 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Introduction of nitrogen with controllable configuration into graphene via vacancies and edges†
Bin
Wang
*a,
Leonidas
Tsetseris
ab and
Sokrates T.
Pantelides
acd
aDepartment of Physics and Astronomy, Vanderbilt University, Nashville, Tennessee 37235, USA. E-mail: bin.wang@vanderbilt.edu
bDepartment of Physics, National Technical University of Athens, GR-15780 Athens, Greece
cDepartment of Electrical Engineering and Computer Science, Vanderbilt University, Nashville, Tennessee 37235, USA
dOak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
First published on 11th October 2013
Abstract
Doping with nitrogen in controllable configurations is very valuable to tailor the properties of graphene. Here we report density-functional theory calculations of chemical reactions of ammonia, a widely used nitrogen source, at vacancies and edges of graphene, through which we explore strategies to achieve N-doped graphene with optimized properties. We show that at different defects, ammonia reacts to form nitrogen impurities in distinct configurations, i.e. graphitic-N at single vacancies, pyridinic- or pyrrolic-N at divacancies, pyrrolic-N at armchair edges, and N in a four-member ring at zigzag edges. Moreover, different nitrogen-related defect configurations introduce distinct changes in the electronic structure of graphene. By calculating the core level shift of C1s electrons, we find configuration-dependent redistribution of electrons around the N-dopant. A discussion of how to achieve optimized doping and enhanced chemical reactivity in experiments is included.
Introduction
Graphene exhibits unusual physical properties that can be exploited for various applications,1 such as electronic and optoelectronic devices,2,3 biosensing and imaging,4 and energy conversion and storage.5 Similar to semiconductors, chemical functionalization serves as a promising way to tune the properties of graphene in a desirable manner.6–8 One of the most studied and widely used impurities for functionalization of graphene is nitrogen.9Introduction of nitrogen in low concentration can result in the opening of an energy band gap,10–12 enhanced carrier densities with minor reduction of the mobility,13 and reduction of sheet resistance due to the enhanced carrier densities.14 The latter effect is important for the use of graphene as a transparent electrode.14 Nitrogen-doped (N-doped) graphene has also shown high charging/discharging rate and large capacity in lithium ion batteries,15 remarkable efficiency for oxygen reduction in fuel cells,16–19 enhanced capacitance for supercapacitors,20,21 and improved electrocatalytic activity for biosensing.22 Moreover, nitrogen impurities provide favourable binding sites for other chemical groups to achieve further functionalization.23–25 Importantly, many of these desirable properties depend on the exact nature of nitrogen configuration. For example, nitrogen-induced doping could be either n-type or p-type.26 Charge redistribution around nitrogen-related species in graphene27–30 is also dictated by their arrangement, which, in turn, controls chemical reactivity in oxygen reduction reactions.31–34 Therefore, introduction of nitrogen in controllable configurations is highly desirable in order to optimize doping and catalytic phenomena.
Ammonia is a typical precursor that has been used widely as a nitrogen source in experiments on graphene.9 Nitrogen atoms can be introduced using ammonia plasma,35 through electrical joule heating or thermal annealing of graphene36 and graphene oxide in the presence of ammonia gas,37,38 by adding NH3 gas during chemical vapor deposition (CVD) growth of graphene,10,16,26,30,39 or by arc discharge in a NH3 atmosphere.40,41 Undesirably, in the majority of experiments N atoms eventually appear in different configurations, such as graphitic-N, pyridinic-N and pyrrolic-N.22,26,33,42,43 The nature of N-related structures is influenced by multiple factors, e.g. the catalyst in the chemical vapor deposition, the type and flow rate of the nitrogen and carbon source, and the post-treatment method.9 Although edges and point defects are known to be critical for the introduction of nitrogen,37,44–47 a detailed understanding of the dependence of nitrogen configuration on the type of defects, e.g. vacancies, zigzag or armchair edges, remains elusive. Controllable introduction of nitrogen in a desirable configuration has not yet been achieved.
Here we report extensive first-principles calculations on reaction paths of NH3 dissociation on various graphene defects including single vacancies, divacancies, armchair and zigzag edges. The dissociation reactions enable the introduction of distinct functional groups, namely –NH2, –NH, or –N, in graphitic-N, pyridinic-N, and pyrrolic-N configurations within graphene, and the type of functional groups is determined by the defects, i.e. graphitic-N at single vacancies, pyridinic- or pyrrolic-N at divacancies, pyrrolic-N at armchair edges, and N in a four-member ring at zigzag edges. Electronic structure calculations suggest that only graphitic-N induces n-type doping, while pyridinic-N could result in p-type doping, if the defect is not hydrogenated. Calculations of core level shifts of C1s electrons show distinguishable charge redistribution patterns around different N-related defects. A graphitic-N impurity donates electrons to carbon atoms, with the largest donation amplitude at the nearest ones. Other nitrogen configurations induce negative charge (electron donation) at the nearest carbon, but positive charge at the second nearest neighbours. Overall, the present systematic study on NH3 dissociation of at various defect sites on graphene suggests that one can achieve optimized doping and catalytic reactivity by introducing nitrogen in a controllable manner.
Methods
Spin-polarized density functional calculations were carried out using the VASP package.48 The PBE-GGA exchange-correlation functional49 was used. Electron–core interactions were described by ultrasoft Vanderbilt pseudopotentials50 in the calculation of reaction barriers to speed up the calculations. The cutoff energy was set to 400 eV, and the Brillouin zone was sampled with a single k-point at the Γ point. Tests with a finer 3 × 3 × 1 k-grid showed that reaction energies are converged within less than 20 meV.For the calculations of the projected density of states, we used pseudopotentials based on the projector augmented wave (PAW) method51,52 and a k-mesh of 5 × 5 × 1 grid centred at the Γ point. The Gaussian smearing method (width = 0.2 eV) was used. In the text, all the barrier calculations are based on ultrasoft pseudopotentials. We double-checked some reactions and found that calculations using PAW pseudopotentials show almost identical values for the enthalpy change.
The supercells contained 160 C atoms for calculations of reactions at vacancies, 96 C atoms for reactions at a zigzag edge, and 90 C atoms for reactions at an armchair edge. For such supercells, dopants are separated by about 2 nm from their periodic images in neighboring cells. Test calculations with even larger supercells with 640 carbon atoms (or ∼200 C for graphene nanoribbons) for the study of reactions at vacancies in the basal plane showed that the reaction energies are well converged (within 30 meV) with respect to supercell size.
The graphene layer and the impurities were free to relax until the self-consistent forces reached 0.02 eV Å−1. The nudged elastic band method53 with 16 images connecting the initial and final states was used to find the saddle points, which were identified by a vibrational analysis showing the existence of single normal mode associated with an imaginary frequency. The calculations of core level shifts were performed based on the initial state approximation and the results were checked with the transition state model.54
Results and discussions
Fig. 1 shows the reaction path of an NH3 molecule at a zigzag graphene edge. Such an edge has been observed, for example, under high vacuum conditions in a TEM chamber.55,56 The NH3 molecule first dissociates to form a NH2 group and one H atom bonded to the nearest edge carbon (IS in Fig. 1). In a second step, an NH group is formed by overcoming a small reaction barrier (IMS1). Further dissociation becomes difficult, since the two nearest active carbon atoms at the edge are both occupied. The calculated migration barrier of the H atom at the zigzag edge is 1.9 eV, so H hopping is activated at a moderate temperature. Once the previously dissociated H migrates away, breakup of the NH group is allowed. Formation of either a dangling N (IMS3) or an N atom in a four-membrane ring (FS) can be achieved. The calculation suggests that at room temperature, the dominant species of NH3 dissociation at a zigzag edge is the NH group, while at high temperatures dangling N or four-member-ring-N are formed. The reaction under a realistic condition should also depend on the concentration of NH3. High concentration may cause partial dissociation of NH3 due to the occupation of the active carbon at the edge. | ||
| Fig. 1 Dissociation of NH3 at a zigzag edge of graphene. A NH3 molecule dissociates to form NH2 (IS) and the most stable nitrogen configuration in a four-member ring (FS). Only the edge carbon atoms are displayed in IS, FS and the intermediate structures (IMS). The insets on the right show the transition states (TS). Carbon, nitrogen and hydrogen atoms are colored in grey, blue, and green, respectively. | ||
Differing from the zigzag edge, the reactive carbon atoms form pairs at an armchair edge. Fig. 2 shows the dissociation of NH3 at an armchair edge. The NH3 molecule first adsorbs at one carbon site at the edge. The molecule dissociates thus to an NH2 group and a H atom at the nearest edge carbon with a small reaction barrier of 0.2 eV and an energy gain of 2.8 eV. Further decomposition leads to a pyrrolic-N configuration, wherein the NH group is bridged between two edge carbon atoms forming a five-member ring, while the other two H atoms bind to two nearest carbon atoms. The associated reaction barrier is 1.5 eV. The cluster defect is energetically favoured over a configuration with the released H atom further away from the pyrrolic-N site. Because of the small reaction barrier, the NH3 reaction at an armchair edge likely reaches the pyrrolic-N configuration.
 | ||
| Fig. 2 Dissociation of NH3 at an armchair graphene edge. The decomposition reaction introduces pyrrolic-N. The stable structures (IS, FS, IMS) are shown schematically, while the transition states (TS) are displayed. | ||
It should be noted that here nitrogen is introduced through chemical reactions of NH3 at the reactive edges. If the edge atoms are saturated, e.g. by hydrogen atoms, the dissociation of NH3 needs to overcome a reaction barrier that is larger than its adsorption energy.44 Here we focus on the fresh edges and vacancies, which dominate under high-vacuum conditions and during non-equilibrium high-temperature growth.55–57
On the graphene basal plane, nitrogen may be introduced through reactions at carbon vacancies. Fig. 3 shows one such multi-step reaction at a single vacancy. A NH3 molecule physisorbs on graphene and overcomes a 0.2 eV reaction barrier to form a chemisorbed NH3 species (IMS1 in Fig. 3), which, in turn, dissociates to a bonded H atom and a NH2 species (IMS2). Subsequently, the NH2 group changes its configuration by bridging the nitrogen atom between two carbon atoms with an energy gain of 1.9 eV (IMS3) and a reaction barrier of 0.1 eV. The desirable reaction is desorption of a H2 molecule and formation of a graphitic-N. However, a reaction barrier of 2 eV has to be overcome for such a step. An alternative reaction is dissociation of the NH2 species to a NH group and another H atom forming a CH2 group (FS:CH2 in Fig. 3), which has a reaction barrier of only 0.2 eV. The corresponding overall energy gain is 6 eV. One should note that the energy gain for the formation of graphitic-N through desorption of a H2 molecule is by 0.3 eV larger than that of the sequence of reactions that lead to the NH–CH2 species (structure FS:CH2 in Fig. 3). The former reactions, however, has a much larger reaction barrier.
 | ||
| Fig. 3 Dissociation of NH3 at a single vacancy in graphene. The initial (IS), intermediate (IMS) and final (FS) states are shown schematically, while the transition states (TS) are displayed. | ||
Fig. 4 shows the reaction path for the transformation of the NH–CH2 group into a graphitic-N configuration through desorption of a H2 molecule. The NH group first dissociates to an H atom bonded to a nearby carbon and a pyridinic-N. Recombination of two hydrogen atoms (H1 and H2 in Fig. 4) forms a H2 molecule that desorbs. The overall reaction barrier to reach the graphitic-N configuration is 2.3 eV, which suggests that annealing at high temperature is needed in order to activate the process. The rather large reaction barrier also agrees with our previous molecular dynamics calculations, in which we observed the NH–CH2 species, but not the graphitic-N configuration within a short timescale (a few ps) at 1500 K.46 The difficulty to remove hydrogen impurities may also explain the reduced conductance of N-doped graphene.47
 | ||
| Fig. 4 Transformation of a NH–CH2 defect complex (FS:CH2) into a graphitic-N configuration (FS:Graphitic-N). H1 and H2 recombine forming a H2 molecule and the third hydrogen atom remains on the surface. The corresponding transition states are shown in Fig. S1 in the ESI.† | ||
In irradiated graphene, both divacancies and single vacancies have been widely observed.58 Removal of two neighboring carbon sites from a graphene sheet creates dangling bonds in four proximal C atoms. For this reason, one would expect that the reaction of NH3 is different between the divacancy and single vacancy cases. Fig. 5 shows the steps that lead to the dissociation of NH3 at a so-called 5-8-5 divacancy. The NH3 molecule breaks up to a NH2 group and a bonded H atom at one reactive carbon with a reaction barrier of 0.3 eV and a large energy gain of 2.0 eV (IMS1 in Fig. 5). The reaction barrier is comparable to the adsorption energy of NH3 on graphene. The NH2 moiety then dissociates to a NH group and an H atom that binds to another reactive carbon by overcoming a reaction barrier of 1.1 eV (IMS2 in Fig. 5). The other released H atom migrates away, while, in the process, the remaining NH group relaxes to form a pyrrolic-N structure as a bridge between two carbon atoms (structure FS in Fig. 5). In such a way, all the reactive carbon atoms around the divacancy are passivated by either a hydrogen atom or an N atom, and the overall energy gain is 3.8 eV.
 | ||
| Fig. 5 Dissociation of NH3 at a divacancy in graphene. | ||
Although desorption of a hydrogen molecule from a single vacancy may be achievable at an elevated temperature due to a reaction barrier of 2.3 eV (Fig. 4), H2 desorption is rather difficult in the case of the divacancy. We checked several possible reaction paths for removal of hydrogen atoms. Desorption of two H atoms from two C–H bonds is highly endothermic with a large reaction barrier (∼4 eV). Fig. 6 shows the reaction path with the lowest reaction barrier, wherein the pyrrolic-N structure first transforms into a pyridinic-N geometry, followed by release of a H2 molecule. The overall activation energy for the process is 3.1 eV, a significant reaction barrier. Moreover, differing from the exothermic reactions at a single vacancy, desorption of hydrogen at a divacancy costs energy of 1.0 eV. Therefore, high-temperature annealing is needed to remove the hydrogen impurities and create bare or hydrogenated pyridinic-N.
 | ||
| Fig. 6 Change of a pyrrolic-N configuration to a pyridinic-N structure at a divacancy through desorption of a H2 molecule. The structures of the transition states are shown in Fig. S2, ESI.† | ||
It should be noted that the 5-8-5 divacancy dominates in the Ar+ irradiated graphene,59 but may also reconstruct under electron irradiation to a more stable structure through bond rotation with a very large reaction barrier of 5–6 eV.60 Stabilization of IS in Fig. 5 by 1 eV due to the reconstruction58 should prohibit the NH3 dissociation.
Table 1 summarizes all the reaction barriers of the rate-determining steps for reactions of NH3 that lead to partial or full dissociation of the molecule at edges and vacancies. The calculated reaction of NH3 at vacancies and edges suggests that different N configurations can be introduced depending on the type of the defects, i.e. graphitic-N at single vacancies, pyrrolic-N and pyridinic-N at divacancies, pyrrolic-N at armchair edges, and N in a four-member ring at zigzag edges. Henceforth, we show that these varied nitrogen configurations modify the electronic properties of graphene in a very different manner.
| Partial dissociationa | Complete dissociationb | Removal of hydrogen | |
|---|---|---|---|
| a The first step of the dissociation resulting in the NH2 group. b Formation of the most stable nitrogen configuration. c Formation of NH2 at a zigzag edge is barrierless. | |||
| Zigzag edge | 0.0c | 1.9 | |
| Armchair edge | 0.2 | 1.5 | |
| SV | 0.2 | 2.0 | 2.3 |
| DV (585) | 0.3 | 1.1 | 3.1 |
Fig. 7 depicts the electronic density of states (DOS) of graphene with nitrogen in different configurations. Single vacancies are known to act as acceptors and lead to p-type doping of graphene.61,62 In agreement with experiments,26 the graphitic-N structure donates electrons to the host system, resulting in n-type doping in graphene, i.e. the Dirac point shifts to lower energies than the Fermi energy (blue curve in Fig. 7a) In contrast, the pyridinic-N site behaves as an electron acceptor (red curve in Fig. 7b), resulting in p-type doping shown by the upshifted DOS. If the pyridinic-N defect is stabilized by a H atom, which is likely to happen under realistic conditions, the doping effect is reduced significantly. The pyrrolic-N configuration (orange dashed curve in Fig. 7b) does not induce electron/hole doping. Therefore, among all possible reaction products of NH3 dissociation at mono- and di-vacancies of graphene, only the graphitic-N structure leads to significant n-type doping, while the hydrogenated defects show largely reduced doping effect despite the nitrogen configuration (dashed curves in Fig. 7). If hydrogen can be removed by high-temperature annealing, the pyridinic-N may lead to p-type doping. However, due to the dangling bonds, bare pyridinic-N is very reactive and may exist only in high vacuum.
 | ||
| Fig. 7 Projected density of states onto carbon atoms in N-doped graphene. Nitrogen atoms in different configurations are schematically shown as insets. | ||
At the armchair and zigzag edges, the covalent binding of nitrogen and hydrogen cause minor doping effects as shown in Fig. S3 (ESI).† It should be noted that for nitrogen impurities, we only consider the species created from the calculated dissociation of NH3 at the edges instead of substitutional nitrogen as in some previous publications,63–66 in which it has been shown that the doping may also depend on the symmetry and width of the ribbons and the position of the dopant in the ribbon.
Redistribution of electrons in N-doped graphene is suggested to be critical for its enhanced chemical reactivity for the oxygen reduction reaction in fuel cells,27,67 although the catalytic sites, graphitic-N and/or pyridinic-N, for the oxygen reactions remain unclear.9 Because of this ambiguity, we have performed calculations of the core level shift (CLS) of C1s electrons. The results are shown in Fig. 8. They were obtained within the so-called initial state approximation, wherein the core level is calculated without the removal of a core electron. The calculated CLS can be used to evaluate the local charge redistribution. Removal of electrons from carbon results in weaker electronic screening and smaller binding energy for the 1s electrons. In contrast, if carbon accepts electrons, the enhanced electronic screening increases the binding energy for the C1s core electrons.
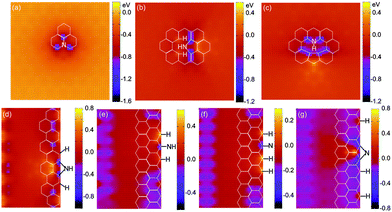 | ||
| Fig. 8 Core level shifts (CLS) of the C1s orbital around various N-related defects in graphene (a–c) and nanoribbons (d–g). The reference level (CLS = 0) is the energy of the C1s state in pristine graphene. Here negative values indicate increased binding energies. | ||
Despite the nitrogen configurations, carbon atoms always withdraw electrons from an adjacent N site. The binding energy thus increases (here negative values indicate increased binding energies) by ∼1 eV for C1s electrons. The value is in agreement with pertinent experimental data on the core level shift of sp2-bonded carbon atoms that are adjacent to a nitrogen atom in N-doped graphene9 and carbon nanotubes.68
However, close inspection of these configurations reveals that nitrogen atoms in varied configurations induce different charge redistribution. In the graphitic-N case of Fig. 8a, there is a negative core level shift (increased binding energy for core electrons) at the three carbon atoms adjacent to the nitrogen impurity, a clear signature of enhanced electronic screening at those carbon atoms due to charge transfer from the nitrogen.28 Differing from the graphitic-N, the pyrrolic-N structure created by NH3 dissociation at a divacancy induces decreased C1s core level binding energy for the second-nearest carbon neighbour (Fig. 8b). If high-temperature annealing is used to remove H atoms and form pyridinic-N geometries (either –N or –NH), the carbon in the pentagon ring still displays decreased C1s binding energy (Fig. 8c). Similarly, at armchair and zigzag edges (Fig. 8d–g), all the newly formed nitrogen species lead to negatively charged carbon at the nearest neighbour, and positively charged carbon at the second-nearest carbon neighbour. The carbon atoms that are bonded with hydrogen species show decreased C1s binding energy, indicating a positive charge as well. Moreover, the carbon atoms at a zigzag edge are more negatively charged compared to those at an armchair edge. Such different spatial charge redistribution may result in variation of the local work function, valuable for optimal charge transfer in contacts to photovoltaics or nanoelectronic devices.
The next question is how to introduce nitrogen in a desirable configuration. In principle, defects can be introduced into graphene in a controllable manner using ion and electron irradiation.69 For example, Ar+ irradiation creates mostly single vacancies in graphite70 but 5-8-5 divacancies in epitaxial graphene on SiC.59 By introducing NH3 and high temperature annealing, through reaction of NH3 at single vacancies on the basis of the aforementioned reaction path, graphitic-N can be controllably introduced into the pre-irradiated graphene. Moreover, once graphitic-N is introduced, post-treatment using electron radiation can transform graphitic-N into pyridinic-N since the carbon atom adjacent to the nitrogen is easier to remove.71 If divacancies are created directly in graphene, pyrrolic-N should dominate due to its high stability.
Prolonged irradiation by the electron beam can create large holes in graphene.55 In this case, edges dominate, and pyrrolic-N or N in four-member-rings can be introduced. Indeed, experiments show that under prolonged plasma treatment, the pyrrolic-N portion increases and the graphitic-N portion decreases.42 Therefore, by intentionally creating corresponding defects and controlling temperature, the nitrogen configuration can be tuned. In a similar way, other functional groups such as metal atoms can also be introduced.72
In principle, the concentration of the vacancies can be controlled by an irradiation dose, which may in turn allow introduction of high concentrations of nitrogen in graphene. However, interaction between defects leads to aggregation of vacancies,58 so that the maximum concentration of nitrogen introduced using the proposed strategy remains to be explored. Recent experiments using low energy ion implantation have shown that nitrogen or boron can be introduced into graphene with rather high concentrations (>10%).73 Such ion implantation introduces vacancies as well,73,74 so that it may be possible to get defect-free doping by combining implantation with the method proposed here.
Conclusions
In conclusion, we have analyzed various dissociation paths of NH3 at single vacancies, divacancies, armchair and zigzag edges of graphene. Through these reactions, a number of distinct functional groups, namely –NH2, –NH, and –N, can be introduced in the graphitic-N, pyridinic-N and pyrrolic-N configurations. In particular, graphitic-N structures can be created through ammonia dissociation at single vacancies, while pyrrolic-N sites can be introduced at divacancies and armchair edges. By controlling the type of the defects using electron or ion irradiation, one may be able to tailor the exact configuration of nitrogen-related functional impurities. In this way, one can optimize doping levels and electron redistribution for specific applications, such as electronic devices and catalysts in fuel cells.Acknowledgements
This work was supported by DTRA Grant no. HDTRA1-10-1-0016 and the William A. and Nancy F. McMinn Endowment at Vanderbilt University. The calculations were performed at ORNL's Center for Computational Sciences and the Air Force Research Laboratory DoD Supercomputing Resource Center.Notes and references
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- P. Avouris, Nano Lett., 2010, 10, 4285–4294 CrossRef CAS PubMed.
- Q. L. Bao and K. P. Loh, ACS Nano, 2012, 6, 3677–3694 CrossRef CAS PubMed.
- Y. X. Liu, X. C. Dong and P. Cheng, Chem. Soc. Rev., 2012, 41, 2283–2307 RSC.
- N. G. Sahoo, Y. Z. Pan, L. Li and S. H. Chan, Adv. Mater., 2012, 24, 4203–4210 CrossRef CAS.
- S. T. Pantelides, Y. S. Puzyrev, L. Tsetseris and B. Wang, MRS Bull., 2012, 37, 1187–1194 CrossRef CAS.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- J. E. Johns and M. C. Hersam, Acc. Chem. Res., 2013, 46, 77–86 CrossRef CAS PubMed.
- H. B. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- D. C. Wei, Y. Q. Liu, Y. Wang, H. L. Zhang, L. P. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- D. Usachov, O. Vilkov, A. Grüneis, D. Haberer, A. Fedorov, V. K. Adamchuk, A. B. Preobrajenski, P. Dudin, A. Barinov, M. Oehzelt, C. Laubschat and D. V. Vyalikh, Nano Lett., 2011, 11, 5401–5407 CrossRef CAS PubMed.
- A. Lherbier, A. R. Botello-Méndez and J.-C. Charlier, Nano Lett., 2013, 13, 1446–1450 CAS.
- A. Lherbier, X. Blase, Y.-M. Niquet, F. Triozon and S. Roche, Phys. Rev. Lett., 2008, 101, 036808 CrossRef.
- J. O. Hwang, J. S. Park, D. S. Choi, J. Y. Kim, S. H. Lee, K. E. Lee, Y.-H. Kim, M. H. Song, S. Yoo and S. O. Kim, ACS Nano, 2012, 6, 159–167 CrossRef CAS PubMed.
- Z.-S. Wu, W. C. Ren, L. Xu, F. Li and H.-M. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- D. S. Deng, Y. Chen, Y. G. Chen, Y. L. Li, R. Y. Li, X. L. Sun, S. Y. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 Search PubMed.
- C. H. Choi, M. W. Chung, H. C. Kwon, S. H. Park and S. I. Woo, J. Mater. Chem. A, 2013, 1, 3694–3699 CAS.
- Q. Q. Li, S. Zhang, L. M. Dai and L. S. Li, J. Am. Chem. Soc., 2012, 134, 18932–18935 CrossRef CAS PubMed.
- F. M. Hassan, V. Chabot, J. D. Li, B. K. Kim, L. Ricardez-Sandoval and A. P. Yu, J. Mater. Chem. A, 2013, 1, 2904–2912 CAS.
- K. Gopalakrishnan, A. Govindaraj and C. N. R. Rao, J. Mater. Chem. A, 2013, 1, 7563–7565 CAS.
- Y. Wang, Y. Y. Shao, D. W. Matson, J. H. Li and Y. H. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- L.-S. Zhang, X.-Q. Liang, W.-G. Song and Z.-Y. Wu, Phys. Chem. Chem. Phys., 2010, 12, 12055–12059 RSC.
- G. Kim and S.-H. Jhi, ACS Nano, 2011, 5, 805–810 CrossRef CAS PubMed.
- T. Schiros, D. Nordlund, L. Pálová, D. Prezzi, L. Y. Zhao, K. S. Kim, U. Wurstbauer, C. Gutiérrez, D. Delongchamp, C. Jaye, D. Fischer, H. Ogasawara, L. G. M. Pettersson, D. R. Reichman, P. Kim, M. S. Hybertsen and A. N. Pasupathy, Nano Lett., 2012, 12, 4025–4031 CrossRef CAS PubMed.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- J. C. Meyer, S. Kurasch, H. J. Park, V. Skakalova, D. Künzel, A. Groß, A. Chuvilin, G. Algara-Siller, S. Roth, T. Iwasaki, U. Starke, J. H. Smet and U. Kaiser, Nat. Mater., 2011, 10, 209–215 CrossRef CAS PubMed.
- D. H. Deng, X. L. Pan, L. Yu, Y. Cui, Y. P. Jiang, J. Qi, W.-X. Li, Q. Fu, X. C. Ma, Q. K. Xue, G. Q. Sun and X. H. Bao, Chem. Mater., 2011, 23, 1188–1193 CrossRef CAS.
- L. Y. Zhao, R. He, K. T. Rim, T. Schiros, K. S. Kim, H. Zhou, C. Gutiérrez, S. P. Chockalingam, C. J. Arguello, L. Pálová, D. Nordlund, M. S. Hybertsen, D. R. Reichman, T. F. Heinz, P. Kim, A. Pinczuk, G. W. Flynn and A. N. Pasupathy, Science, 2011, 333, 999–1003 CrossRef CAS PubMed.
- H. Niwa, K. Horiba, Y. Harada, M. Oshima, T. Ikeda, K. Terakura, J. I. Ozaki and S. Miyata, J. Power Sources, 2009, 187, 93–97 CrossRef CAS PubMed.
- R. L. Liu, D. Q. Wu, X. L. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, J. Mater. Chem., 2010, 20, 7491–7496 RSC.
- Z. Q. Luo, S. Lim, Z. Q. Tian, J. Z. Shang, L. F. Lai, B. MacDonald, C. Fu, Z. X. Shen, T. Yu and J. Y. Lin, J. Mater. Chem., 2011, 21, 8038–8044 RSC.
- Y.-C. Lin, C.-Y. Lin and P.-W. Chiu, Appl. Phys. Lett., 2010, 96, 133110 CrossRef.
- X. R. Wang, X. L. Li, L. Zhang, Y. Yoon, P. K. Weber, H. L. Wang, J. Guo and H. J. Dai, Science, 2009, 324, 768–771 CrossRef CAS PubMed.
- X. L. Li, H. L. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Joroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110–3116 CrossRef CAS PubMed.
- R. T. Lv, Q. Li, A. R. Botello-Méndez, T. Hayashi, B. Wang, A. Berkdemir, Q. Z. Hao, A. L. Elías, R. Cruz-Silva, H. R. Gutiérrez, Y. A. Kim, H. Muramatsu, J. Zhu, M. Endo, H. Terrones, J.-C. Charlier, M. Pan and M. Terrones, Sci. Rep., 2012, 2, 586 CrossRef PubMed.
- L. S. Panchakarla, K. S. Subrahmanyam, S. K. Saha, A. Govindaraj, H. R. Krishnamurthy, U. V. Waghmare and C. N. R. Rao, Adv. Mater., 2009, 21, 4726–4730 CAS.
- N. Li, Z. Wang, K. Zhao, Z. Shi, Z. Gu and S. Xu, Carbon, 2010, 48, 255–259 CrossRef CAS PubMed.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- L. Tsetseris and S. T. Pantelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155446 CrossRef.
- F. Cervantes-Sodi, G. Csányi, S. Piscanec and A. C. Ferrari, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 165427 CrossRef.
- B. Wang and S. T. Pantelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245403 CrossRef.
- B. D. Guo, Q. Liu, E. Chen, H. Zhu, L. Fang and J. R. Gong, Nano Lett., 2010, 10, 4975–4980 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- W. Olovsson, C. Göransson, T. Marten and I. A. Abrikosov, Phys. Status Solidi B, 2006, 243, 2447–2464 CrossRef CAS.
- G. O. Girit, J. C. Meyer, R. Erni, M. D. Rossell, C. Kisielowski, L. Yang, C.-H. Park, M. F. Crommie, M. L. Cohen, S. G. Louie and A. Zettl, Science, 2009, 323, 1705–1708 CrossRef PubMed.
- X. T. Jia, M. Hofmann, V. Meunier, B. G. Sumpter, J. Campos-Delgado, J. M. Romo-Herrera, H. Son, Y.-P. Hsieh, A. Reina, J. Kong, M. Terrones and M. S. Dresselhaus, Science, 2009, 323, 1701–1705 CrossRef CAS PubMed.
- X. Y. Zhang, J. Xin and F. Ding, Nanoscale, 2013, 5, 2556–2569 RSC.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed.
- M. M. Ugeda, I. Brihuega, F. Hiebel, P. Mallet, J.-Y. Veuillen, J. M. Gómez-Rodríguez and F. Ynduráin, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 121402 CrossRef.
- J. Kotakoski, A. V. Krasheninnikov, U. Kaiser and J. C. Meyer, Phys. Rev. Lett., 2011, 106, 105505 CrossRef CAS.
- J. J. Palacios and F. Ynduráin, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 245443 CrossRef.
- B. Wang and S. T. Pantelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 165438 CrossRef.
- S. S. Yu, W. T. Zheng, Q. B. Wen and Q. Jiang, Carbon, 2008, 46, 537–543 CrossRef CAS PubMed.
- B. Biel, X. Blase, F. Triozon and S. Roche, Phys. Rev. Lett., 2009, 102, 096803 CrossRef.
- E. Cruz-Silva, Z. M. Barnett, B. G. Sumpter and V. Meunier, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155445 CrossRef.
- J. Jiang, J. Turnbull, W. C. Lu, P. Boguslawski and J. Bernholc, J. Chem. Phys., 2012, 136, 014702 CrossRef PubMed.
- L. P. Zhang and Z. H. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- S. H. Lim, H. I. Elim, X. Y. Gao, A. T. S. Wee, W. Ji, J. Y. Lee and J. Lin, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045402 CrossRef.
- A. V. Krasheninnikov and K. Nordlund, J. Appl. Phys., 2010, 107, 071301 CrossRef.
- M. M. Ugeda, I. Brihuega, F. Guinea and J. M. Gómez-Rodríguez, Phys. Rev. Lett., 2010, 104, 096804 CrossRef CAS.
- T. Susi, J. Kotakoski, R. Arenal, S. Kurasch, H. Jiang, V. Skakalova, O. Stephan, A. V. Krasheninnikov, E. I. Kauppinen, U. Kaiser and J. C. Meyer, ACS Nano, 2012, 6, 8837–8846 CrossRef CAS PubMed.
- A. V. Krasheninnikov, P. O. Lehtinen, A. S. Foster, P. Pyykkö and R. M. Nieminen, Phys. Rev. Lett., 2009, 102, 126807 CrossRef CAS.
- U. Bangert, W. Pierce, D. M. Kepaptsoglou, Q. Ramasse, R. Zan, M. H. Gass, J. A. Van den Berg, C. B. Boothroyd, J. Amani and H. Hofsäss, Nano Lett., 2013, 13, 4902–4907 CrossRef CAS PubMed.
- E. H. Åhlgren, J. Kotakoski and A. V. Krasheninnikov, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115424 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Structures of transition states and density of states of N-doped graphene nanoribbons are presented. See DOI: 10.1039/c3ta13610h |
| This journal is © The Royal Society of Chemistry 2013 |