Proteinaceous microspheres for targeted RNA delivery prepared by an ultrasonic emulsification method†
Ulyana
Shimanovich
a,
Dror
Eliaz
b,
Sally
Zigdon
b,
Vadim
Volkov
b,
Adva
Aizer
b,
Artur
Cavaco-Paulo
c,
Shulamit
Michaeli
b,
Yaron
Shav-Tal
b and
Aharon
Gedanken
*a
aDepartment of Chemistry and Kanbar Laboratory for Nanomaterials, Bar-Ilan University Center for Advanced Materials and Nanotechnology, Bar-Ilan University, Ramat-Gan 52900, Israel. E-mail: ushimanovich@yahoo.com; gedanken@mail.biu.ac.il
bThe Mina and Everard Goodman Faculty of Life Sciences and Institute of Nanotechnology, Bar-Ilan University, Ramat-Gan, 52900, Israel. E-mail: droreliaz@gmail.com; sally.zigdon@gmail.com; vadimvolkov@013.net; darzia@gmail.com; michaes@mail.biu.ac.il; Yaron.Shav-Tal@biu.ac.il
cUniversity of Minho, Textile Engineering Department, P-4800058 Guimaraes, Portugal. E-mail: artur@det.uminho.pt
First published on 22nd October 2012
Abstract
In the present work we used sonochemically prepared proteinaceous BSA spheres as a novel RNA-delivery system. The preparation of RNA-loaded BSA spheres was accomplished using an environmental friendly method termed the “ultrasonic emulsification method”. It was demonstrated that ultrasonic waves do not cause the RNA chains to degrade and the RNA molecules remain untouched. The BSA–RNA complex was successfully introduced into mammalian (human) U2OS osteosarcoma cells and Trypanosoma brucei parasites. Using PVA coating of the RNA–BSA spheres we have achieved a significant increase in the number of microspheres penetrating mammalian cells. The mechanism of RNA encapsulation and the structure of the RNA–BSA complex are reported.
Introduction
In the present report we tried to expand our previous work reporting on the sonochemical encapsulation of RNA molecules inside BSA spheres.1 The main goal of the present work is to unveil the mechanism of the RNA encapsulation and the structure of the RNA–BSA complex. Moreover, we applied surface modifications of the microspheres and succeeded in controlling the quantity of penetrated RNA–BSA spheres and in achieving targeted delivery.2–5RNA encapsulation and delivery is a new field of research that has coalesced during the last decade from independent studies on various organisms. Proteins are found to be excellent candidates for RNA carrier container (capsule) construction due to their biocompatibility and non-toxicity.6–9
In the present study, ultrasonic waves were used for the formation of proteinaceous microspheres (PMs) and the simultaneous encapsulation of RNA in those spheres. Micrometer sized gas-filled or liquid-filled microspheres could be produced from various kinds of proteins10–13 by using an ultrasonic emulsification method. We have recently shown that native RNA and DNA molecules could be nanospherized using high-intensity ultrasonic waves.14,15 The possibility to direct the size scale of BSA–RNA capsules from nanoscale sizes to microscopic sizes by changing fundamental parameters such as the lengths of RNA chain or organic solvent:aqueous solution ratio was demonstrated. In the current manuscript we mostly used the term “microsphere” to refer to fundamental studies on the formation of RNA loaded BSA spheres and how the surface modifications of those spheres affect their penetration into cells. Bovine serum albumin is widely used for the preparation of proteinaceous microspheres due to its availability in a pure form and its biodegradability, nontoxicity, and nonimmunogenicity. The mechanism of the sonochemical formation of microspheres has been discussed previously.16–19 According to the mechanism of the sonochemical formation of PMs, the microspheres are formed by chemically cross-linking cysteine residues of the protein, which undergo oxidation by a HO2˙ radical formed inside or around a micron-sized gas bubble or a non-aqueous droplet. The formation of S–S bonds is the chemical cross-linking process that is responsible for the formation of the microspheres, and is a direct result of the chemical effects of ultrasound radiation on an aqueous medium.
The preparation of BSA microspheres and the encapsulation of RNA in the spheres were achieved by a one-step process, starting with an aqueous solution of native BSA protein and RNA, over layered by oil (soya bean oil). For the encapsulation studies, different types of RNAs were used: (1) Cy3 labeled RNA; (2) Cy3-6FAM labeled RNA; and (3) SLRNA (spliced leader total RNA extracted from Leptomonas collosoma parasites). In the current work we used proteinaceous microspheres for RNA delivery into mammalian U2OS osteosarcoma cells20 and Trypanosoma brucei (T.b.) parasites.21 Moreover, using polymer for the PM coating, we succeeded in targeting the PMs and in introducing the RNA encapsulated inside BSA into the nucleus of the mammalian cells. Two types of coatings22 were used for studying the target-delivery of RNA: (1) polymeric coating and (2) metal coating. For polymeric coating polyvinyl alcohol (PVA) and polyethyleneimine (PEI) were used. Due to the neutral charge of PVA, the PVA coated RNA–BSA complex enables a larger number of spheres to penetrate the U2OS cells. When the positively charged PEI (a biocompatible polymer) was used for the PMs coating, the nucleus of the human cancer cells was successfully targeted with RNA loaded BSA PMs. The Fe2O3 (metal) coating was used to follow and detect RNA-loaded spheres inside the cells by transmission electron microscopy (TEM). The influence of the high-intensity ultrasonic waves on the length and biological activity of RNA molecules was studied using the “primer extension” technique. The RNA-loaded microspheres were analyzed and characterized by SEM (scanning electron microscopy, JSM-840, JEOL), fluorescence light microscopy (Apo-Tome microscope, Zeiss and NIKON 90i fluorescent microscope), confocal microscopy (Confocal Zeiss), DLS (dynamic light scattering, Marwen) measurements, and TEM (transmission electron microscopy) analysis. The mechanism of RNA encapsulation was studied using confocal microscopy.
Experimental
Preparation of RNA-loaded BSA spheres
The following RNA molecules were encapsulated inside the BSA spheres: (1) Cy3-RNA, (2) 5′-Cy3-RNA-3′-6FAM, and (3) SLRNA (splice leader total RNA). Splice leader RNA was extracted from Leptomonas collosoma cells using a Trizol (Sigma Aldrich) reagent. Cy3-RNA and Cy3-6FAM labeled RNAs were purchased from “Synthessa Bioscience”. Cy3 is a reactive (red) fluorescent marker with an excitation wavelength of 650 nm and emission at 670 nm. 6FAM is a reactive (green) fluorescent marker with an excitation wavelength of 492 nm and emission at 517 nm.The preparation of the RNA-loaded BSA spheres followed the typical synthesis of PMs, as described previously.23 In brief, 6.7 mL of an organic solvent (soya bean oil) was layered over a 10 mL water solution of 5% w/v BSA protein (bovine serum albumin protein, Sigma-Aldrich) and 1% w/v purified RNA. The volume ratio between the 5% (w/v) BSA aqueous solution and soya bean oil was kept constant at 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, respectively. The molar ratio between the RNA and BSA protein was 1:50 (w/w), respectively. RNA-loaded PMs were synthesized with a high-intensity ultrasonic probe (Sonic and Materials, VC-600, 20 kHz, 0.5 in. Ti horn, at 78% amplitude). The volume of the acoustic chamber was 25 mL and the total volume of all the ingredients was 16.7 mL. The bottom of the high-intensity ultrasonic horn was positioned at the aqueous–organic interface, employing an acoustic power of ≈45 W cm−2 with an initial temperature of 22 °C in the reaction cell. The sonication process lasted for 3 min at 22 °C using an ice-cooling bath to maintain the low temperature. The product was separated from the mother solution. The separation was accomplished within a few minutes due to the lower density of the microspheres, relative to the density of water. To obtain a more complete separation of the proteinaceous microspheres from the mother solution, the separation flasks were placed in a refrigerator (4 °C) for 24 h. After the separation, the residual aqueous phase and the organic solvent (oil) were removed and the product was resuspended in water.
:2, respectively. The molar ratio between the RNA and BSA protein was 1:50 (w/w), respectively. RNA-loaded PMs were synthesized with a high-intensity ultrasonic probe (Sonic and Materials, VC-600, 20 kHz, 0.5 in. Ti horn, at 78% amplitude). The volume of the acoustic chamber was 25 mL and the total volume of all the ingredients was 16.7 mL. The bottom of the high-intensity ultrasonic horn was positioned at the aqueous–organic interface, employing an acoustic power of ≈45 W cm−2 with an initial temperature of 22 °C in the reaction cell. The sonication process lasted for 3 min at 22 °C using an ice-cooling bath to maintain the low temperature. The product was separated from the mother solution. The separation was accomplished within a few minutes due to the lower density of the microspheres, relative to the density of water. To obtain a more complete separation of the proteinaceous microspheres from the mother solution, the separation flasks were placed in a refrigerator (4 °C) for 24 h. After the separation, the residual aqueous phase and the organic solvent (oil) were removed and the product was resuspended in water.
Assessment of quality of encapsulated RNA inside BSA microspheres. The extraction of RNA from BSA spheres
The integration of the encapsulated RNA inside BSA spheres was performed for SLRNA molecules due to their sequence and properties which allow us to compare at the same time the encapsulated SLRNA molecules and delivered SLRNA into T.b. parasites. The encapsulated SLRNA molecules were regenerated from the BSA spheres using a phenol:chloroform extraction method and further precipitated with ethanol according to the manufacturer's instructions. In addition, SLRNA molecules were extracted using the TRI reagent (Trizol, Sigma-Aldrich) from T.b. parasites which were previously incubated for 3 hours with BSA spheres carrying SLRNA (Leptomonas collosoma spliced Leader RNA). The quality of the extracted RNA molecules was assessed by a primer extension technique and compared to the untreated RNA.Preparation of PVA, PEI and Fe2O3 coated RNA–BSA spheres
For the preparation of the polymer coated RNA–BSA spheres polyvinylalcohol (PVA) (10000 Mw, Sigma) and branched polyethyleneimine (600 Mw, Sigma) were used. The branched PEI with low molecular weight polymer was chosen due to its non-toxicity towards cells. 0.05 g of PVA/PEI was dissolved in 10 mL of double distilled water (DDW). To assist in the dissolution of the polymer in water it was sonicated for 30 min in a sonication bath. After this pretreatment, 1 mL of the polymeric solution was added to 1 mL of the RNA loaded spheres and the mixture was shaken at room temperature for 20 min. The product was washed twice with 1 mL DDW. The efficiency of the polymeric coating of the spheres was checked by measuring the change in electrical charge on the surface of the coated spheres. In addition in order to reassure the attachment of the polymers to the surface of PMs, the polymers were coupled with FITC dye and the green signal emitted from the surface of the coated PMs was detected by fluorescence microscopy.
For the preparation of the metal coated spheres, nanostructured iron oxide (Fe2O3)22,24 particles were synthesized. Soya bean oil was layered over an aqueous solution of iron acetate (Fe(CH3CO2)2 95%-Sigma, 7.66 × 10−3 M) and 5% w/v BSA and RNA aqueous solution. The mixture was then sonicated for 3 min (following the general procedure for PMs preparation). The Fe2O3 particles were found to be attached to the outer surface of the RNA-loaded PMs. The concentration of Fe3+ ions on the surface was 7–9% w/w. The percentage of iron ions on the surface of the spheres was determined by destructing the spheres and potassium iodide was used for the quantitative analysis of Fe3+. The exact synthesis and characterization of iron oxide-coated proteinaceous microspheres was previously discussed.22 In order to identify the iron oxide coating on the BSA microspheres' surface, the coated microspheres were analyzed using a transmission electron microscope. The products (iron coated RNA–BSA microspheres) were introduced into T.b. parasites and U2OS cells. The parasites and mammalian cells containing iron coated BSA PMs were analyzed and characterized by light microscopy (Apo-Tome) and transmission electron microscopy (TEM).
Determination of RNA concentration in the BSA microspheres
The amount of RNA loaded molecules in the microspheres was determined using UV spectrophotometric measurements (NanoDrop1000 UV-spectrophotometer; the RNA absorption peak was measured at 260 nm) by subtracting the amount of RNA remaining in the aqueous solution after sonication, from the initial amount of RNA.In addition, the number of RNA molecules which were encapsulated inside the BSA spheres was also calculated by a different method. First, the total number of spheres in the resulting solution was found. For this calculation we took into account the initial volume of mother solution in the reaction cell (water solution of BSA and RNA + organic solvent = 16.7 mL), the volume of the microsphere resulting from the sonochemical reaction (5 mL), and the average size of BSA microspheres (2.5 μm). These numbers served for calculating the number of BSA molecules which construct the walls of the BSA spheres (molecular weight of BSA protein = 67 kDa was taken into account). After finding out the number of BSA spheres formed in the sonication process, we calculated the number of RNA molecules encapsulated inside the BSA spheres. We compared the results obtained by these two methods.
Determination of RNA localization in BSA spheres
For determining the exact shape and localization of the RNA molecules in the BSA spheres, Cy3 labeled RNA and Cy3-6FAM labeled RNA were used. The procedure for encapsulation of Cy3-RNA and Cy3-RNA-6FAM is described above. The samples were analyzed using fluorescence microscopy (Apo-Tome, Zeiss), CD (Circular Dichroism, Applied BioPhysics) and confocal microscopy (Confocal Zeiss microscope), and 3D images were reconstructed from z-stack confocal cuts using the “Imaris” image analysis program (Bitplane, Switzerland).Introduction of RNA-loaded PMs into Trypanosoma brucei parasites and human cells
In order to introduce the RNA-loaded PMs into T.b. parasites and human U2OS cells, coated (polymeric coating or metal coating) and uncoated BSA–RNA spheres were used.T.b. parasites were maintained in a BACK medium. The parasites were incubated with PMs for 3 hours at 37 °C. After the incubation, the parasites were washed 3 times with a 1× PBS buffer solution in order to remove the residual PMs. Nuclei were counterstained with DAPI. The parasites were cross-linked on a glass-slide with 4% paraformaldehyde and then microscopically analyzed.
Human U2OS cells were maintained in low glucose DMEM (Biological Industries, Kibbutz Beit Haemek, Israel) containing 10% fetal bovine serum (Hy-Clone Laboratories, Logan, UT). The cells were grown on a glass slide overnight. PMs were added to the cells and incubated overnight at 37 °C. After the incubation, the cells were washed 5 times with a 1× PBS buffer solution in order to remove the PMs that did not penetrate the cells. The cells were fixed with 4% paraformaldehyde. The nuclei were counterstained with DAPI, and coverslips were mounted on mounting medium. The samples were analyzed using wide-field fluorescence microscopy. 3D images were reconstructed using the “Imaris” image analysis program.
Instrumentation
Dynamic light scattering (DLS) measurements and z-potential measurements were carried out on an ALV/CGS-3 compact goniometer system equipped with an ALV/LSE-5003 light scattering electronic and multipurpose titrator (MPT2), a multiple digital correlator, and a 632.8 nm JDSU laser 1145P. DLS and z-potential experiments were carried out in a doubly diluted, as-separated PM solution, i.e. the PMs removed after sonication were diluted with an equal amount of double-distilled water (DDW). Each measurement took 10 s; particle distribution and electrical charge distribution were obtained by averaging ten DLS measurements.For light and fluorescence microscopy (Apo-Tome microscope, Zeiss), samples were prepared by depositing the aqueous dispersions, without further purification, onto a glass slide. The RNA-loaded BSA microspheres were analyzed and characterized by SEM (scanning electron microscopy, JSM-840, JEOL), fluorescence and light microscopy (Apo-Tome microscope, Zeiss), confocal microscopy (Confocal, Zeiss microscope) and DLS (dynamic light scattering) measurements. For confocal microscopy analysis the following excitation and fluorescence wavelengths were used: “Rhod.” (red fluorescent signal; excitation peak at 550 nm, emission at 573 nm) and GFP (green fluorescent signal; excitation at 489 nm, emission at 509 nm). The fluorescence signals were observed using the following lasers: HeNe 543 nm and the argon line at 488, 514 nm.
Wide-field fluorescence images were obtained using the Image-J system based on a NIKON 90i fully motorized microscope (60× Plan Apo objective, 1.42 numerical apertures [NA]), rapid wavelength switching, and driven by the Image-J software. A 3D volume of the cell containing NPs (135z-slices of 0.30 μm; total height, 40 μm) was imaged and reconstructed using the “Imaris” image analysis software.
For transmission electron microscopy (TEM), samples were prepared by depositing the aqueous dispersions on a gold-coated TEM Cu grid (300 strands per inch). TEM measurements were performed on a JEOL JEM-100CX and a 200 kV FEI TECNAI F20 S-TWIN apparatus with a field emission source connected to an EDAX detector (USTEM, Vienna University of Technology). For scanning electron microscopy analysis the samples were coated with a thin layer of carbon after the deposition of the spheres on the grid.
Results and discussion
The morphology of sonochemically prepared RNA-loaded BSA spheres was studied using electron microscopy (SEM). Fig. 1(a) shows BSA spheres loaded with SLRNA molecules with relatively smooth surfaces and an average size of about 1.5 μm. An enlarged image of a hollow BSA sphere is presented in the inset of Fig. 1(a). The average size of the microspheres was confirmed by DLS measurements. The changes in size of RNA loaded spheres vs. length of RNA chain are presented elsewhere.1 The morphology of the surface of RNA loaded PMs changed after the polymer coating was applied. Fig. 1(b) depicts a large PVA coated sphere, where an increase in roughness of the surface is clearly observed as compared with the uncoated sphere presented in Fig. 1(a). The same rough surface was observed for PEI coated PMs (see Fig. 1(c)). For both Fig. 1(b) and (c) enlarged images of coated BSA spheres are presented in the inset of each image. When polymer coatings were applied to the PMs the average size and surface charge were changed. | ||
| Fig. 1 Electron microscopy (SEM) image of: (a) an uncoated PM; enlarged image of a hollow uncoated sphere is presented in the inset of the image; (b) PVA coated PM; an enlarged image of the coated surface is presented in the inset of the image; (c) PEI coated PMs; an enlarged image of the coated sphere with rough surface is presented in the inset of the image. Scale bar = 4 micrometers. | ||
The polymeric coating was designed to neutralize the negative charge of the RNA loaded BSA spheres. Indeed, a dramatic change in microspheres' charge was detected as a result of the polymer deposition on the surface of the spheres. The charge of the surface of RNA loaded PMs changed from −40 meV to 0 meV for PVA coated spheres and to +20 meV for PEI coated spheres. The change in the average size of uncoated and coated spheres was detected by DLS measurements and was found to be around 30 nm for both polymers, from 1500 nm to 1530 nm, respectively. However, the polydispersity index of the average size of the coated as well as uncoated spheres was found to be relatively high (see ESI†, Table S1) which means that small spheres are present in the resulting solution as well. It was previously demonstrated1 how the average size of the spheres could be manipulated by changing the RNA length (use of smaller RNA chains leads to formation of small nanosized BSA spheres). The results of changes in sizes and surface electrical charge are summarized in Table S1 in the ESI.†
The efficiency of the sonochemical method in encapsulating RNA within the PMs was studied using spectrophotometric analysis. The amount of BSA and RNA molecules was assayed using a NanoDrop 1000 spectrophotometer at 280 nm and 260 nm, respectively. The number of RNA molecules encapsulated inside the BSA spheres was found using the following calculation. First the total number of spheres which were produced during the sonochemical reaction (3 min) was calculated. 5 mL of loaded BSA spheres were produced from 16.7 mL mother solution (water solution of BSA, SLRNA and soya bean oil) with an average size of 1.5 μm. The total number of loaded PMs in the 5 mL resulting solution was found to be approximately 6.1 × 1011 spheres. Then the number of BSA molecules (with initial concentration 5 mg mL−1 and molecular weight 67 kDa) constructing a single microsphere was calculated using the following equation: 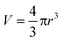 . The number of BSA molecules that construct a single sphere is found to be 1.82 × 106 molecules (when the percentage of spheridized BSA molecules is 90%). Finally, the number of SLRNA molecules (95 nucleotides) encapsulated within PMs was calculated and found to be 6 × 106 molecules per sphere (with efficiency of RNA encapsulation 60%, and assuming an equal distribution of the RNA molecules among the BSA spheres). The ratio between the number of BSA molecules constructing a single microsphere wall and the number of RNA molecules encapsulated inside the single BSA sphere is 1:3, respectively. This ratio remains constant and was found to be independent of the length of the RNA chain. The initial ratio (in mother solution) between RNA molecules and BSA protein molecules is 1:50.
. The number of BSA molecules that construct a single sphere is found to be 1.82 × 106 molecules (when the percentage of spheridized BSA molecules is 90%). Finally, the number of SLRNA molecules (95 nucleotides) encapsulated within PMs was calculated and found to be 6 × 106 molecules per sphere (with efficiency of RNA encapsulation 60%, and assuming an equal distribution of the RNA molecules among the BSA spheres). The ratio between the number of BSA molecules constructing a single microsphere wall and the number of RNA molecules encapsulated inside the single BSA sphere is 1:3, respectively. This ratio remains constant and was found to be independent of the length of the RNA chain. The initial ratio (in mother solution) between RNA molecules and BSA protein molecules is 1:50.
The structure of the RNA-loaded PMs was studied using confocal microscopy. For this purpose we have employed Cy3-RNA and Cy3-RNA-6FAM, RNA molecules with fluorescent tags. The fluorescent tag “Cy3” emits a red fluorescent signal, while green fluorescence is emitted by the “6FAM” tag (wavelengths of excitations and emissions are described above in the Experimental section). Cy3 labeled RNA formed a sphere encapsulated inside FITC-BSA (fluorescein isothiocyanate labeled bovine serum albumin protein; 488/520) microspheres. The same is true for the red-RNA (Cy3-RNA) that was found to be the inner sphere of the green-BSA (FITC-BSA) microcapsule. The example of Cy3-RNA encapsulated within FITC-BSA is presented in Fig. 2(a). Fig. 2(a) clearly shows the green walls of the FITC-BSA spheres and the red color which is emitted from the inner part of the green sphere (red color relates to red-RNA). To find the exact localization of RNA inside the BSA sphere the Cy3-RNA-6FAM was used. Cy3-RNA-6FAM is a RNA molecule which is tagged with two different colors (red and green) on each end. The green tag (6FAM) emits a much stronger fluorescent signal than Cy3. The role of the double tagged RNA (red tag-RNA-green tag) is to study if the RNA molecules are localized as “brushes” (RNA molecules located parallel to each other and perpendicular to the BSA shell) or are organized in a “non-brush” organization, namely, the RNA molecules are organized parallel to the BSA membrane. The illustration of the localization of FITC-BSA and Cy3-RNA-6FAM inside the sphere is presented in Fig. S2 in the ESI.† We clearly observed 2 green fluorescent rings. The red fluorescent signal, which was emitted from Cy3, was very weak. The “brush” organization would yield 3 fluorescent rings: the first ring is green, and is related to the green protein shell. The second and the third rings are related to the 2 ends of the RNA. The observation of two rings (see Fig. S2(b) in the ESI†) suggests that the Cy3-RNA-6FAM is situated perpendicular to the BSA shell in what is called “brush” formation.
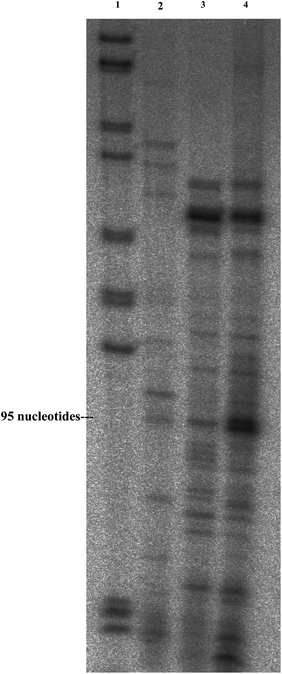 | ||
| Fig. 2 Primer extension (polyacrylamide gel). Primer extension was performed with a radiolabeled oligonucleotide complementary to the SLRNA. Corresponding cDNA was separated on a 6% sequencing gel titration. Column 1 indicates marker; 2 – cDNA obtained from RNA delivered to T. brucei parasites; 3 – cDNA obtained from RNA extracted from PMs; and 4 – cDNA obtained from untreated total tRNA. | ||
In order to find out the mechanism of RNA encapsulation the following experiments were performed. An aqueous solution containing FITC labeled BSA protein and Cy3-RNA was prepared. The BSA and RNA molecules tend to aggregate upon aging to yield a BSA–RNA complex. The confocal image of a BSA–RNA aggregate is presented in Fig. S3 in the ESI.†
When the soya bean oil layer was added to the aqueous solution of FITC–BSA–Cy3–RNA, the BSA protein and RNA molecules formed droplets. Upon sonication of the aqueous solution of BSA–RNA and the soya bean oil, BSA microspheres filled with RNA were formed (synthesis is described in the Experimental part). The mechanism of RNA loaded BSA spheres formation is presented in Scheme 1, where the first step is BSA–RNA coupling.
 | ||
| Scheme 1 Schematic presentation of RNA loaded BSA microsphere formation. A = FITC labeled BSA protein; B = Cy3 labeled RNA molecule; intermediate = droplet of BSA–RNA aggregates in water–oil solution; and microsphere = FITC-BSA microsphere loaded with Cy3-RNA. | ||
The importance of ultrasonic waves in the microsphere formation and stability of the formed microspheres was studied. We have tried to form microspheres emulsifying the initial solution (water solution of BSA, RNA and soya bean oil) by vortexing. In the resulting solution BSA–RNA microspheres were obtained. The major difference between sonochemically prepared microspheres and microspheres prepared by vortex emulsification is in the stability of the product: more than 6 months after sonication compared to 1 day for the product obtained by vortexing. The BSA–RNA spheres do not tend to aggregate due to their negative charge. Moreover, there is another difference in microsphere structures prepared using these two different methods. The walls of sonochemically prepared RNA-loaded BSA microspheres consist of BSA protein, while the RNA molecules were found in the inner part of the BSA sphere as described above. On the other hand, the walls of BSA–RNA microspheres prepared by vortex emulsification were constructed from a mixture of BSA and RNA molecules.
Why the hydrophilic RNA molecules are found in the inner part of the hydrophobic environment inside the BSA sphere? To answer this question, the interactions between the protein (BSA) and the nucleic acid (RNA) molecules were studied. The majority of the interactions between protein residues and RNAs occur on accessible surface area of the proteins (ASA) where the polar and non-polar amino acid groups of the protein are exposed and available to create polar/hydrogen/van der Waals contacts and other types of interactions.25 Under high intensity ultrasonic treatment there was no covalent bond formation between the BSA and RNA. However, the formation of hydrogen bonds between the aromatic amino acids of the BSA and nucleoside bases of RNA was detected. Those hydrogen bonds (hydrophobic interactions) are responsible for the initial formation of the BSA–RNA complex. When the ultrasonic waves are passed through an aqueous–oil biphasic solution it leads to creation of microscopic oil in water droplets. Then, the BSA–RNA complex stabilizes those oil droplets and act in this case as a surfactant. Both BSA and RNA molecules are negatively charged molecules. Due to the big difference in sizes between the BSA protein molecules and RNA molecules, RNA molecules tend to be localized in the inner part of the BSA sphere (“mass transfer effect”) where the charged amino acids of the BSA protein are facing the aqueous medium. However, the initial stabilization of oil in water droplets by the BSA–RNA complex does not clarify the fact that the RNA molecules fill the inner part of the BSA spheres (phenomenon observed for the first hour after preparation of the sphere). We offer two possible explanations for the above described phenomenon: (1) a small amount of water is encapsulated inside the inner part of the spheres and (2) the labeling of RNA molecules with Cy3 fluorescent tag might affect the solubility of RNAs in a less hydrophilic environment and their sequestration to the oil phase due to the presence of two aromatic rings and a polymethine chain. Anyway, both factors stabilize the RNA in the inner part of the sphere only for the first hour and then the RNA molecules are pushed towards the protein wall carried by the hydrophobic oil forming a double wall sphere structure.
As a result, after ultrasonic emulsification of aqueous BSA–RNA–oil solution double wall spheres were obtained where the internal wall was made of RNA molecules and external wall was made of BSA protein.
The most important question in the present study is whether the encapsulated RNA molecules upon their release from the PMs inside the living cells stay intact, and are they biologically active? To answer this question we examined if the sonochemical radiation of an aqueous solution of BSA, RNA and soya bean oil is degrading the RNA molecule. In addition, we checked if the RNA delivered to the cell remains intact and biologically active. For this purpose total RNA extracted from Leptomonas collosoma parasites was used. First, the PMs loaded with total RNA were destroyed by the TRI reagent (TRIZOL) and the RNA molecules were extracted and analyzed by the primer extension technique with an SLRNA specific probe. In addition, the SLRNA loaded PMs were introduced into Trypanosoma brucei parasites and RNA was extracted from parasites using the TRI reagent and analyzed by the same primer extension technique.
This technique is used to determine the 5′ end of the RNA. The function of the reverse transcriptase is to synthesize cDNA which is complementary to RNA using a specific primer. The length of synthesized cDNA should be identical to the initial RNA molecule. The extracted RNA was incubated with radioactive end-labelled anti-sense primer. After primer extension the products were separated on 6% polyacrylamide/7 M urea gels and visualized by autoradiography. By separating the product on a polyacrylamide gel, it is possible to determine the exact size of the RNA, and especially monitor if degradation has occurred. The extracted RNAs (RNA extracted from PMs and RNA extracted from T. brucei parasites) were compared with native RNA.
The results are presented in Fig. 2 and show that the sonochemical encapsulation does not lead to the degradation of the RNA during the formation of the RNA loaded BSA sphere, since exactly the same cDNA products were obtained.
In addition, the intensities of the cDNA bands were compared and the percentage of the delivered and encapsulated RNA was illuminated. The results indicate that 53% of SLRNA present in the sample was encapsulated inside PMs and 32% of SLRNA was successfully delivered to T. brucei parasites.
The delivering of RNA into cells and parasites is a complicated process since it involves the use of different mediators. In the current study we used different types of coatings to study RNA delivery. First Fe2O3 coated spheres were introduced into T. brucei parasites and U2OS mammalian cells. The metal coating enabled detection of the microspheres inside the parasites and mammalian cells. The microspheres inside the parasites and mammalian cells were identified by detecting the presence of iron oxide on the microsphere membrane inside the cells by EDS measurements. Changes in the morphology of the T. brucei or the U2OS mammalian cells due to metal coating of the microsphere surface were not detected. Moreover, after coating the spheres with iron oxide, their surface charge (measured by z-potential) changed from −40 meV to −13 meV. On the other hand we could not observe changes in the average sizes of uncoated and coated microspheres. Fig. 3 shows TEM images of untreated T. brucei parasites and untreated U2OS cells (Fig. 3(a) and (d)) and T. brucei parasites and U2OS cells incubated with RNA loaded PMs (Fig. 3(b), (c) and (e). The difference between the images presented in Fig. 3(b) and (c) is the size of the parasite presented in the image. The bigger T. brucei parasite presented in Fig. 3(c) is magnified to show the penetrated spheres in a better way. In the inset of each image the enlarged part of parasite/cell is presented.
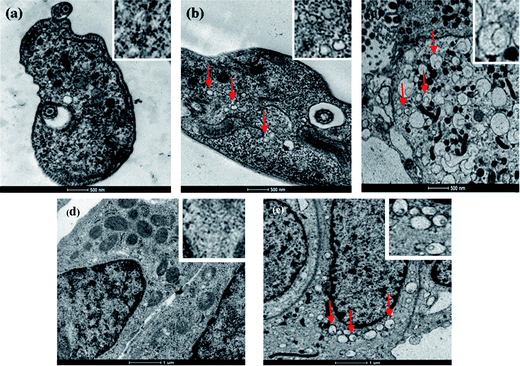 | ||
| Fig. 3 TEM images of the Trypanosoma brucei parasites and mammalian U2OS cells. (a) T.b. parasite without microspheres, the enlarged image of parasite tissue is shown in the inset; (b) Trypanosoma brucei parasite with penetrated microspheres, the enlarged image of parasite tissue with penetrated iron oxide coated PMs is shown in the inset; (c) a large Trypanosoma brucei parasite with penetrated microspheres, the enlarged image of parasite tissue with penetrated iron oxide coated PMs is shown in the inset; (d) mammalian U2OS cells without microspheres, the enlarged image of cell tissue is shown in the inset; and (e) mammalian U2OS cells with penetrated microspheres, the enlarged image of cell tissue with penetrated iron oxide coated PMs is shown in the inset. Some of the spheres inside the parasites and cancer cells are marked with red arrows. The EDS analysis results are shown in Fig. S4 in the ESI.† | ||
It was found that only PMs with sizes of 50–170 nm could penetrate into trypanosome bodies. This is due to the trypanosomes' body size, which is only 5 micrometers. In addition, the entry of bodies into the trypanosome cell is restricted to the flagella pocket that mediates the entry of particles in sizes of ∼100 nm. The entrance of the particles into U2OS cancer cells is due to the endocytosis process. The estimated size of the spheres that are able to penetrate the U2OS mammalian cells can reach 400–500 nm. This is because the size of the mammalian cell is 20 μm. Due to the size difference in the ability to penetrate cells, it is critically important to separate the spheres along their sizes. The separation could be accomplished either by manipulating the average sphere sizes as it was demonstrated previously1 or by filtration of the spheres.
The release of the RNA from the inner part of the BSA spheres occurs via protease digestion of BSA protein inside the parasite and cancer cell body. The kinetics of the release function of BSA spheres was studied in vitro. First, the stability of the spheres in the cell medium was measured. The spheres were found to be stable in cell and parasite media for several days (studies were performed for 3 days, where for all 3 days spheres demonstrated stability). Then, the kinetics of the sphere destruction (RNA release) inside the parasites and cancer cells was studied by the following experiments: the BSA spheres filled with FITC colored oil (FITC-dye which exhibits a green fluorescent signal where the excitation wavelength is at 488 nm and the emission at 520 nm) were internalized inside the parasite and cancer cell bodies. After one hour of incubation (for cancer cells as well as for parasites) a green signal emitted from the BSA spheres loaded with FITC–oil was detected. The signal appears as green points inside the cell body. After several hours the nature of the green signal changed. The green signal was homogeneously spread all over the parasites and cancer cells, which means that the spheres were destroyed and FITC–oil was spread over the cell bodies. For parasites the change in signal was detected after 2.5–3 hours and for cancer cells the pointed-green signal completely disappeared after 24 hours. It is important to notice that for cancer cells the autofluorescent signal started to rise after 5 hours pointing out that some of the spheres were destroyed.
In the next step we introduced uncoated RNA–BSA spheres, PVA and PEI coated spheres into U2OS mammalian cells and the number of penetrated cell spheres was compared. We preferred using mammalian cells in these experiments because of their larger size (20 μm). The coated and uncoated spheres were successfully introduced into the cancer cells without using any mediators, and cells were then analyzed by confocal microscopy. The nucleus of the cancer cells was stained with DAPI which gives a blue fluorescent signal. The fluorescence was collected with DAPI (blue signal with excit. at 359 nm, emis. at 457 nm), GFP (green signal with excit. at 489, emis. at 509 nm) and Rhod. (red signal with excit. at 550 nm, emis. at 573 nm) filters.
The overlap of red and green signals gives a yellow color. Fig. 4 shows images of U2OS cells incubated with uncoated and coated RNA loaded PMs.
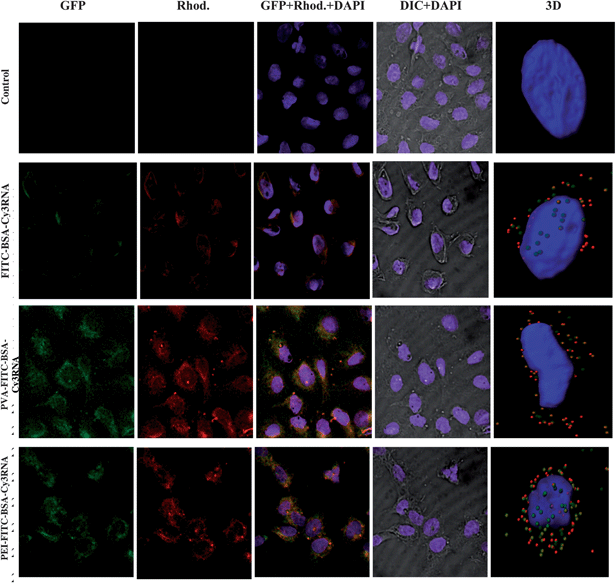 | ||
| Fig. 4 Confocal images of U2OS mammalian cancer cells with RNA loaded PMs. (Control-first row) cancer cells; (FITC-BSA–Cy3RNA-second row) cancer cells incubated with uncoated complex of FITC-BSA–Cy3RNA which exhibits green and red signals; (PVA coated FITC-BSA–Cy3RNA-third row) cancer cells with internalized PVA coated RNA loaded PMs; and (PEI coated FITC-BSA–Cy3RNA-fourth row) cancer cells with internalized PEI coated RNA loaded PMs. Green fluorescent signal is emitted from the FITC-labeled BSA (first column). Red fluorescent signal is emitted from the Cy3 labeled RNA (second column). The overlap of red and green signals gives a yellow color (third column). The nucleus of the cell colored with DAPI dye which emits blue signal (fourth column). The 3d reconstructions of z-stack confocal images are presented in the fifth column. The fluorescent signals passed through the following filters: DAPI, Rhod., GFP and DIC. Scale bar = 5 μm. | ||
The number of RNA–BSA spheres coated with PVA present in the U2OS cells has quadrupled as compared to the uncoated spheres. This is due to surface charge neutralization of RNA loaded PMs by the PVA and PEI coatings. When PMs were coated with PVA polymer the number of penetrated spheres increased dramatically. No green and red fluorescent signals were observed from the empty cancer cells (see Fig. 4-first row). When the cancer cells were incubated with RNA loaded BSA PMs, the spheres have penetrated the cell membrane and were localized near the membrane borders (see Fig. 4-second row). For detection of the exact location of the spheres, z-stack confocal images were reconstructed using “Imaris” image analysis software and presented in column five of Fig. 4. When the RNA loaded PMs were coated with PVA polymer, the surface charge of the spheres was neutralized and a larger amount of spheres penetrated the cell and localized near the nucleus of the U2OS cancer cells. Moreover, the PEI coating allows the RNA loaded PMs to penetrate the nucleus of the cells (see Fig. 4-fourth row, fifth column). The PEI polymer is known from the literature as an excellent agent for gene delivery applications, delivering nucleic acid molecules inside the cell body as well as inside the nucleus of cells. The sizes of BSA–RNA spheres inside the cells vary from 50 to 400 nm. In summary, using different polymeric coatings enabled targeting the delivery of the RNA molecules into the cells, and even into the nucleus.
Conclusion
In the current research we demonstrate a method for the synthesis and surface modifications of sonochemically prepared RNA-loaded PMs. The RNA-loaded spheres were used as a RNA delivery agent. We showed that different coatings could be used for controlled targeted delivery of RNA. Two types of coating were used: metal coating for detection of RNA loaded PMs inside the Trypanosoma brucei parasites and mammalian U2OS cells and polymeric coating for controlling the number of PMs penetrating the cells. Uncoated PMs tend to localize near the cells' membrane. Neutralizing the surface charge of PMs to 0 meV by PVA coating led to the concentration of the penetrated spheres near the nucleus of cancer cells. When the surface charge reached +20 meV (due to PEI coating) the spheres penetrated the nucleus of the cells. For both types of polymeric coatings applied, the number of penetrated PMs increased four times as compared to the number of uncoated spheres. The mechanism of RNA encapsulation and alignment of RNA inside the BSA spheres were studied by confocal microscopy and by using fluorescent tags (Cy3, 6FAM for RNA and FITC for BSA). In further work, we plan to explore the above-described method for siRNA encapsulation and delivery.Acknowledgements
Ulyana Shimanovich thanks Ministry of Science and Technology, Israel for the “Woman in Science” scholarship (3-8219).References
- U. Shimanovich, I. D. Tkacz, D. Eliaz, A. Cavaco-Paulo, S. Michaeli and A. Gedanken, Adv. Funct. Mater., 2011, 21(19), 3659–3666 CrossRef CAS.
- L. Crombez, A. Charnet, M. C. Morris, G. Aldrian-Herrada, F. Heitz and G. Divita, Biochem. Soc. Trans., 2007, 35, 44–46 CrossRef CAS.
- A. D. Judge, V. Sood, J. R. Shaw, D. Fang, K. Mc-Clintock and I. MacLachlan, Nat. Biotechnol., 2005, 23, 457–462 CrossRef CAS.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS.
- N. Unnamalai, B. G. Kang and W. S. Lee, FEBS Lett., 2004, 566, 307–310 CrossRef CAS.
- Y. Han, D. Shchukin, J. Yang, C. R. Simon, H. Fuchs and H. Möhwald, ACS Nano, 2010, 4(5), 2838–2844 CrossRef CAS.
- P. Rijn and A. Böker, J. Mater. Chem., 2011, 21, 16735–16747 RSC.
- X. Yan, J. Blacklock, J. Li and H. Möhwald, ACS Nano, 2012, 6, 111–117 CrossRef CAS.
- X. Yan, J. Li and H. Möhwald, Adv. Mater., 2012, 24, 2663–2667 CrossRef CAS.
- K. S. Suslick and M. W. Grinstaff, J. Am. Chem. Soc., 1990, 112(21), 7807–7809 CrossRef CAS.
- M. W. Grinstaff and K. S. Suslick, Macromolecular Assemblies in Polymeric Systems, ed. P. Stroeve and A. C. Balazs, Am. Chem. Soc., Washington, D. C. , 1992, 493, pp. 218–226 Search PubMed.
- M. Wong and K. S. Suslick, MRS Symp. Proc. v372, ed. D. L. Wilcox, M. Berg, T. Bernat, D. Kellerman and J. Corchran, Matl. Res. Soc., Pitcburg, 1995, pp. 89–94 Search PubMed.
- U. Angel (Shimanovich), D. Matas, S. Michaeli, A. Cavaco-Paulo and A. Gedanken, Chem.–Eur. J., 2010, 16, 2108–2114 CrossRef.
- U. Shimanovich, V. Volkov, D. Eliaz, A. Aizer, S. Michaeli and A. Gedanken, Small, 2011, 7, 1068 CrossRef CAS.
- U. Shimanovich, D. Eliaz, A. Aizer, I. Vayman, S. Micheli, Y. Shav-Tal and A. Gedanken, ChemBioChem, 2011, 12(11), 1678–1681 CrossRef CAS.
- F. Kratz, I. Fichtner, U. Beyer, P. Schumacher, T. Roth and H. H. Fiebig, Eur. J. Cancer., 1997, 33, 784 CrossRef.
- C. Chevalier, A. Saulnier, Y. Benureau, D. Flchet, D. Delgrange, F. Colbre-Garapin, C. Wychowski and A. Martin, Mol. Ther., 2007, 15, 1452–1462 CrossRef CAS.
- R. Prabhu, R. F. Garry and S. Dash, Virol. J., 2006, 3, 100 CrossRef.
- A. Gedanken, Chem.–Eur. J., 2008, 14, 3840–3853 CrossRef CAS.
- D. G. Monroe, F. J. Secreto, M. Subramaniam, B. J. Getz, S. Khosla and T. C. Spelsberg, Mol. Endocrinol., 2004, 19, 1555–1568 CrossRef.
- Z. Wang, J. C. Morris, M. E. Drew and P. T. Englund, J. Biol. Chem., 2000, 275, 40174–40179 CrossRef CAS.
- S. Avivi, I. Felner, I. Novik and A. Gedanken, Biochim. Biophys. Acta, Gen. Subj., 2001, 1527, 123–129 CrossRef CAS.
- A. F. Bell, L. Hecht and L. D. Barron, J. Chem. Soc., Faraday Trans., 1997, 93, 553–562 RSC.
- A. F. Bell, L. Hecht and L. D. Barron, J. Am. Chem. Soc., 1998, 120, 5820–5821 CrossRef CAS.
- S. Jones, D. T. A. Daley, N. Luscombe, H. M. Berman and J. M. Thornton, Nucleic Acids Res., 2001, 29(4), 943–954 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2tb00012a |
| This journal is © The Royal Society of Chemistry 2013 |