhibitor-free THF (1 mg ml−1) at room temperature. The columns contained Phenomenex Phenogel 5 μm beads with 500, 104 and 106 Å pore sizes. The flow rate was 1 ml min−1. Potentiometric titration was conducted using a Mettler Toledo DL15 titrator. Measurements were performed on 40 ml of a 1 wt% non-crosslinked dispersion using standardized NaOH solutions. All pKa values reported here are apparent values. Optical microscopy was conducted with an Olympus BX41 microscope and white transmitted light. Dispersions were deposited on SEM stubs by evaporation at room temperature. Thin sections of gel were rapidly frozen in liquid nitrogen and residual moisture removed by freeze-drying. Rheology measurements were performed at 25 °C using a TA AR-G2 rheometer and a 250 μm gap with a 20 mm diameter steel plate at 25 °C. All strain-sweep measurements were conducted at a frequency of 1 Hz. All frequency-sweep measurements were conducted at a strain of 0.1%. A particle concentration of 5.0 wt% was used for these studies. DLS measurements were performed on a light scattering photometer consisting of a 50 mW He/Ne laser, operating at 633 nm, with a standard avalanche photodiode (APD) and 90° detection optics connected to a Malvern Zetasizer Nano ZS90 autocorrelator. The measurements were carried out at pH values ranging from 5.4–8.0 and at a constant solution concentration of 0.1 wt%. The dispersions were treated with 1.0 M NaOH solution until the desired pH value was reached, then allowed to equilibrate for 15 min. The hydrodynamic diameter (Dh, in nm) of the particles was measured at 90° using a Malvern Zetasizer Nano ZS90. At least 10 correlation functions were analyzed per sample, at each pH value, in order to obtain an average measurement. The volume swelling ratio, Q, of the PMMA-MAA and PEA-MAA particles, functionalized with either CYS or DTP, was estimated using Q = (Dh/Dh(collapse))3, where Dh and Dh(collapse) are the hydrodynamic diameters of the particles at a given pH (well below the particles' pKa values) and in the collapsed non-swollen state, respectively. All Dh(collapse) values were measured at the lowest pH values tested. The degree of neutralization, α, of the available RCOOH groups was calculated using the Henderson–Hasselbalch equation: α = 1/(10pKa−pH + 1).3 Results and discussion
Synthesis and characterisation of the copolymers
PMMA-MAA and PEA-MAA were synthesized by free-radical polymerisation of the appropriate mixture of monomers in the presence of AIBN using a previously reported method.52 The copolymers were characterized by GPC and 1H NMR. The weight average molecular weight and polydispersity of the copolymers were determined by GPC (Fig. S2, ESI†). The copolymers had molecular weights ranging from 18120 to 40390 g mol−1 (Table 1). GPC analyses gave monomodal distributions with polydispersities ranging from 1.75 to 1.96. The copolymers' structural compositions were determined from their 1H NMR spectra in DMSO-d6 (Fig. S3, ESI†). The compositions of the PMMA-MAA samples were calculated by examining the relative ratios of signal intensities of the methoxyl protons of MMA groups and the methyl and methylene protons of both MMA and MAA groups. The composition of the PEA-30MAA polymer was determined from the ratios of the signal intensities of the methylene protons of the ethyl ester units to the methylene protons of both EA and MAA groups (see Fig. S3, ESI†). The experimental values obtained by 1H NMR were in good agreement with the theoretical values (Table 1).
| Composition | M w (g mol−1) | PDIa | Mol% MAA | pKac | ||
|---|---|---|---|---|---|---|
| Theor. | Exper.b | Exper.c | ||||
| a Determined by GPC. b Determined by 1H NMR. c Determined by potentiometric titration. | ||||||
| PMMA-29MAA | 40390 |
1.89 | 30 | 29 | 34.3 | 7.3 |
| PMMA-38MAA | 31130 |
1.89 | 40 | 38 | 38.5 | 6.9 |
| PMMA-65MAA | 23410 |
1.96 | 70 | 65 | 68.3 | 6.3 |
| PEA-30MAA | 20370 |
1.75 | 30 | 30 | 26.1 | 6.2 |
| PEA-65MAA | 18120 |
1.85 | 70 | 65 | 66.9 | 5.6 |
Preparation and characterisation of PMMA-MAA and PEA-MAA non-crosslinked particles
Dispersions of PMMA-29MAA-, PMMA-38MAA-, PMMA-65MAA-, PEA-30MAA- and PEA-65MAA-based non-crosslinked particles were prepared by the sequential emulsification-solvent evaporation protocol described previously (Scheme 1a).21 An important discovery in this work was that the rate of addition of the copolymer solution greatly affected the dispersion stability. In this study an optimized addition rate of the CH2Cl2–MeOH/copolymer solution was established (10 ml min−1), which was much lower than that employed previously53 and resulted in the formation of homogeneous, colloidally stable particle dispersions (Fig. 1a). These dispersions remained stable for at least two months after their preparation. After the crosslinking step, highly elastic gels were formed when the pH was raised by treatment with sodium hydroxide solution. Optical micrographs of PMMA-38MAA dispersions taken after CH2Cl2 evaporation are presented in Fig. 1a. Some of the collapsed particles are with doughnut-like morphology, which is often reported for hollow particles.54 A representative SEM image of non-crosslinked PEA-30MAA particles is shown in Fig. 1b. Both the optical (Fig. 1a) and SEM images of all non-crosslinked dispersions studied indicated collapsed objects of various sizes and shapes. The particles' diameters range from less than 1 μm up to around 5 μm. The polydispersity of the samples was due to the turbulent shear flow during the high shear mixing.21,39 If required, the polydispersity could be reduced by fractionation.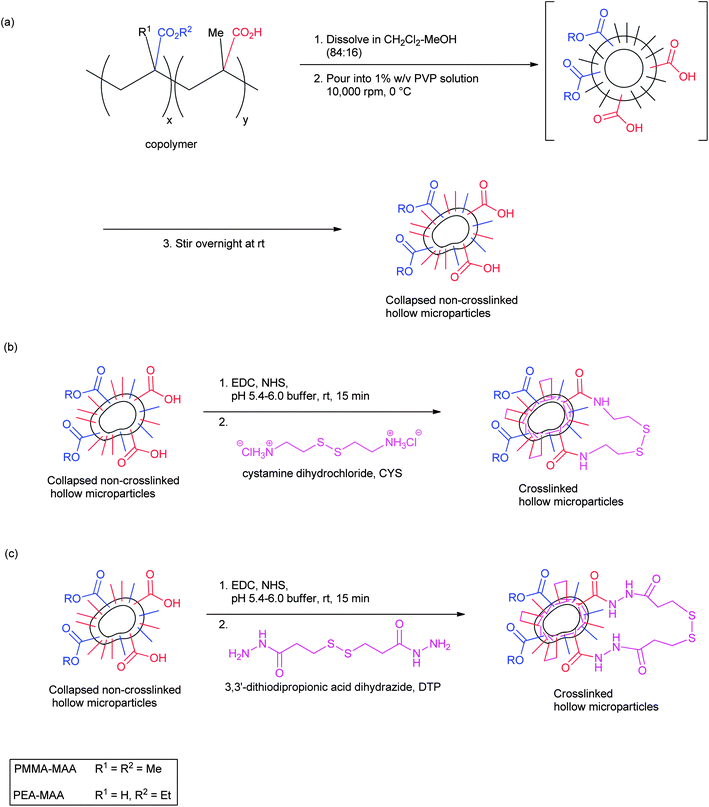 | ||
| Scheme 1 Method for preparation of pH-responsive, crosslinked doughnut-like particles: (a) depicts the preparation of the non-crosslinked particles; (b) and (c) depict the preparation of crosslinked particles using CYS and DTP, respectively. For clarity the PVP stabilizer is not shown. | ||
 | ||
| Fig. 1 (a) and (b) show an optical micrograph and SEM image for PMMA-38MAA (pH = 6.5) and PEA-30MAA (pH = 6.0), respectively. (c) and (d) show an optical micrograph and SEM image of PMMA-20MAA/9CYS (pH = 6.5) and PEA-10MAA/20DTP (pH = 6.0), respectively. | ||
The formation of the PMMA-MAA and PEA-MAA particles is mainly controlled by the amphiphilic nature of the copolymers. The formation of spherical core–shell micelles has been reported for both random and crosslinked PMMA-MAA and PEA-MAA, poly(styrene-b-methacrylic acid), poly(S-b-MAA), poly(isobutylene-b-methacrylic acid), poly(IB-b-MAA), poly(styrene-b-acrylic acid) and poly(S-b-AA) copolymers in aqueous solution.55–57 In the present study, PMMA-MAA and PEA-MAA particles were prepared using the solvent evaporation approach. This type of technique has been previously used to manufacture solid polymeric microparticles or core–shell micelles from amphiphilic block copolymers (such as poly(n-butyl methacrylate)-b-poly(2-methacryloyloxyethyl phosphorylcholine)).58–60 The solvent evaporation approach has also been used to generate hollow particles.61,62 For the earlier investigated PMMA-MAA and PEA-MAA copolymer analogues, the formation of hollow particles shortly after emulsification was attributed to the polymers' precipitation at the CH2Cl2 droplet/water interface due to transfer of methanol to the aqueous phase.21 Unfortunately, based on the optical and SEM images of the present PMMA-MAA and PEA-MAA non-crosslinked particles (Fig. 1) it is difficult to unambiguously determine the particle morphologies present. However, a coexistence of hollow particles and solid particles is favoured in the aqueous dispersions of the new PMMA-MMA and PEA-MAA. This is supported by the doughnut-like particles highlighted in Fig. 1a and c. However, the gels formed from the crosslinked particles, under physiological conditions, are shown to have high elasticities and ductilities, which was the ultimate aim of this research.
The molar percentages (mol%) of MAA in the PMMA-MAA and PEA-MAA non-crosslinked particles were measured by potentiometric titration. The values calculated by potentiometric titration and those estimated from the 1H NMR spectra of the copolymers were in reasonable agreement with each other and with the theoretical values (Table 1). The pH vs. degree of neutralisation (α) plots for PMMA-MAA and PEA-MAA dispersions are shown in Fig. S4, ESI.† The pKa values decrease as the MAA content increases (see Table 1). The pKa values for the PMMA-MAA particles were higher than those of PEA-30MAA, which is proposed to be the result of lower chain flexibility due to the higher glass transition temperature of poly(MMA).57,63
Preparation and characterisation of crosslinked particles
The particles were crosslinked by bis-amide formation through their carboxylic acid groups with the symmetrical diamines CYS or DTP, using EDC-mediated coupling methodology (Scheme 1b and c).64–66 It is important to note that although the amide bonds formed are stable, the crosslinking itself is reversible due to the presence of labile disulphide bonds within the crosslinker molecules.26,27,67 In all crosslinking reactions, the same molar proportions of reagents (EDC, NHS, CYS or DTP) relative to the number of available COOH groups were employed. Elemental analysis measurements revealed that the proportion of available RCOOH groups which were successfully reacted with either CYS or DTP were 5–31% for the MMA-containing copolymers and 9–67% for the PEA-MAA (Table 2). These results were lower than the theoretically predicted values which would correspond to a 100% chemical yield (see the Experimental section). It was found that the extent of crosslinking achieved was highly dependent upon the pH of the reaction mixture and this effect has been reported for related systems.64–66 For example, a previously reported investigation into the reaction between carboxylic acids and EDC, as well as subsequent amide formation, showed that a pH of 3.5–4.5 was optimal for the activation of carboxyl groups by EDC.59 However, the amine groups of CYS (pKa of CYS = 8.35)68 would be extensively protonated under such acidic conditions and therefore less reactive,65,67 but as the pH increases toward neutrality the percentage of successfully activated carboxyl groups falls sharply. In contrast, the terminal nitrogen atoms of hydrazides have been found to retain their nucleophilicity at acidic pH values which would render amines relatively unreactive.26,49,69 This allowed the highest extent of crosslinker incorporation to be achieved for DTP (Table 2, entry 7). However, we show below that these high levels of DTP incorporation were not beneficial in terms of the resulting gel's mechanical properties.| Entry | Copolymer | RCONHRa (%) | Crosslinked copolymer particles composition | Q max |
|
G ′max (kPa) |
|
|---|---|---|---|---|---|---|---|
a Percentage of RCONHR formed.
b Swelling ratio at 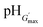 , In each case this value of Q corresponded to the maximum swelling ratio.
c pH at which gels with G′max are formed.
d The maximum storage modulus value.
e Yield strain at , In each case this value of Q corresponded to the maximum swelling ratio.
c pH at which gels with G′max are formed.
d The maximum storage modulus value.
e Yield strain at 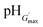 . The swelling ratio for entry 8 was not determined, as the particles were already swollen at the lowest pH tested (pH 5.4). For entries 1, 2, 4 and 8 the . The swelling ratio for entry 8 was not determined, as the particles were already swollen at the lowest pH tested (pH 5.4). For entries 1, 2, 4 and 8 the 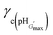 values were not determined because they exceeded the maximum γ values used in the experiments. values were not determined because they exceeded the maximum γ values used in the experiments.
|
|||||||
| 1 | PMMA-29MAA | 31 | PMMA-20MAA/9CYS | 5.9 | 7.0 | 8.7 | — |
| 2 | 14 | PMMA-25MAA/4DTP | 11.4 | 7.0 | 56.3 | — | |
| 3 | PMMA-38MAA | 37 | PMMA-24MAA/14CYS | 3.4 | 7.0 | 5.4 | 17.7 |
| 4 | 5 | PMMA-36MAA/2DTP | 11.9 | 7.0 | 100.0 | — | |
| 5 | PMMA-65MAA | 31 | PMMA-45MAA/20DTP | 4.7 | 6.5 | 0.2 | 43.7 |
| 6 | PEA-30MAA | 33 | PEA-20MAA/10CYS | 11.5 | 6.5 | 7.3 | 53.9 |
| 7 | 67 | PEA-10MAA/20DTP | 8.4 | 6.5 | 1.4 | 18.3 | |
| 8 | PEA-65MAA | 9 | PEA-59MAA/6DTP | — | 6.5 | 24.2 | — |
Consideration of the pKa values of the non-crosslinked particles was of critical importance during the crosslinking reactions (Scheme 1b and c), as the dispersions dissolved when the solution pH reached their corresponding pKa values. For PMMA-29MAA and PMMA-38MAA particles (Table 2, entries 1–4) the crosslinking reactions were performed in pH 6.0 phosphate buffer, whereas, due to their relatively low pKa values of 5.6–6.3, PMMA-65MAA, PEA-30MAA and PEA-65MAA particles, (Table 2, entries 5–8) were crosslinked in pH 5.4 buffer. The collapsed crosslinked PMMA-20MAA/9CYS and PEA-10MAA/20DTP particles were visualized by optical microscopy (Fig. 1c) and SEM (Fig. 1d). The collapsed morphology was also apparent for the respective parent non-crosslinked particles (see Fig. 1a and b). Some of the particles folded inwards upon themselves, a feature which has been previously observed for other related systems.21,39,54
Dynamic light scattering data
The sizes of the non-crosslinked and crosslinked particles were investigated by DLS as a function of the solution pH. The measurements were carried out at pH values ranging from 5.4–8.0. Examples of size distributions for PMMA-38MAA-, PEA-30MAA- and PMMA-29MAA-based non-crosslinked particles are shown in Fig. 2a as well as in Fig. S5a, ESI.† The Dh values are summarized in Table S1, ESI.† For the dispersions of non-crosslinked PMMA-29MAA (Fig. S5a, ESI†), PMMA-65MAA and PEA-30MAA, the distributions were monomodal at all pH values tested. For the PMMA-38MAA (Fig. 2a) and PEA-65MAA dispersions two modes particles slow (Dslowh) and fast (Dfasth) (small particles) were apparent at pH 6.0–6.5. The data in Table S1 and Fig. S5a, ESI† show that upon increasing the pH any large particles which were present gradually dissociated into smaller aggregates and eventually the particles dissolved.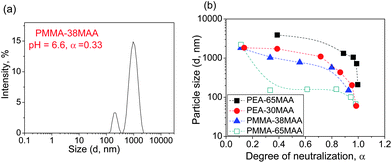 | ||
| Fig. 2 Size distribution function data for the parent non-crosslinked particles are shown in (a). The Dh as a function of α are shown in (b). For PEA-65MAA and PMMA-38MAA, only the Dslowh are considered. | ||
Fig. 2b shows the variation of the particle size (Dh) with α. The data in Fig. 2b show that for all systems, particle size decreases as the value of α increases, resulting in complete dissolution of the non-crosslinked particles at 100% neutralisation (α = 1). The increasing solubility of the particles with increasing pH is due to the ionisation of RCOOH groups along the polymer chain, which causes the polymer chains to repel each other due to their mutual negative charges and thus weaken their hydrophobic interactions.70,71 Eventually (at high α) the polymer chains became hydrophilic enough to overcome the intermolecular forces between adjacent water molecules and dissolve. The particles shrink in size due to individual chains undergoing this process of dissolution.72 As will be shown below, this process can lead to particle swelling rather than disintegration if interchain crosslinks are present. For PMMA-38MAA and PEA-30MAA the Dh decreases gradually with α over the entire course of neutralisation, reflecting the comparatively lower solubility of these copolymers. The Dhvs. α curves exhibit a gentle slope in α, ranging from ∼0.1 to 0.8 and the slope becomes steeper as α increases until complete neutralisation is reached. This suggests that the particles may undergo a conformational transition during the course of neutralisation.57,73 For PMMA-65MAA and PEA-65MAA the particle size decreased sharply as α increased, indicating that dissociation of the carboxylic acid groups for these particles leads to a correspondingly greater increase in copolymer chain solubility compared to their less hydrophilic, (meth)acrylate-rich counterparts. Compared to the PMMA-65MAA, the particles formed from PEA-65MAA are larger, which could be due to the greater flexibility of PEA compared to PMMA segments.
Compared to the non-crosslinked particles, dispersions of particles crosslinked with CYS or DTP showed an initial sharp increase in the particles' diameter, i.e., pH-triggered swelling (see Fig. 3). This was followed by a peak in Dh and then a subsequent decrease at higher pH values after the Dh reached its maximum value. This was due to pH-triggered dissolution (see Fig. 3c and d). Representative crosslinked particle size distributions for PMMA-24MAA/14CYS, PEA-20MAA/10CYS and for PMMA-25MAA/4DTP are presented in Fig. 3a and b and in Fig. S5b, ESI.† The corresponding Dh are summarized in Table S1, ESI.† For the crosslinked particle dispersions containing MMA, the distributions were monomodal at all pH values tested. For the PEA-20MAA/10CYS- (Fig. 3b), PEA-10MAA/20DTP- and PEA-59MAA/6DTP-based crosslinked particles the distributions were typically bimodal or an intermediate mode was also apparent. Although the fast and intermediate particles appear to swell to a lesser extent than the slow particles, similar variations in their Dh values with changes in the solution pH were clearly observed (see Table S1, ESI†).
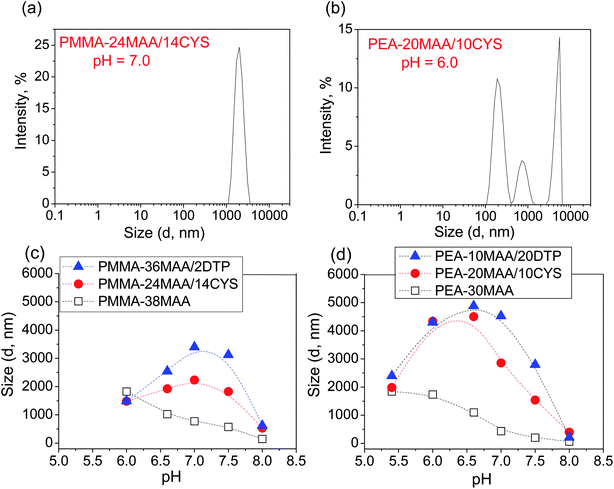 | ||
| Fig. 3 Effect of pH on the Dh of crosslinked and non-crosslinked particles. Selected size distribution functions for crosslinked particles are shown in (a) and (b). The Dh as a function of pH for (c) PMMA-38MAA, PMMA-36MAA/2DTP and PMMA-24MAA/14CYS and (d) PEA-30MAA, PEA-10MAA/20DTP and PEA-20MAA/10CYS. For PMMA-38MAA and PEA-20MAA/10CYS, Dh refers specifically to Dslowh only. | ||
For the particles crosslinked with either CYS or DTP, the maximum swelling values (Qmax) are presented in Table 2. For the PMMA-20MAA/9CYS, PMMA-25MAA/4DTP, PMMA-24MAA/14CYS and PMMA-36MAA/2DTP particles Qmax is observed at pH 7.0 (Fig. 3c), whereas for the PMMA-45MAA/20DTP, PEA-20MAA/10CYS and PEA-10MAA/20DTP this maximum is shifted downwards to pH 6.5 (Fig. 3d). This correlates well with the pKa values of the corresponding (parent) non-crosslinked particles (see Table 1). The Q values for all crosslinked particle dispersions at various pH values are listed in Table S1, ESI.† Swelling ratios of up to 11.5 and 11.9 for CYS- and DTP-functionalized particles, respectively, were measured. Unfortunately, for PEA-59MAA/6DTP the swelling ratio was not measured as the particles were already swollen at pH 5.4 (Table S1, ESI,† entry 13).
The crosslinked particles initially expand to swollen hydrated particles as the pH increases and this process is driven by electrostatic repulsion between negatively charged carboxylate groups. Whereas the non-crosslinked particles gradually disintegrate into unimers or small clusters when this electrostatic repulsion exceeds the hydrophobic attraction between MMA or EA groups, the polymer chains of the swollen crosslinked particles are effectively held together by their crosslinks (see Fig. 3c and d). However, disulphide groups, such as those present in CYS and DTP, are known to be readily cleaved under alkaline conditions.74,75 As a consequence the crosslinked particles also succumb to disintegration and for this reason the pKa values of the crosslinked particles (necessary in order to correlate solution pH with α values) were not determined.
Close examination of the pH-dependent swelling data results (Fig. 3 and Table 2) reveals some important general trends. Firstly, between particles of similar composition such as PMMA-20MAA/9CYS and PMMA-24MAA/14CYS, the larger swelling ratio (of about 5.9 at pH 7.0) was observed for the former, which features a lower level of crosslinking (see Table 2, entries 1 and 3). The same trend is present for the dispersions of PMMA-36MAA/2DTP, PMMA-25MAA/4DTP and PMMA-45MAA/20DTP where the larger swelling ratio (of about 11.9 at pH 7.0) is found for the PMMA-36MAA/2DTP particles (see Table 2, entries 2, 4 and 5). This is due to the increased expansion of the polymer chains as the extent of the crosslinking decreases.27 Particles composed of EA-containing copolymers were found to swell more at a given pH than their structurally isomeric MMA-based counterparts. This can be explained by their relatively lower pKa values, which results in a correspondingly greater extent of carboxyl group ionisation at a particular pH. For example, compare PMMA-20MAA/9CYS with PEA-20MAA/10CYS, where the EA-copolymer particles, with an approximately equivalent percentage of functionalized COOH groups, swell to almost twice the volume (see Table 2, entries 1 and 6). This difference also has a contribution from the increased chain mobility due to more flexible EA segments. The same trend occurs for the dispersions of PMMA-45MAA/20DTP and PEA-10MAA/20DTP, which feature swelling ratios of 4.7 and 8.4, respectively (Table 2, entries 5 and 7). Furthermore, particles crosslinked with DTP swell to a larger extent than those which are crosslinked with a comparable proportion of CYS, presumably due to the increased length of the DTP molecule. For example, for PMMA-24MAA/14CYS the swelling ratio at pH 7.0 was 3.4, whereas for PMMA-45MAA/20DTP particles at the same pH, the value was 4.7 (Table 2, entries 3 and 5). This is particularly illustrative of the lowered restriction toward the particles' expansion afforded by DTP, as the notably higher level of crosslinking, which reduces swelling ratios, has been adequately compensated for. The effect of the crosslinker structure (CYS versus DTP) upon the mechanical properties of the gels is discussed below.
Particle gel morphology and dynamic rheology
For the PMMA-29MAA and PMMA-38MAA particles crosslinked with either CYS or DTP strong physical gels were formed at particle concentrations of only 5 wt% and in the pH range of 7.0–7.5, which is highly desirable for future potential application in vivo. Gelation occurred at pH 6.5–7.0 for PMMA-45MAA/20DTP, PEA-20MAA/10CYS, PEA-10MAA/20DTP and PEA-59MAA/6DTP. Formation of physical gels under these conditions occurred when the effective volume fraction occupied by the swollen particles exceeded that which permits inter-particle movement.21,38,39 The particles swelled to the point of inter-particle contact and this physically locked them in their locations. At this point the gels were able to distribute load throughout the network of physically interconnected particles.Representative SEM images of freeze-dried gels formed from swollen PMMA-24MAA/14CYS, PMMA-36MAA/2DTP and PMMA-25MAA/4DTP gels, at pH 7.0, are shown in Fig. 4 and in Fig. S6, ESI.† All gels studied feature a highly porous interconnected morphology, similar to what has been previously reported for analogous systems.21,38,39 Their pore sizes range from 1 to 5 μm and are comparable to those observed in the corresponding crosslinked particle precursors (Fig. 1). Compared to gels derived from PMMA-20MAA/9CYS, PEA-10MAA/20DTP, PMMA-45MAA/20DTP, PEA-20MAA/10CYS and PMMA-24MAA/14CYS (Fig. 4c), those gels which are formed from PMMA-36MAA/2DTP (Fig. 4a and b), PEA-59MAA/6DTP and PMMA-25MAA/4DTP (Fig. S6, ESI†) particles had much thicker pore walls and high interconnectivities. We stress that these new PMMA-36MAA/2DTP, PMMA-25MAA/4DTP and PEA-59MAA/6DTP gels also gave outstanding elasticity values (see below).
 | ||
| Fig. 4 SEM images of freeze-dried (a) and (b) PMMA-36MAA/2DTP and (c) PMMA-24MAA/14CYS gels formed at pH 7.0. | ||
The mechanical properties of the physical gels were evaluated by dynamic rheology measurements. Dispersions of the crosslinked copolymer particles (5 wt% concentration) were subjected to frequency-sweep studies from 0.1 to 100 Hz at a constant strain of 0.1%. The variation of G′ and tanδ (tanδ = G′′/G′) with the oscillatory frequencies for PMMA-24MAA/14CYS, PMMA-36MAA/2DTP at pH 7.0 and PEA-20MAA/10CYS, PMMA-45MAA/20DTP physical gels at pH 6.5 are shown in Fig. 5a and b. The data are characterized by G′ exhibiting a plateau in the whole frequency range which is indicative of a stable crosslinked network. Within the frequency range 1–10 Hz all systems behaved as elastic gels with tanδ values less than 1.0, i.e., G′ > G′′ (Fig. 5b). At higher frequencies, from 20–100 Hz, the structures did not have time to relax upon application of the deformation stress which resulted in tanδ values increasing to greater than 1.0 and occurrence of network breakdown. The observed differences in G′ arise from the different number density of elastically effective chains in the gels which correspond well with those obtained from the strain-sweep test (Fig. 5c and d). The lowest G′ values were observed for the heavily crosslinked PMMA-45MAA/20DTP. For this physical gel the frequency at which tanδ = 1.0 (a critical frequency, fcrit) occurred was about 20 Hz, compared to 80–90 Hz for the other PMMA-containing gels. This corresponds to a critical relaxation time (τcrit = 1/fcrit) of about 0.05 s for PMMA-45MAA/20DTP and around 0.011–0.013 s for rearrangements of the PMMA-24MAA/14CYS and PMMA-36MAA/2DTP physical gels. The data for PEA-20MAA/10CYS had a higher fcrit (about 100 Hz) and a shorter τcrit (<0.01 s). This shorter relaxation time corresponds to an increased rate at which the inter-particle network repairs under dynamic strain and is consistent with the greater mobility of the PEA chains.57,63 For the PMMA-containing gels the data are consistent with a decrease in chain mobility as the extent of crosslinking increases.
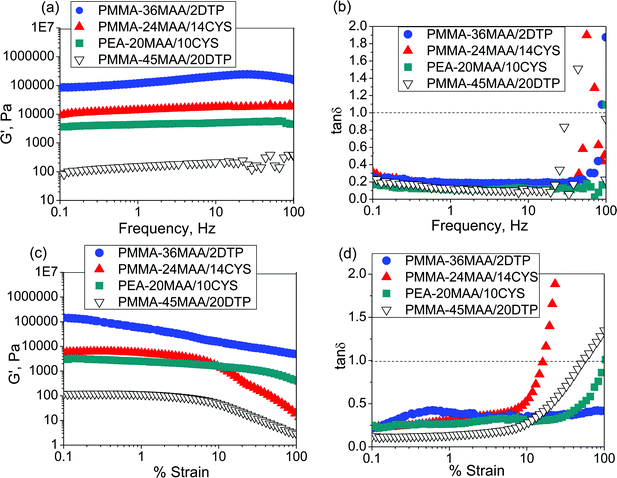 | ||
| Fig. 5 Frequency-sweep data (a) and (b) and strain-sweep data (c) and (d) for PMMA-36MAA/2DTP, PMMA-24MAA/14CYS, PEA-20MAA/10CYS and PMMA-45MAA/20DTP at pH = 7.0. The frequency-sweep data were measured at a strain of 0.1%. The strain-sweep measurements were conducted at a frequency of 1 Hz. | ||
Strain-sweep dynamic rheology experiments were performed on the crosslinked particle gels over a broad pH range (from pH 6.0–11.0). Fig. 5c and d show data for PMMA-24MAA/14CYS, PMMA-36MAA/2DTP, PEA-20MAA/10CYS and PMMA-45MAA/20DTP measured at pH = 7.0. For strains (γ) less than 10%, the gels had tanδ values less than 1.0. The critical yield strain (γc) is the γ value corresponding to tanδ = 1.0. When γ is greater than γc more energy is dissipated (as heat) than stored and the gel is considered to be fluid. Throughout the strain range studied the concentrated dispersions of PMMA-24MAA/14CYS, PMMA-45MAA/20DTP, PEA-20MAA/10CYS (Fig. 5c and d) and PEA-10MAA/20DTP particles at pH = 6.5 (Fig. S9e and f, ESI†) showed both fluid-like and gel-like behaviour. At low deformations the dispersions were more elastic, with tanδ < 1.0 (G′ > G′′), whereas at higher deformations the response was clearly non-elastic. However, under the same conditions, the behaviour of the PMMA-36MAA/2DTP dispersion was distinctly different and showed entirely elastic responses at all strain percentages. Whilst there was a modest decrease in G′ for PMMA-36MAA/2DTP, with γ values greater than 0.15% (Fig. 5c) and a corresponding increase in tanδ (Fig. 5d), the values for tanδ remained well below 1.0 over the entire strain range studied (up to 100%). Elastic behaviour over the entire range of strain percentages tested was also observed for the PMMA-25MAA/4DTP, PMMA-20MAA/9CYS and PEA-59MAA/6DTP particle gels at pH = 7.0 and 6.5, respectively (see Fig. S8c, d, g, h, S9c and d, ESI†).
The variations of G′ and tanδ with pH measured at 1% strain for PMMA-24MAA/14CYS, PMMA-20MAA/9CYS, PMMA-36MAA/2DTP, PMMA-45MAA/20DTP, PEA-10MAA/20DTP and PEA-20MAA/10CYS are shown in Fig. 6 and S7, ESI.† The full set of data for all the gels are shown in Fig. S8 and S9, ESI.† Similar to what has been previously reported for analogous systems,21,39 the initial increase in pH resulted in an increase in G′ (Fig. 6a and b) and a decrease in tanδ (Fig. S7, ESI†). This is attributed to the swelling of the particles as the pH increased. As the particles swelled, and impinged on each other, a greater inter-particle contact area resulted and this, in turn, increased the number density of elastically effective chains within each particle that contributed to stress distribution. Gels with an optimum elastic response were observed at pH 6.5–7.5, which is the ideal pH range for future physiological applications. In this pH region maximum values of G′ (G′max) and minimum tanδ values were observed. Values for the G′max of each gel and the corresponding pH value 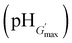 are shown in Table 2. Interestingly, the
are shown in Table 2. Interestingly, the 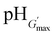 values correspond to the pH where Qmax occurs, demonstrating a strong correlation between maximum particle swelling and gel elasticity. Further increases in pH resulted in a decrease in G′ (Fig. 6a and b) and an increase in tanδ (Fig. S7, ESI†) as a consequence of decreased particle swelling and particle interpenetration. The particles partially deswelled due to the increased ionic strength. This decreased the extent of inter-particle contact and hence the number density of elastically effective chains. Interestingly, the corresponding decreases in G′ were more pronounced for PMMA-24MAA/14CYS and PMMA-45MAA/20DTP than for the related PMMA-36MAA/2DTP, PMMA-20MAA/9CYS gels (Fig. 6a). The first two systems were more highly crosslinked and had lower respective Qmax values than the latter. This implies that interparticle molecular entanglements (i.e., inter-particle contact area at the particle peripheries) were more extensive for the more highly swollen (PMMA-36MAA/2DTP and PMMA-20MAA/9CYS) particle gels. Interestingly, those gels were also less susceptible to pH-triggered dissolution. For those (physical) particle gels a decrease in crosslink density actually increased gel elasticity. This is opposite to the trend predicted from rubber elasticity theory and is a unique feature of these pH-responsive particle gels. Similar trends were observed for the PEA-containing gels. As seen in Fig. 6b the decrease in G′ was more pronounced for PEA-10MAA/20DTP compared to PEA-20MAA/10CYS. The PEA-20MAA/10CYS gel was less extensively crosslinked and also had a lower Qmax value.
values correspond to the pH where Qmax occurs, demonstrating a strong correlation between maximum particle swelling and gel elasticity. Further increases in pH resulted in a decrease in G′ (Fig. 6a and b) and an increase in tanδ (Fig. S7, ESI†) as a consequence of decreased particle swelling and particle interpenetration. The particles partially deswelled due to the increased ionic strength. This decreased the extent of inter-particle contact and hence the number density of elastically effective chains. Interestingly, the corresponding decreases in G′ were more pronounced for PMMA-24MAA/14CYS and PMMA-45MAA/20DTP than for the related PMMA-36MAA/2DTP, PMMA-20MAA/9CYS gels (Fig. 6a). The first two systems were more highly crosslinked and had lower respective Qmax values than the latter. This implies that interparticle molecular entanglements (i.e., inter-particle contact area at the particle peripheries) were more extensive for the more highly swollen (PMMA-36MAA/2DTP and PMMA-20MAA/9CYS) particle gels. Interestingly, those gels were also less susceptible to pH-triggered dissolution. For those (physical) particle gels a decrease in crosslink density actually increased gel elasticity. This is opposite to the trend predicted from rubber elasticity theory and is a unique feature of these pH-responsive particle gels. Similar trends were observed for the PEA-containing gels. As seen in Fig. 6b the decrease in G′ was more pronounced for PEA-10MAA/20DTP compared to PEA-20MAA/10CYS. The PEA-20MAA/10CYS gel was less extensively crosslinked and also had a lower Qmax value.
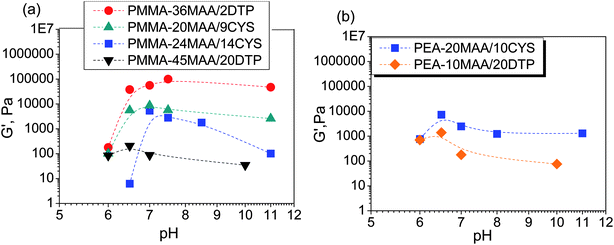 | ||
| Fig. 6 Variation of G′ with pH for PMMA-MAA and PEA-MAA-based crosslinked particles. | ||
The values of G′max for these PMMA- and PEA-containing gels are up to factors of 10 and 2 greater, respectively, compared to the related gels which were reported previously.21,39 Along with the optimized particle compositions, which resulted in formation of highly elastic MAA-containing physical gels, another reason for these high G′max values is the improved dispersion stability achieved by the slower feed employed during particle preparation. A particle gel that forms from well dispersed, swollen particles, will have a more uniform distribution of elastically effective chains per unit volume than a gel that forms from aggregated particles. A higher number density of elastically effective chains will, in turn, increase G′. For the PMMA-MAA-containing crosslinked particle gels the G′max values were found to decrease in the following order: PMMA-36MAA/2DTP > PMMA-25MAA/4DTP > PMMA-20MAA/9CYS > PMMA-24MAA/14CYS > PMMA-45MAA/20DTP. This also corresponds to an increasing extent of crosslinking and decreasing swelling ratio. For the PMMA-36MAA/2DTP gel G′max was 100.0 kPa, compared to 56.3 kPa for PMMA-25MAA/4DTP, 8.7 kPa for PMMA-20MAA/9CYS, 5.4 kPa for the PMMA-24MAA/14CYS and 0.2 kPa for PMMA-45MAA/20DTP (Table 2). The results correlate well with the DLS data, where larger pH-dependent swelling ratios (4.9–11.9) were observed for the PMMA-36MAA/2DTP compared to the PMMA-25MAA/4DTP (4.2–11.4), PMMA-20MAA/9CYS (4.6–5.9), PMMA-24MAA/14CYS (2.3–3.4) and PMMA-45MAA/20DTP (1.2–4.7) (see Table S1, ESI†). The same trend was observed for the PEA-MAA-containing gels where the highest G′max values of 24.2 kPa were observed for the gel derived from the lightly crosslinked PEA-59MAA/6DTP. The decrease in G′ can be rationalized by the increased crosslink concentration, which reduces the particles' swelling ratios (Table 2) and therefore the level of inter-particle contact, which was an integral part of gel formation.
An unexpected and new finding in this study was that functionalisation of PMMA- and PEA-containing particles with the DTP crosslinker caused a significant increase in G′max and a decrease in tanδ, relative to their CYS-functionalized analogues (Fig. 6 and S7, ESI†). The G′max values for the DTP-functionalized gels are up to factors of 11 greater than for the CYS-functionalized gels. Furthermore, the relatively thick-walled morphology observed by SEM for PMMA-36MAA/2DTP and PMMA-25MAA/4DTP gels (Fig. 4a, b and S6, ESI†) implies that the walls have higher elasticity and toughness than the thinner walls observed for their CYS-functionalized analogue (Fig. 4c). The remarkable rheology data for the DTP-crosslinked gels require further comment. Our data demonstrate for the first time that the identity of the crosslinker exerts a major influence upon the extent of particle swelling and particle-based gel elasticity. This may be rationalized by considering the molecular structures of the two crosslinkers. It is further proposed that the formation of stronger physical gels from the DTP-based crosslinked structures is associated with the different conformational constraints and rigidities of the CYS and DTP crosslink units. Due to the restricted rotation in the C–N bonds76–78 of the two amides at each end of the DTP molecule, it possesses less freedom of movement compared to the CYS crosslinked structure and is therefore able to confer a more rigid, extended conformation upon the elastically effective chains. This may in turn increase G′. Further study regarding the effect of crosslinker structure and physical gel elasticity is required in order to fully explore the potential benefits of this type of modification.
Fig. 7 shows the variation of γc with pH for the gels. In many cases the γc values were particularly high and exceeded the maximum γ values used in the experiments, which prevented determination of those values. This was the case for the highly elastic PMMA-36MAA/2DTP, PMMA-25MAA/4DTP, PMMA-20MAA/9CYS, PEA-59MAA/6DTP gels (Fig. S8 and S9, ESI†). Initially, γc for PMMA-24MAA/14CYS, PMMA-45MAA/20DTP and PEA-20MA/10CYS increased with pH, up until pH 7.0, followed by a decrease (Fig. 7). For each of these gels the maximum γc value was recorded at pH 6.5–7.0. Compared to the PMMA-24MAA/14CYS and PMMA-45MAA/20DTP, the lightly crosslinked PEA-20MAA/10CYS gel was more ductile and also had higher elasticity. The 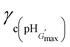 for the highly elastic gels such as PMMA-25MAA/4DTP, PMMA-36MAA/2DTP, PEA-59MAA/6DTP and PMMA-20MAA/9CYS exceeded 100% (Table 2). These gels were formed from the most lightly crosslinked particles and were able to deform rapidly and reversibly in response to the strain, whereas for the heavily crosslinked systems (e.g., PMMA-24MAA/14CYS and PEA-10MAA/20DTP) a decreased rearrangement ability of the polymer segments in response to strain would have reduced their ductility, as observed by experiment.
for the highly elastic gels such as PMMA-25MAA/4DTP, PMMA-36MAA/2DTP, PEA-59MAA/6DTP and PMMA-20MAA/9CYS exceeded 100% (Table 2). These gels were formed from the most lightly crosslinked particles and were able to deform rapidly and reversibly in response to the strain, whereas for the heavily crosslinked systems (e.g., PMMA-24MAA/14CYS and PEA-10MAA/20DTP) a decreased rearrangement ability of the polymer segments in response to strain would have reduced their ductility, as observed by experiment.
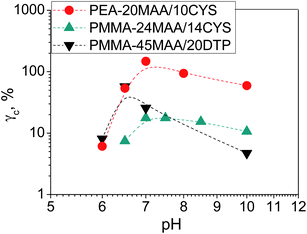 | ||
| Fig. 7 Variation of γc with pH for PMMA-MAA and PEA-MAA-based crosslinked particles. | ||
Particle composition–gel properties relationship
Numerous researchers have established a linear relationship between the value of G′ and a hydrogel's swelling ratio.27,79 These literature data are in good agreement with the swelling ratio studies which we have carried out with the new PMMA-MAA and PEA-MAA crosslinked particle gels. The swelling is pH-triggered and is caused by electrostatic repulsion between negatively charged carboxylate groups on neighboring polymer chains, which are effectively held together by their crosslinks. The non-crosslinked PMMA-MAA and PEA-MAA dispersions did not swell. Instead, they gradually disintegrated into unimers or small clusters when this electrostatic repulsion exceeded the hydrophobic attraction between MMA or EA groups (see DLS results, Fig. 3c and d). The new PMMA-MAA and PEA-MAA crosslinked particle-based gels were prepared under physiological pH conditions by employing biodegradable DTP or CYS as crosslinkers which were incorporated into the particle shells using carbodiimide-mediated amide formation. A conceptual model is proposed to explain the process of gelation for PMMA-MAA and PEA-MAA functionalized particles. Gelation occurs from sufficiently concentrated PMMA- or PEA-containing crosslinked particle dispersions (Fig. 8a) upon raising the pH to a level where particle swelling is maximized and the particles' shells begin to come into contact. The process of gelation clearly involves a system-wide structural transformation from discrete, crosslinked swollen particles (Fig. 8b), to a continuous undifferentiated, physical network of crosslinked copolymer chains (Fig. 8c). For both PMMA- and PEA-containing crosslinked particle gels the G′max values were found to decrease as the extent of crosslinking increased and the corresponding swelling ratio decreased (Table 2, compare entries 2, 4 and 5; 1 and 3; 6 and 7). Furthermore, the ductilities of these highly elastic gels were found to exceed 100% (Table 2). The decreases in gel elasticity and ductility could be attributed to a reduction in flexibility of the polymer chains due to the increased extent of crosslinks and, therefore, the level of particle interpenetration, which is an integral part of the gel formation process. The lightly crosslinked gels, which displayed the highest mechanical strengths, are able to deform rapidly and reversibly in response to an applied strain, unlike the heavily crosslinked systems. Interestingly, gels derived from DTP-crosslinked particles feature considerably higher mechanical strengths than their CYS-functionalized counterparts (see Table 2). Furthermore, the DTP-functionalized gels feature a thicker-walled morphology which implies a more efficient use of the crosslinker (see SEM images, Fig. 4 and S6, ESI†). The extra amide bonds (which feature restricted rotation, similar to alkene bonds)80 within the DTP molecule impart an increased degree of rigidity to the interchain linkages. This would, in turn, lead to a greater resistance toward compressive stress forces which are trying to push the chains closer together. The extra length of the DTP molecule compared to CYS also imparts an increased level of ductility to the DTP-functionalized particles' gels.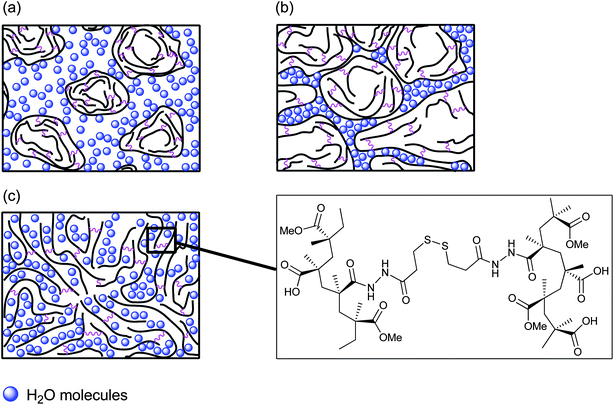 | ||
| Fig. 8 Representation of (a) PMMA-MAA and PEA-MAA crosslinked particles in non-swollen state at pH < 6.5–7.0; (b) PMMA-MAA and PEA-MAA crosslinked particles in swollen state at physiological pH; (c) the process of gelation for the PMMA-MAA/DTP crosslinked particles at pH = 6.5–7.0. | ||
4 Conclusions
In the present study we explored new types of high porosity, high elasticity and ductility PMMA-MAA- and PEA-MAA-based pH-responsive particle gels, which are potentially suitable for future use in minimally-invasive tissue repair applications. The gels were formed from concentrated dispersions of the particles, which were crosslinked with either CYS or DTP, in the physiological pH range. The CYS-crosslinked particles formed physical gels featuring much higher elasticities than those reported previously.21,39 Moreover, we have described for the first time the preparation and properties of particles derived from PMMA- and PEA-based copolymers crosslinked with DTP. The resulting gels were found to display excellent mechanical properties. We have also reported the pH-dependent study of the non-crosslinked and crosslinked particles using DLS. We have established control over the particles' properties, as well as the mechanical stability, interconnected porosity and ductility of their gels by varying the copolymer composition, the chemical structure of the crosslinker and the extent of crosslinking. Compared to the physical gels crosslinked with CYS, the DTP-crosslinked gels had both higher elasticity and ductility values. Furthermore, in this study for the first time particle-based physical gels with a G′max value of 100 kPa were prepared (with a yield strain greater than 100%). Compared to the CYS-crosslinked particle gels, PMMA-25MAA/4DTP, PMMA-36MAA/2DTP and PEA-59MAA/6DTP gels featured thicker pore walls and improved interconnectivity. This latter property is anticipated to encourage tissue growth, in addition to allowing the rapid diffusion of nutrients and oxygen in vivo. Because these gels can be formed under physiological pH conditions and the PMMA-containing systems contain components generally regarded as clinically safe, this work will bring their future application as injectable gels for soft tissue repair much closer.Acknowledgements
The authors gratefully acknowledge the EPSRC, the School of Materials and the School of Medicine, University of Manchester, for funding this project.Notes and references
- J. Elisseeff, K. Anseth, D. Sims, W. McIntosh, M. Randolph and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3104–3107 CrossRef CAS.
- S. M. Richardson, J. M. Curran, R. Chen, A. Vaughan-Thomas, J. A. Hunt, A. J. Freemont and J. A. Hoyland, Biomaterials, 2006, 27, 4069–4078 CrossRef CAS.
- X. Liu, X. Jin and P. X. Ma, Nat. Mater., 2011, 10, 398–406 CrossRef CAS.
- A. H. Milani, A. J. Freemont, J. A. Hoyland, D. J. Adlam and B. R. Saunders, Biomacromolecules, 2012, 13, 2793–2801 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- J. K. Oh, D. J. Siegwart, H. Lee, J. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5945 CrossRef CAS.
- D. S. Benoit, M. P. Schwartz, A. R. Durney and K. S. Anseth, Nat. Mater., 2008, 7, 816–823 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS.
- A. S. Hoffman, J. Controlled Release, 1987, 6, 297–305 CrossRef CAS.
- S. Rimmer, S.-P. Wilshaw, P. Pickavance and E. Ingham, Biomaterials, 2009, 30, 2468–2478 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- M. Dadsetan, K. E. Taylor, C. Yong, Ž. Bajzer, L. Lu and M. J. Yaszemski, Acta Biomater., 2013, 9, 5438–5446 CrossRef CAS.
- L.-H. Han, J. H. Lai, S. Yu and F. Yang, Biomaterials, 2013, 34, 4251–4258 CrossRef CAS.
- D. Buenger, F. Topuz and J. Groll, Prog. Polym. Sci., 2012, 37, 1678–1719 CrossRef CAS.
- H. Tai, D. Howard, S. Takae, W. Wang, T. Vermonden, W. E. Hennink, P. S. Stayton, A. S. Hoffman, A. Endruweit, C. Alexander, S. M. Howdle and K. M. Shakesheff, Biomacromolecules, 2009, 10, 2895–2903 CrossRef CAS.
- P. D. Thornton, S. M. R. Billah and N. R. Cameron, Macromol. Rapid Commun., 2013, 34, 257–262 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2005, 38, 8155–8162 CrossRef CAS.
- N. V. Tsarevski and K. Matyjaszewski, Macromolecules, 2005, 38, 3087–3092 CrossRef.
- J. K. Oh, C. Tang, H. Gao, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 5578–5584 CrossRef CAS.
- R. Bird, T. Freemont and B. R. Saunders, Soft Matter, 2012, 8, 1047–1057 RSC.
- C. Li, J. Madsen, S. P. Armes and A. I. Lewis, Angew. Chem., Int. Ed., 2006, 45, 3510–3513 CrossRef CAS.
- Y. Kakizawa, A. Harada and K. Kataoka, J. Am. Chem. Soc., 1999, 121, 11247–11248 CrossRef CAS.
- Y. Kakizawa, A. Harada and K. Kataoka, Biomacromolecules, 2001, 2, 491–497 CrossRef CAS.
- Y. Lee, H. J. Chung, S. Yeo, C. H. Ahn, H. Lee, P. B. Messersmith and T. G. Park, Soft Matter, 2010, 6, 977–983 RSC.
- X. Z. Shu, Y. Liu, Y. Luo, M. C. Roberts and G. D. Prestwich, Biomacromolecules, 2002, 3, 1304–1311 CrossRef CAS.
- K. Ghosh, X. Z. Shu, R. Mou, J. Lombardi, G. D. Prestwich, M. H. Rafailovich and R. A. F. Clark, Biomacromolecules, 2005, 6, 2857–2865 CrossRef CAS.
- T. Kissel, Y. Li and F. Unger, Adv. Drug Delivery Rev., 2002, 54, 99–134 CrossRef CAS.
- S. Lee and M. Vert, Macromolecules, 2003, 36, 8008–8014 CrossRef.
- S. Halacheva, S. Rangelov and V. M. Garamus, Macromolecules, 2007, 40, 8015–8021 CrossRef CAS.
- S. Halacheva, S. Rangelov and C. Tsvetanov, J. Phys. Chem. B, 2008, 112, 1899–1905 CrossRef CAS.
- S. Halacheva, S. Rangelov and Ch. Tsvetanov, Macromolecules, 2008, 41, 7699–7705 CrossRef CAS.
- Y. Cheng, X. Luo, G. F. Payne and G. W. Rubloff, J. Mater. Chem., 2012, 22, 7659–7666 RSC.
- C.-C. Wang, K.-C. Yang, K.-H. Lin, H.-C. Liu and F.-H. Lin, Biomaterials, 2011, 32, 7118–7126 CrossRef CAS.
- A. Vashist, S. Shahabuddin, Y. K. Gupta and S. Ahmad, J. Mater. Chem. B, 2013, 1, 168–178 RSC.
- M. A. Rice, K. R. Waters and K. S. Anseth, Acta Biomater., 2009, 5, 152–161 CrossRef CAS.
- R. Liu, A. H. Milani, T. J. Freemont and B. R. Saunders, Soft Matter, 2011, 7, 4696–4704 RSC.
- R. Bird, S. Tungchaiwattana, T. Freemont and B. R. Saunders, Soft Matter, 2012, 8, 3062–3066 RSC.
- R. Bird, T. J. Freemont and B. R. Saunders, Chem. Commun., 2011, 47, 1443–1445 RSC.
- A. N. Zelikin, Q. Li and F. Caruso, Chem. Mater., 2008, 20, 2655–2661 CrossRef CAS.
- F. Caruso, A. Caruso and H. Mohwald, Science, 1998, 282, 1111–1114 CrossRef CAS.
- O. J. Cayre and S. Biggs, J. Mater. Chem., 2009, 19, 2724–2728 RSC.
- G. Li, G. Liu, E. T. Kang, K. G. Neoh and X. Yang, Langmuir, 2008, 24, 9050–9055 CrossRef CAS.
- D. G. Shchukin, G. Sukhorukov and H. Möhwald, Angew. Chem., Int. Ed., 2003, 42, 4472–4475 CrossRef CAS.
- G. Sukhorukov, A. Fery and H. Möhwald, Prog. Polym. Sci., 2005, 30, 885–897 CrossRef CAS.
- M. Yang, J. Ma, C. Zhang, Z. Yang and Y. Lu, Angew. Chem., Int. Ed., 2005, 44, 6727–6730 CrossRef CAS.
- B. L. Seal, T. C. Otero and A. Panitch, Mater. Sci. Eng., R, 2001, 34, 147–230 CrossRef.
- G. D. Prestwich, D. M. Marecak, J. F. Marecek, K. P. Vercryusse and M. R. Ziebell, J. Controlled Release, 1998, 53, 93–103 CrossRef CAS.
- G. D. Prestwich, J. F. Marecak, K. P. Vercruysse, K. R. Kirker and J. S. MacMaster, in New Frontiers in Medical Sciences: Redefining Hyaluronan, ed. G. Abatangelo, Portland Press, London, 2000, pp. 181–194 Search PubMed.
- K. P. Vercruysse, D. M. Marecak, J. F. Marecek and G. D. Prestwich, Bioconjugate Chem., 1997, 8, 686–694 CrossRef CAS.
- Z. Rodriguez-Docampoa and S. Otto, Chem. Commun., 2008, 5301–5303 RSC.
- S. Kiatkamjornwong and S. Tessiri, J. Appl. Polym. Sci., 2002, 86, 1829–1837 CrossRef CAS.
- R. Bird, Complied PhD Thesis, University of Manchester, Faculty of Engineering and Physical Sciences, 2012.
- T. Chen, B. Du and Z. Fan, Langmuir, 2012, 28, 11225–11231 CrossRef CAS.
- L. F. Zang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef.
- L. F. Zhang, R. J. Barlow and A. Eisenberg, Macromolecules, 1995, 28, 6055–6066 CrossRef CAS.
- C. Wang, P. Ravi, K. C. Tam and L. H. Gan, J. Phys. Chem. B, 2004, 108, 1621–1627 CrossRef CAS.
- G. Tse, D. Blankschtein, A. Shefer and S. Shefer, J. Controlled Release, 1999, 60, 77–100 CrossRef CAS.
- M. J. Alonso, S. Cohen and H. Bernstein, in Nanoparticulate Drug Carrier Technology. Microparticulate Systems for Delivery of Proteins and Vaccines, Marcel Decker, New York, 1996, pp. 203–242 Search PubMed.
- Z. Jiao, N. Liu and Z. Chen, Pharm. Dev. Technol., 2012, 17, 164–169 CrossRef CAS.
- N. Li, G. Ye, Y. He and X. Wang, Chem. Commun., 2011, 47, 4757–4759 RSC.
- S. H. Im, U. Jeong and Y. Xia, Nat. Mater., 2005, 4, 671–675 CrossRef.
- J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Block, CRC Polymer Handbook, 4th edn, Wiley, New York, 1999 Search PubMed.
- J. W. Kuo, D. A. Swann and G. D. Prestwich, Bioconjugate Chem., 1991, 2, 232–241 CrossRef CAS.
- N. Nakajima and Y. Ikada, Bioconjugate Chem., 1995, 6, 123–130 CrossRef CAS.
- T. Pouyani, K. R. Kirker and G. D. Prestwich, Bioconjugate Chem., 1994, 5, 339–347 CrossRef CAS.
- P. Bulpitt and D. Aeschlimann, J. Biomed. Mater. Res., 1999, 47, 152–169 CrossRef CAS.
- I. H. Pitman and I. J. Morris, Aust. J. Chem., 1979, 32, 1567–1573 CrossRef CAS.
- Y. Luo, K. R. Kirker and G. D. Prestwich, Bioconjugate Chem., 1994, 5, 339–347 CrossRef.
- E. Kokufuta, B. Wang, R. Yoshida, A. R. Khokhlov and M. Hirata, Macromolecules, 1998, 31, 6878–6884 CrossRef CAS.
- O. E. Philippova, D. Hourdet, R. Audebert and A. R. Khokhlov, Macromolecules, 1997, 30, 8278–8285 CrossRef CAS.
- S. Halacheva, G. J. Price and V. M. Garamus, Macromolecules, 2011, 44, 7394–7404 CrossRef CAS.
- P. Ravi, C. Wang, K. C. Tam and L. H. Gan, Macromolecules, 2003, 36, 173–179 CrossRef CAS.
- S. S. Thakur and P. Balaram, J. Am. Soc. Mass Spectrom., 2009, 20, 783–791 CrossRef CAS.
- A. K. Galande, J. O. Trent and A. F. Spatola, Biopolymers, 2003, 71, 534–551 CrossRef CAS.
- W. D. Philips, J. Chem. Phys., 1955, 23, 1363–1364 CrossRef.
- H. F. Gilbert, Peptide Bonds, Disulphide Bonds and Properties of Small Peptides, in Encyclopedia of Life Sciences (ELS), John Wiley & Sons, Ltd, Chichester, 2010 Search PubMed.
- A. H. Lewin and M. Frucht, Org. Magn. Reson., 1975, 7, 206–225 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS.
- R. Quintanilla-Licea, J. F. Colunga-Valladares, A. Caballero-Quintero, C. Rodríguez-Padilla, R. Tamez-Guerra, R. Gómez-Flores and N. Waksman, Molecules, 2002, 7, 662–673 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available:1H NMR and 13C NMR spectra of DTP in DMSO-d6; 1H NMR spectra (DMSO-d6) and GPC traces for some of the PMMA-MAA and PEA-MAA copolymers; potentiometric titration data for non-crosslinked PMMA-MAA and PEA-MAA particles; a table showing the variation of the hydrodynamic diameter (Dh, in nm) with pH for non-crosslinked and crosslinked particles; size distribution functions for non-crosslinked and crosslinked PMMA-MAA-containing particles taken at different pH values; SEM images of freeze-dried PMMA-25MAA/4DTP gels formed at pH 7.0; strain sweep data for all PMMA-MAA and PEA-MAA crosslinked particle gels studied. See DOI: 10.1039/c3tb20758g |
| This journal is © The Royal Society of Chemistry 2013 |