 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoparticle amplification via photothermal unveiling of cryptic collagen binding sites
Justin H. Lo†a, Geoffrey von Maltzahn†a, Jacqueline Douglassb, Ji-Ho Parkc, Michael J. Sailorde, Erkki Ruoslahtifg and Sangeeta N. Bhatia*ah
aHarvard-MIT Division of Health Sciences and Technology, 77 Massachusetts Ave. 76-453, Cambridge, MA 02139, USA. E-mail: sbhatia@mit.edu
bDepartment of Chemical Engineering, MIT, Cambridge, Massachusetts 02139, USA
cDepartment of Bio and Brain Engineering, Korea Advanced Institute of Science and Technology, Yuseong-gu, Daejeon 305-701, Korea
dMaterials Science and Engineering Program, University of California, San Diego, La Jolla, CA 92093, USA
eDepartment of Chemistry and Biochemistry, Department of Bioengineering, Department of Nanoengineering, University of California, San Diego, La Jolla, CA 92093, USA
fCancer Research Center, Sanford-Burnham Medical Research Institute, La Jolla, CA 92037, USA
gCenter for Nanomedicine and Department of Cell, Molecular and Developmental Biology, University of California, Santa Barbara, CA 93106, USA
hHoward Hughes Medical Institute, Department of Electrical Engineering and Computer Science, MIT, David H. Koch Institute for Integrative Cancer Research, MIT, Department of Medicine, Brigham and Women's Hospital, Boston, MA 02115, USA
First published on 4th June 2013
Abstract
The success of nanoparticle-based cancer therapies ultimately depends on their ability to selectively and efficiently accumulate in regions of disease. Outfitting nanoparticles to actively target tumor-specific markers has improved specificity, yet it remains a challenge to amass adequate therapy in a selective manner. To help address this challenge, we have developed a mechanism of nanoparticle amplification based on stigmergic (environment-modifying) signalling, in which a “Signalling” population of gold nanorods induces localized unveiling of cryptic collagen epitopes, which are in turn targeted by “Responding” nanoparticles bearing gelatin-binding fibronectin fragments. We demonstrate that this two-particle system results in significantly increased, selective recruitment of responding particles. Such amplification strategies have the potential to overcome limitations associated with single-particle targeting by leveraging the capacity of nanoparticles to interact with their environment to create abundant new binding motifs.
Introduction
Nanoparticle delivery strategies for targeting cancer have great potential for improving cancer therapy by accumulating at sites of disease while sparing healthy tissues. Advances in active targeting of tumor receptors have yielded particles which home to particular cancer-associated markers rather than solely relying upon passive selectivity derived from physical parameters.1–3 However, such targeting remains restricted by several factors, including limited receptor availability,4 receptor heterogeneity amongst tumors, imperfect selectivity due to normal expression of receptors on healthy cells, and constraints on penetration depth such as binding site barriers5 and dense, fibrous matrix. Many natural processes such as the immune response and downstream signalling from growth factors overcome similar hurdles through amplification schemes at the molecular and cellular level. Because of their versatility, nanoparticles can be engineered to participate in signal amplification as a means of overcoming biological limitations on targeting.Nanoparticle amplification has been used to great effect for ultra-sensitive cancer diagnostics, such as using gold nanosphere-mounted DNA bar codes for PCR-based detection of scarce proteins.6 Our group has recently begun exploring the possibility of harnessing amplification in the tumor setting through modalities including (1) dynamic nanoparticle-mediated upregulation of targetable surface receptors via the cellular machinery7 and (2) integration of nanoparticles into the self-amplifying coagulation cascade to generate targetable microclots.8 However, the possibility of exploiting the tumor environment as a substrate for amplified targeting has not yet been explored.
We hypothesize that amplification of nanoparticle binding can be mediated by selective disruption of tumor-associated extracellular matrix (ECM), thus incorporating a manner of “stigmergic” coordination, in which insoluble environmental cues left by a signalling population recruit an overwhelming responding population. ECM molecules adopt intricate superstructures that display rigid, repeating epitopes, and physical interruption of these structures can alter mechanosensitive binding sites. For instance, tensile disruption of fibronectin fibers has been shown to functionally eliminate binding epitopes recognized by pathogenic bacteria.9 Simultaneously, formerly cryptic motifs can emerge,10 serving as plentiful yet disease-specific binding sites.
In this work, we have constructed an amplification system around the denaturation of collagen, in which the signalling population consists of gold nanorods that accumulate near collagen and, as a result of near-infrared laser-induced surface plasmon resonance, denature collagen fibrils. These fibrils are then targeted by a responding population of fibronectin fragment-functionalized iron oxide nanoparticles that actively target denatured but not intact collagen (Fig. 1).
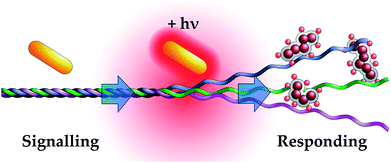 | ||
| Fig. 1 Schematic of nanoparticle amplification via photothermal denaturation of collagen. Near-infrared irradiation induces surface plasmon resonance in gold nanorods, locally heating collagen and exposing cryptic epitopes. Fibronectin fragment-conjugated nanoworms recognize denatured collagen but not intact fibrils, allowing them to ‘Respond’ to gold nanorod activity. This process produces specific, amplified accumulation of responding nanoparticles at sites of gold nanorod localization. | ||
Results
The tumor milieu and associated stroma are rich in extracellular matrix molecules, which present an abundant source of potential binding sites for nanoparticle targeting. Amongst the myriad of ECM components, Type I collagen is ubiquitous and implicated in promoting malignancy.11 Moreover, as a frequent target of structural remodeling, collagen naturally possesses the malleability to permit signal propagation through a change in physical state. The denaturation threshold of collagen's quarternary structure lies near physiologic temperature,12 suggesting that denaturation of the native triple helix into partially unwound configurations via non-ablative supraphysiologic heating may be a feasible mechanism for generating an amplified physical signal. The folded and denatured states present contrasting epitopes which permit unique recognition of the denatured state; for instance, glycine residues are only revealed in denatured collagen,10 altering the spatial configuration of residues involved in the ionic interactions thought to mediate fibronectin binding.13,14To construct a collagen-centric amplification system out of nanoparticles, we developed a signalling mechanism to induce local hyperthemia and paired this with a secondary signal-responsive mechanism that would recognize and accumulate at binding sites revealed by hyperthermia. We selected PEG-protected gold nanorods as the signalling component, as they can heat the local environment through near infrared laser-mediated surface plasmon resonance (Fig. 2A and B). PEGylated gold nanorods are amenable to use as cancer therapy because of their small size, favorable circulation half-life of ∼17 hours,15 and peak excitation wavelength in the first near-infrared optical window (Fig. 2A), a range of wavelengths to which skin and other tissues are more permissive compared to visible or ultraviolet light.16
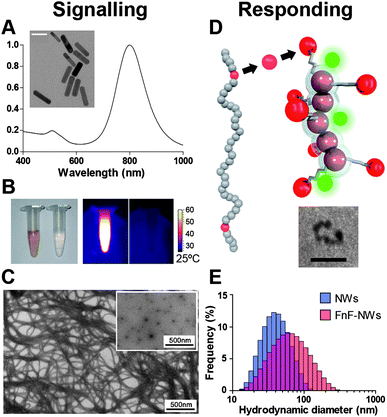 | ||
| Fig. 2 Nanoparticle components comprising the signalling and responding modules. (A) Absorbance spectrum of gold nanorods. Inset: TEM of gold nanorods, scale bar: 40 nm. (B) Left: visible-light photograph of gold nanorod solution (left) and saline solution (right). Right: thermographic image contrasting heating response of gold nanorods (light) and saline solution (right) to near-infrared laser (λ = 808 nm). (C) TEM images of intact collagen fibers and collagen denatured by heating (inset). (D) The gelatin-binding proteolytic fragment of fibronectin (FnF, red) is used as a targeting domain, conjugated to a dextran-coated iron oxide nanoworm (NW) bearing near-infrared fluorescent tracking dye (green). Inset: TEM of iron oxide NW. Scale bar: 20 nm. (E) Size distributions of nanoworms conjugated to dye only (blue) or dye and FnF (red), as measured by dynamic light scattering. | ||
Heating collagen disrupts its tri-helical tertiary structure (Fig. 2C), revealing cryptic epitopes which represent a tumor-specific target for therapeutic secondary particles. We used a 45 kDa gelatin-binding fibronectin fragment (FnF) to target denatured collagen (Fig. 2D). To establish the minimal temperature that enables FnF recognition of the cryptic collagen I epitopes, we measured the temperature-dependent binding of unconjugated FnF to collagen-coated wells in a 96-well plate, compared to the binding of albumin control (Fig. 3A). Binding of FnF and albumin to uncoated plates was minimal and not temperature-dependent.
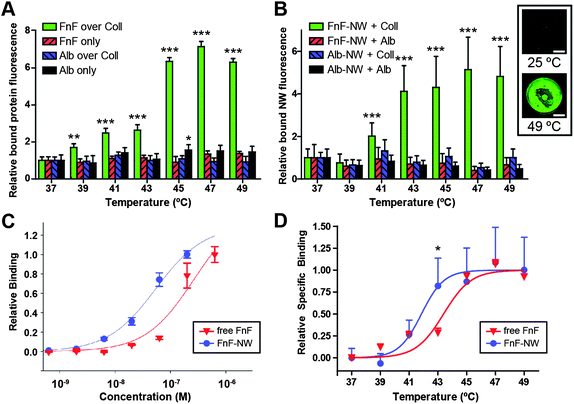 | ||
| Fig. 3 Characterization of the responding nanoparticles. (A) Relative binding of fluorescently labeled fibronectin fragment (FnF) or bovine serum albumin (Alb) to wells coated in collagen (Coll) or left uncoated, as a function of the temperature to which the collagen-coated or uncoated wells were subjected prior to protein addition. Temperature was regulated by water bath. Asterisks in (A) and (B) indicate statistical significance by two-way ANOVA with Bonferroni post test across temperatures, comparing against 37 °C (*: p < 0.05, **: p < 0.01, ***: p < 0.001). Error bars indicate standard deviation. (B) Relative temperature-dependent binding of FnF-conjugated nanoworms (FnF-NW) and albumin-conjugated nanoworms (Alb-NW) to collagen- or albumin-coated wells. Temperature was regulated by water bath. Inset: representative scans of fluorescent FnF-NWs (green) bound to collagen incubated at room temperature or 49 °C. Scale bar: 2 mm. (C) Normalized binding curves contrasting relative binding to denatured collagen as a function of FnF or FnF-NW concentration. (D) Normalized temperature-dependent binding curves, combined from (A) and (B), contrasting the relative binding of free FnF and FnF-NW to collagen heated to different temperatures. Asterisk indicates p < 0.05 by two-way ANOVA with Bonferroni post test (FnF-NWs vs. free FnF). Curve fit is a variable-slope sigmoidal dose response. | ||
Since practical applications would incorporate FnF as a targeting moiety rather than a standalone polypeptide, we generated representative responding nanoparticles that embody the typical size and multivalency of a potential therapeutic or diagnostic nanoparticle. For the backbone of these particles, we used aminated dextran-coated iron oxide nanoworms (NWs), which are elongated particles consisting of linear chains of approximately spherical cores.17 The NW backbone was selected because of its paramagnetic properties, demonstrated to be useful for MRI diagnostic applications. NWs with amine-conjugated near-infrared tracking fluorophores (VivoTag-S750) were functionalized with either FnF or albumin. Using dynamic light scattering, we determined that NWs with covalently linked FnF (FnF-NWs) had an average hydrodynamic diameter of 52.2 nm, versus 36.5 nm for NWs with near-infrared dye only, a size increase consistent with the addition of 45 kDa polypeptides over the surface of the nanoparticles (Fig. 2E).18
The temperature-dependent binding of the functionalized nanoworms was examined (in an analogous setup to the free protein experiment) to confirm their binding response to idealized collagen heating (Fig. 3B; see inset for representative imaging). To determine the effects of multivalency, we measured concentration-dependent binding of free FnF versus FnF-NW on a molar basis of protein or NW cores (Fig. 3C). These curves demonstrated a leftward shift along the concentration axis, indicating higher denatured collagen binding affinity for the FnF-NW formulation relative to free FnF. Furthermore, when the FnF and FnF-NW temperature-dependent binding curves from Fig. 3A and B are normalized for specific binding and compared head-to-head, there is an offset between the binding curves, with half-maximal binding temperatures of 43.4 °C for free FnF and 41.7 °C for FnF-NW (Fig. 3D).
Combining the signalling and responding components, we characterized the behavior of the full two-nanoparticle system. We introduced gold nanorods over collagen or control substrate (bovine serum albumin) in a 96-well plate and irradiated the wells to 45 ± 1 °C at steady-state, with nanorod concentrations and laser intensities based on experimental kinetic heating curves including those displayed in Fig. 4A. The laser power density required to heat the nanorods to the target temperature under these conditions was <0.5 W cm−2. After removal of the nanorods, the wells were then incubated with nanoworms conjugated to FnF or control albumin. Under these conditions, we observed a statistically significant six-fold increase in binding of the FnF-NWs to collagen over baseline unheated controls (p < 0.001), whereas combinations with control substrate or control targeting moieties showed no such increase (Fig. 4B). To demonstrate basic spatial specificity of the two-particle system, we added gold nanorods to specific wells of a collagen-coated 96-well plate in a pattern (Fig. 4C, inset; nanorod-containing wells denoted by red fill) and irradiated all wells equally with the near-infrared laser. Responding nanoparticles only accumulated appreciably in wells with nanorods, confirming that there was minimal heating independent of the nanorods (Fig. 4C).
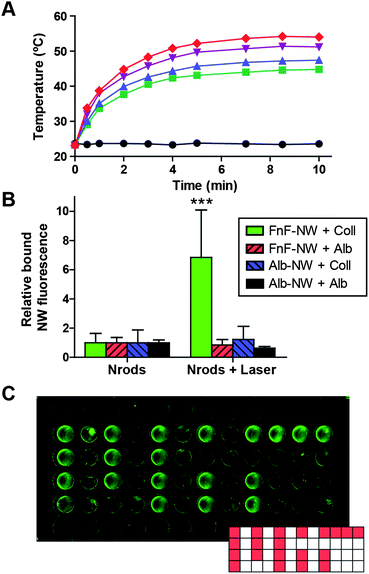 | ||
| Fig. 4 Amplification of nanoparticle accumulation through stigmergy. (A) Representative heating kinetics of gold nanorods at different concentrations (black circles: 0.0, green squares: 12.5, blue triangles: 17.7, purple triangles: 33.9, red diamonds: 70.6, all in μg mL−1), under constant irradiation by an 808 nm laser (∼120 mW cm−2). (B) Conjugated nanoworm binding to collagen or albumin substrates exposed to either unirradiated nanorods (left) or nanorods irradiated by near-infrared (NIR) laser to 45 °C (right). ***indicates p < 0.001 by two-way ANOVA with Bonferroni post test for irradiated vs. unirradiated. (C) Collagen-coated 96-well plate with nanoworms added only to wells in a pattern based on the “MIT” logo. The entire plate was irradiated by near-infrared laser as in (B), and all wells in the image were subsequently incubated with FnF-NWs. | ||
Discussion
Coordinating amplification via ECM modification offers a mechanism for specific accumulation of targeted nanoparticles by refashioning the environment to display epitopes that are not normally accessible under physiologic conditions. In comparison with other methods of mediating nanoparticle binding, this approach does not require differential expression or upregulation of a receptor, nor homogeneous receptor expression. We measured a 7.2× increase in binding with free FnF to water bath-heated collagen as well as a 6.8× increase in binding of FnF-NW to gold nanorod-heated collagen after normalizing to stringently control for any targeting conferred by the fraction of collagen which is unwound or accessible at 37 °C. These increases were specific to FnF and FnF-NWs, and neither control substrates nor untargeted particles contributed to binding (Fig. 3A and B). Consequently, non-specific accumulation in areas exposed only to the near-infrared laser or only to gold nanorods is minimal (Fig. 4B). The fold binding increase we observed corresponds with the magnitude of responses reported in previous investigations of peptide-targeted NWs.19The consistency in binding enhancement across our different experimental configurations speaks to the modularity of the targeting moiety and its potential for use with a therapeutic responding particle rather than a diagnostic one such as the iron oxide NWs. The changes in collagen structure induced by either water bath or gold nanorod heating were not rapidly reversed when hyperthermia was withdrawn, as the responding particles in these experiments were only added after plates were cooled to 4 °C, suggesting that the system is forgiving towards temporal spacing between the two particle populations. This contrasts with amplification schemes which rely upon direct particle-to-particle interaction or catalysis, or the transient accumulation of soluble signalling intermediates. These findings corroborate evidence in the literature that the collagen I denaturation–renaturation process displays significant hysteresis, with much slower renaturation, and that reformed collagen fibrils have significant representation of homotrimers and partially unwound gelatin.12
The practicality of harnessing the instability of collagen fibrils was borne out in our temperature-dependent binding studies, wherein the binding of free FnF showed half-maximal binding at ∼43.4 °C, while the FnF-NWs reached half-maximal binding at ∼41.7 °C. Importantly, these target temperatures lie within the bounds of mild hyperthermia previously described for non-ablative cancer therapy to enhance drug uptake.20 The lower temperature threshold for functionalized NWs versus free peptide (Fig. 3D) is favorable because it brings the temperatures required for optimal binding down from temperatures that could induce counterproductive coagulation. The basis of the binding at lower temperatures – and hence, with fewer binding sites – may be multivalent presentation of the FnF, which is supported by the lower concentration threshold for binding seen in FnF-NWs versus free FnF (Fig. 3C). Therefore, the use of nanoparticles as the responding element confers improved binding characteristics on account of multivalency.
On the signalling side of the amplification scheme, the need for nanoparticle “antennae” for the outwardly straightforward task of inducing hyperthermia is for the purposes of confining heat-mediated changes to a local site of disease, promoting specificity and safety. Because laser-induced heating only occurs where the nanorods have accumulated, as demonstrated in Fig. 4C, and because accumulated heat generally dissipates poorly from tumors,21 employing temperatures in the range of 40–45 °C in the tumor represents reduced risk of hyperthermic damage to surrounding tissue compared to local thermoablative tumor therapy.22,23 The choice of local heating rather than whole-body hyperthermia, modeled here by the water bath, helps to avoid the side-effects or mortality associated with elevating the entire body over 42.5 °C,21 and the 122–376 mW cm−2 laser power intensity used in our experiments (measurements not shown) is below or comparable to the conservative 330 mW cm−2 limit on occupational exposures to 808 nm light up to 3 × 104 s.24
Gold nanorod-mediated photothermal denaturation of ECM components as a mechanism of indirect signal amplification also presents potential benefits in future translation of such a system into animals. The TEM images of heat-induced unwinding of collagen (Fig. 2C) suggest the possibility of modifying the ECM in a manner which may promote greater transport through the barrier-like tumor stroma, which has been a strategy pursued previously through enzymatic25,26 and receptor-based27 means. In addition, the application of hyperthermia itself induces changes outside of ECM denaturation, and we predict that recruitment of the responding particles would be further enhanced in vivo due to transient vascular changes associated with tumor hyperthermia.28
Conclusions
We have therefore designed and demonstrated a two-nanoparticle system in which a signalling population enables the amplified recruitment of a secondary responding species through modification of ECM collagen I. Each nanoparticle is individually simple but specialized for its role: the gold nanorods can effect local hyperthermia, and the multivalent fibronectin fragment nanoworms show significant binding specificity for collagen denatured at temperatures just above a high fever. Splitting these functions into separate particles presents the opportunity for intermediate amplification, as we have shown in vitro. We believe that the assembly of nanoparticles into cooperative systems that convert ubiquitous ECM components into unique binding sites can build upon traditional active targeting strategies to enhance tumor-specific nanoparticle accumulation.Materials and methods
Preparation of gold nanorods
Bare gold nanorods (30-10-808, Nanopartz, Loveland, CO), 10 nm axial diameter × 41 nm length, were concentrated via centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g. Pellets were consolidated and reacted with 250 μM 5 kDa PEG-thiol (Laysan Bio, Inc., Arab, AL), and excess PEG and surfactant were removed by overnight dialyzation in 5 kDa Spectra/Por cellulose ester membrane (Spectrum Labs, Rancho Dominguez, CA) against ddH2O. Finally, PEGylated nanorods were concentrated to 100 OD (1 cm path, 808 nm, equivalent to 3.6 mg mL−1 and 5.9 × 1013 nanorods per mL) using a 100 kDa Amicon Ultra spin filter column at 4000g (EMD Millipore, Billerica, MA).
000g. Pellets were consolidated and reacted with 250 μM 5 kDa PEG-thiol (Laysan Bio, Inc., Arab, AL), and excess PEG and surfactant were removed by overnight dialyzation in 5 kDa Spectra/Por cellulose ester membrane (Spectrum Labs, Rancho Dominguez, CA) against ddH2O. Finally, PEGylated nanorods were concentrated to 100 OD (1 cm path, 808 nm, equivalent to 3.6 mg mL−1 and 5.9 × 1013 nanorods per mL) using a 100 kDa Amicon Ultra spin filter column at 4000g (EMD Millipore, Billerica, MA).Fluorophore-conjugated proteins
45 kDa fibronectin proteolytic fragment (F0162, Sigma-Aldrich, St. Louis, MO) or bovine serum albumin (Sigma-Aldrich), dissolved in 25 mM HEPES salt buffer, pH 7.5, was mixed with a 2:1 molar ratio of VivoTag-S750 (Perkin-Elmer, Waltham, MA) and allowed to shake for 2 hours at room temperature. Labeled product was dialyzed against 1× phosphate-buffered saline (PBS), pH 7.1, to remove excess dye.Fluorophore-labeled iron oxide nanoworms
Iron oxide nanoworms (NWs) were synthesized as previously reported17 and aminated using 20% vol/vol ammonium hydroxide, with excess ammonia removed by dialysis. Nanoparticles were labeled with near-infrared fluorophore (VivoTag-S750) at a 10:1 molar ratio of dye:NW in 1× PBS for 2 hours at room temperature. Excess fluorophore was removed by centrifugation in a 100 kDa spin filter column at 4000g.Protein-conjugated nanoworms
Following fluorophore labeling, remaining free amines on NWs were activated using a homobifunctional NHS-PEO5-NHS linker (Thermo Scientific, Waltham, MA) at a 500:1 linker:NW ratio. After shaking at room temperature for 30 minutes, excess linker was removed using 100 kDa spin filter. NWs were then added to fibronectin fragment or albumin solutions for a final 7:1 peptide:NW molar ratio. Reaction was allowed to proceed overnight at room temperature while shaking. Excess peptide was removed by 100 kDa spin filter and resuspended in 1× PBS to 1 mg mL−1 for use in experiments. Hydrodynamic diameter was determined using dynamic light scattering (Zetasizer Nano ZS90, Malvern, Worcestershire, UK).Collagen-coated plates
Black (3603, Corning, Corning, NY) or white (3610, Corning) clear-bottom 96-well plates were filled with 100 μL per well of 32 μg mL−1 sterile rat collagen or bovine serum albumin diluted in carbonate-bicarbonate buffer (pH 9.6), then incubated overnight at 4 °C. Uncoated wells were filled with buffer only.Water bath experiments
Collagen/albumin-coated black clear-bottom plates were sealed by adhesive acetate plate sealers and then immersed for 10 min in a stirred water bath of appropriate feedback-regulated temperature. Plates were then cooled for 10 min at 4 °C to simulate the partial refolding occurring when heat is withdrawn.12 Collagen, bovine serum albumin solution, or buffer was aspirated and replaced with either protein–fluorophore conjugate (40 μL at 3 μg mL−1 or 10 μg mL−1 by protein mass, uniform within each experiment) or NW–fluorophore–protein conjugate (40 μL at 25 μg mL−1 of core NW mass, Mw ∼ 115 kDa per core). After two sequential PBS rinses, plates were read using a LI-COR Odyssey Imaging System at λ = 800 nm (LI-COR Biosciences, Lincoln, NE), and signal was integrated per well (n = 3 per condition) using built-in software. Resulting temperature-dependent binding curves were fit to a generic variable-slope sigmoidal curve of the form.where B = binding magnitude, S is the slope, T = temperature, T1/2 is the temperature at half-maximal binding, and Bmax and Bmin are fixed to 1 and 0, respectively, to account for normalization and baselines.
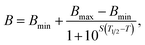
Nanorod heating experiments
0.5 μL of 100 OD (3.5 mg mL−1) PEGylated nanorods were added per well in collagen-coated white plates, diluted into the collagen solution. Wells receiving laser treatment were irradiated with 808 nm laser for 10 min at ∼300 mW cm−2; power levels were confirmed by handheld laser power meter (Edmund Optics, Barrington, NJ). The entire plate was cooled for 10 min at 4 °C prior to aspiration of the collagen solution and replacement with NW–fluorophore–protein conjugate (40 μL at 25 μg mL−1). The remainder of the procedure was identical to the water bath experiments above. n = 4 per condition.TEM imaging
Gold nanorods or collagen solutions were dispensed onto TEM grids. Staining and imaging was performed by the W. M. Keck Microscopy Facility at the Whitehead Institute.Acknowledgements
The authors dedicate this paper to the memory of Officer Sean Collier for his caring service to the MIT community and his sacrifice. This work was supported by the NIH (BRP: R01CA124427-01), NIH/NCI (U54CA119349, U54CA119335, and the Alliance Challenge Project/MIT-Harvard Center of Cancer Nanotechnology Excellence: U54CA151884), Packard Fellowship (1999-1453), and Marie-D. & Pierre Casimir-Lambert Fund. J. H. L. acknowledges support from the NIH/NIGMS MSTP program (T32GM007753). G. v. M. acknowledges support from the Whitaker Foundation and the National Science Foundation. We gratefully acknowledge Dr Shelley J. Coldiron and Dr Christian Schoen at Nanopartz for developing the CTAB-nanorods used in this work; Dr Yoel Fink for generously lending the FLIR infrared thermographic camera used in these experiments. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.Notes and references
- R. H. Begent, M. J. Verhaar, K. A. Chester, J. L. Casey, A. J. Green, M. P. Napier, L. D. Hope-Stone, N. Cushen, P. A. Keep, C. J. Johnson, R. E. Hawkins, A. J. Hilson and L. Robson, Nat. Med., 1996, 2, 979–984 CrossRef CAS.
- O. C. Farokhzad, J. Cheng, B. A. Teply, I. Sherifi, S. Jon, P. W. Kantoff, J. P. Richie and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6315–6320 CrossRef CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS.
- E. Ruoslahti, S. N. Bhatia and M. J. Sailor, J. Cell Biol., 2010, 188, 759–768 CrossRef CAS.
- G. M. Thurber, M. M. Schmidt and K. D. Wittrup, Adv. Drug Delivery Rev., 2008, 60, 1421–1434 CrossRef CAS.
- J. M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS.
- J. H. Park, G. von Maltzahn, M. J. Xu, V. Fogal, V. R. Kotamraju, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 981–986 CrossRef CAS.
- G. von Maltzahn, J. H. Park, K. Y. Lin, N. Singh, C. Schwoppe, R. Mesters, W. E. Berdel, E. Ruoslahti, M. J. Sailor and S. N. Bhatia, Nat. Mater., 2011, 10, 545–552 CrossRef CAS.
- M. Chabria, S. Hertig, M. L. Smith and V. Vogel, Nat. Commun., 2010, 1, 135 CrossRef.
- P. Fratzl, Collagen: Structure and Mechanics, Springer, New York, 2008 Search PubMed.
- T. Armstrong, G. Packham, L. B. Murphy, A. C. Bateman, J. A. Conti, D. R. Fine, C. D. Johnson, R. C. Benyon and J. P. Iredale, Clin. Cancer Res., 2004, 10, 7427–7437 CrossRef CAS.
- E. Leikina, M. V. Mertts, N. Kuznetsova and S. Leikin, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1314–1318 CrossRef CAS.
- E. Engvall and E. Ruoslahti, Int. J. Cancer, 1977, 20, 1–5 CrossRef CAS.
- M. Vuento, E. Salonen, K. Osterlund and U. H. Stenman, Biochem. J., 1982, 201, 1–8 CAS.
- G. von Maltzahn, J. H. Park, A. Agrawal, N. K. Bandaru, S. K. Das, M. J. Sailor and S. N. Bhatia, Cancer Res., 2009, 69, 3892–3900 CrossRef CAS.
- M. J. van Gemert, S. L. Jacques, H. J. Sterenborg and W. M. Star, IEEE Trans. Biomed. Eng., 1989, 36, 1146–1154 CrossRef CAS.
- J. H. Park, G. von Maltzahn, L. Zhang, M. P. Schwartz, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2008, 20, 1630–1635 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS.
- J. H. Park, G. von Maltzahn, L. Zhang, A. M. Derfus, D. Simberg, T. J. Harris, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Small, 2009, 5, 694–700 CrossRef CAS.
- C. van Bree, J. J. Krooshoop, R. C. Rietbroek, J. B. Kipp and P. J. Bakker, Cancer Res., 1996, 56, 563–568 CAS.
- R. W. Rowe-Horwege, Encyclopedia of Medical Devices and Instrumentation, John Wiley & Sons, Inc., 2006 Search PubMed.
- K. Y. Lin, A. F. Bagley, A. Y. Zhang, D. L. Karl, S. S. Yoon and S. N. Bhatia, Nano LIFE, 2010, 01, 277–287 CrossRef CAS.
- D. DasGupta, G. von Maltzahn, S. Ghosh, S. N. Bhatia, S. K. Das and S. Chakraborty, Appl. Phys. Lett., 2009, 95, 233701 CrossRef.
- American National Standard for Safe Use of Lasers Ansi Z136.1-2007, 2007.
- T. D. McKee, P. Grandi, W. Mok, G. Alexandrakis, N. Insin, J. P. Zimmer, M. G. Bawendi, Y. Boucher, X. O. Breakefield and R. K. Jain, Cancer Res., 2006, 66, 2509–2513 CrossRef CAS.
- P. P. Provenzano, C. Cuevas, A. E. Chang, V. K. Goel, D. D. Von Hoff and S. R. Hingorani, Cancer Cell, 2012, 21, 418–429 CrossRef CAS.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, O. M. Girard, D. Hanahan, R. F. Mattrey and E. Ruoslahti, Cancer Cell, 2009, 16, 510–520 CrossRef CAS.
- R. Burd, T. S. Dziedzic, Y. Xu, M. A. Caligiuri, J. R. Subjeck and E. A. Repasky, J. Cell. Physiol., 1998, 177, 137–147 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |