 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Role of the transition metal in Grignard metathesis polymerization (GRIM) of 3-hexylthiophene†
Mahesh P. Bhatta, Harsha D. Magurudeniyaa, Prakash Sistaa, Elena E. Sheinab, Malika Jeffries-ELc, Benjamin G. Janesko*d, Richard D. McCulloughe and Mihaela C. Stefan*a
aUniversity of Texas at Dallas, Department of Chemistry, 800 West Campbell Road, Richardson, TX 75080, USA. E-mail: mihaela@utdallas.edu; Fax: +1 972-883-2925; Tel: +1 972-883-6581
bPlextronics. Inc, 2180 WilliamPitt Way, Pittsburgh, PA 15238, USA. E-mail: ESheina@plextronics.com
cIowa State University, Department of Chemistry, 3101C Hach Hall, Ames, IA 50011, USA. E-mail: malikaj@iastate.edu
dTexas Christian University, Department of Chemistry, TCU Box 298860, Forth Worth, TX 76129, USA. E-mail: b.janesko@tcu.edu
eHarvard University, 1350 Massachusetts Avenue, Holyoke Center, Suite 836, Cambridge, MA 02138, USA. E-mail: richard_mccullough@harvard.edu
First published on 9th September 2013
Abstract
Regioregular poly(3-alkylthiophene)s are widely used in organic electronics applications such as solar cells and field effect transistors. Nickel, palladium, and platinum diphenylphosphinoethane complexes were tested as catalysts for the Grignard metathesis (GRIM) polymerization of 2,5-dibromo-3-hexylthiophene and 2-bromo-5-iodo-3-hexylthiophene. Nickel-mediated polymerization generated regioregular, low-polydispersity poly(3-hexylthiophene) with well-defined molecular weight consistent with a “living” chain-growth mechanism. By contrast, palladium-mediated polymerization proceeded by a step-growth mechanism and generated polymers with less than 80% head-to-tail couplings. Platinum-mediated polymerization gave very low molecular weight products. Kinetic and computational results suggested that the nickel catalyst acts as an initiator and remains associated with the growing polymer chain, while palladium dissociates from the growing chain. Computational and experimental evidence was provided for various side reactions of dissociated Pd(0) catalyst, which could yield a step growth mechanism and lower regioirregularity.
Introduction
Poly(3-alkylthiophene)s are the most used semiconducting polymers for organic electronics applications.1,2 The asymmetric 3-alkyl-2,5-dihalothiophene monomer can generate multiple intra-monomer orientations: “head–head”, “head–tail”, and “tail–tail”. Regioregular polymers containing almost exclusively head-to-tail couplings have improved opto-electronic properties as compared to the regioirregular analogues.3 Synthesis of regioregular poly(3-hexylthiophene) (P3HT) utilizing either Kumada or Negishi metal-mediated cross-coupling reactions were first reported in 1992.4–10 Both methods require cryogenic temperatures to generate regioregular polymer. The Grignard metathesis (GRIM) polymerizations was reported by McCullough's group in 1999 and allows the synthesis of regioregular P3HT in large scale without the use of cryogenic reaction conditions.11 Later, McCullough and Yokozawa reported the quasi-“living” nature of GRIM polymerization.11–14Scheme 1 shows the accepted mechanism for the nickel mediated GRIM polymerization. The cyclic chain growing reaction begins with a M(II)–halide complex bound to the growing chain end (7). The transition metal M is typically Ni. The nickel complex undergoes transmetalation with the 5-halomagnesium thiophene monomer (2). Reductive elimination then forms a new thiophene–thiophene bond. The remaining Ni(0)(dppe) complex is speculated to form an “associated pair” with the thiophene chain end. Subsequent oxidative addition to the terminal carbon–halogen bond regenerates the nickel complex.
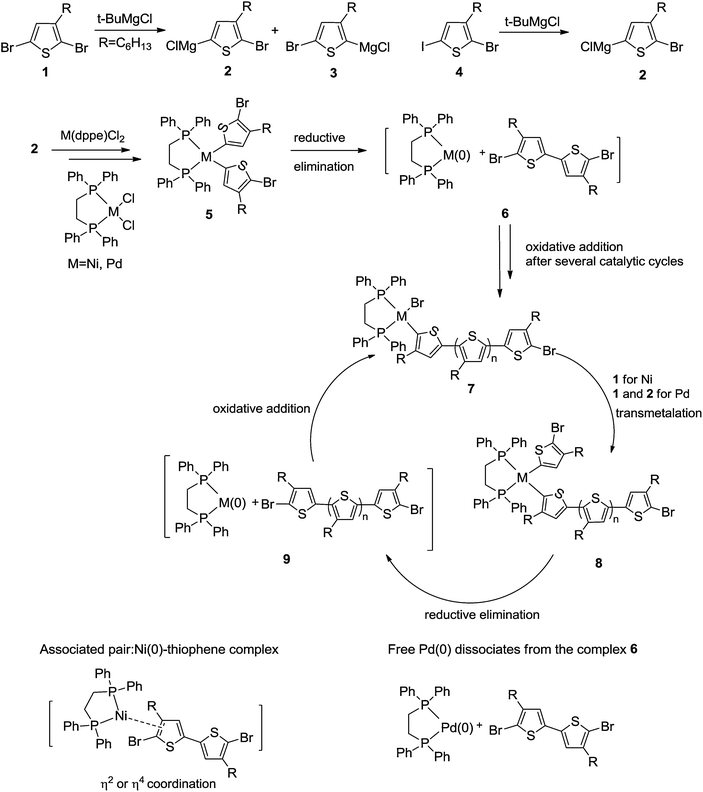 | ||
| Scheme 1 Proposed mechanism of nickel and palladium-mediated Grignard metathesis (GRIM) polymerization (R = C6H13). | ||
McCullough and coworkers demonstrated that GRIM polymerization has some characteristics of a “living” chain growth polymerization. This “living” polymerization enabled the end-functionalization and generated polymers with well-defined molecular weights and relatively narrow polydispersity index.14–17 Yokozawa's group also reported the “living” behavior of nickel catalyzed cross-coupling polymerization of 2-bromo-5-iodo-3-hexylthiophene.13,17 Subsequent work by Luscombe, Kiriy, Locklin, and McNeil18–27 had provided additional experimental evidence for the presence of the associated pair between Ni(0) and the growing chain. Dissociation of free Ni(0) is presumed to yield various side reactions, possibly including inter-chain coupling, and non-“living” behavior.28–30
We investigated the kinetics of GRIM polymerization of 2,5-dibromo-3-hexylthiophene (DBHT) and 2-bromo-5-iodo-3-hexylthiophene (IBHT) monomers with Ni(dppe)Cl2, Pd(dppe)Cl2, and Pt(dppe)Cl2 catalysts. Experimental results indicated that the metal strongly affects the kinetics of polymerization and the regioregularity of synthesized P3HT polymers. Calculations suggested that the metal affects the stability of the associated pair (9) (Scheme 1), but not the subsequent rate of formation of (7).
Experimental
Materials
All reactions were conducted under purified nitrogen, using oven-dried glassware. All glassware was assembled while hot and cooled under nitrogen. Commercial chemicals, purchased from Aldrich Chemical Co., Inc., were used without further purification unless noted otherwise. All solvents were freshly distilled prior to use. Tetrahydrofuran (THF) was distilled from sodium benzophenone ketyl. t-Butylmagnesium chloride (2 M in diethyl ether) and p-dimethoxybenzene were purchased from Aldrich Chemical Co., Inc. and used without further purification. THF was dried over K/benzophenone and freshly distilled prior to use. Catalyst [1,3-bis(diphenylphosphino)ethane]-dichloronickel(II)and[1,3-bis(diphenylphosphino) ethane]-dichloropalladium(II) were purchased from Aldrich and used as received. [1,3-Bis(diphenylphosphino)ethane]-dichloro platinum(II) was synthesized accordingly to a published procedure.31Structural analysis
1H NMR spectra of the P3HT polymers were recorded on a Bruker-500 MHz spectrometer at room temperature. 1H NMR data are reported in parts per million as chemical shift relative to tetramethylsilane (TMS) as the internal standard. Spectra were recorded in CDCl3. GC/MS was performed on an Agilent 6890-5973 GC-MS workstation. The GC column was a Hewlett-Packard fused silica capillary column cross-linked with 5% phenylmethyl siloxane. Helium was the carrier gas (1 mL min−1). The following conditions were used for all GC/MS analyses: injector and detector temperature, 250 °C; initial temperature, 70 °C; temperature ramp, 10 °C min−1; final temperature, 280 °C.Molecular weights of the synthesized polymers were measured by Size Exclusion Chromatography (SEC) analysis on a Viscotek VE 3580 system equipped with ViscoGEL™ columns (GMHHR-M), connected to a refractive index (RI) detectors. GPC solvent/sample module (GPCmax) was used with HPLC grade THF as the eluent and calibration was based on polystyrene standards. Running conditions for SEC analysis were: flow rate = 1.0 mL min−1, injector volume = 100 μL, detector temperature = 30 °C, column temperature = 35 °C. All the polymers samples were dissolved in THF and the solutions were filtered through PTFE filters (0.45 μm) prior to injection.
Polymerization experiment
In a typical experiment, a dry 100 mL three-neck round bottom flask was connected to a nitrogen line and charged with 2,5-dibromo-3-hexylthiophene (1.6 g, 5.0 mmol), p-dimethoxybenzene (internal standard) (0.2 g), and anhydrous THF (10 mL). A 2 M solution of alkyl magnesium chloride (2.5 mL, 5.0 mmol) in diethyl ether (Et2O) was added via a deoxygenated syringe, and the reaction mixture was reacted at room temperature for 2 hours. At this time an aliquot (0.5 mL) was taken out and quenched with water. The organic phase was extracted in Et2O and subjected to GC-MS analysis to determine the composition of the reaction mixture. The main components of the reaction mixture were 2-bromo-5-chloromagnesium-3-hexylthiophene and 5-bromo-2-chloromagnesium-3-hexylthiophene regioisomers. Usually less than 5% of unreacted 2,5-dibromo-3-hexylthiophene was detected by GC-MS analysis. The concentration of 2-bromo-5-chloromagnesium-3-hexylthiophene isomer was estimated to 80 mol%. Before the addition of the catalyst, 40 mL of anhydrous THF was added to the reaction mixture, followed by the addition Ni(dppe)Cl2 (0.04 g, 0.075 mmol). After addition of Ni(dppe)Cl2, aliquots (1 mL) were taken at different time intervals and each was precipitated in methanol (5 mL). For each aliquot a sample was prepared in Et2O (2 mL) and subjected to GC-MS analysis for the determination of concentration of unreacted monomer. After filtration through PTFE filters (0.45 μm), the molecular weight of the polymer samples was measured by SEC with THF as the eluent. Polymerization of 2-bromo-5-iodo-3-hexylthiophene was performed in a similar manner.Chain scissoring experiment
In a 100 mL two neck flask Pd(dppe)Cl2 (10 mg, 0.01 mmol) was dispersed in 10 mL of THF under a nitrogen atmosphere. Sodium metal (1 g, 43 mmol) was added to the reaction mixture and was stirred for 5 hours. At this point, the color of the reaction mixture turned to yellowish green which is due to the formation of Pd(0). A solution of poly(3-hexylthiophene), (100 mg, 0.0025 mmol, Mn = 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), was prepared in 100 mL of freshly distilled THF. The Pd(0) catalyst was cannulated into the solution of P3HT. Magnesium bromide (10 mg, 0.05 mmol) was added to the reaction flask and the reaction mixture was stirred for 12 hours under a nitrogen atmosphere. The reaction was quenched with methanol and the polymer was filtered and purified by using Soxhlet extractions with methanol, hexane and chloroform. No polymer was collected from the methanol and hexane fractions. All the polymer was collected from the chloroform fraction.
000 g mol−1), was prepared in 100 mL of freshly distilled THF. The Pd(0) catalyst was cannulated into the solution of P3HT. Magnesium bromide (10 mg, 0.05 mmol) was added to the reaction flask and the reaction mixture was stirred for 12 hours under a nitrogen atmosphere. The reaction was quenched with methanol and the polymer was filtered and purified by using Soxhlet extractions with methanol, hexane and chloroform. No polymer was collected from the methanol and hexane fractions. All the polymer was collected from the chloroform fraction.Computations
All calculations used the Gaussian 09 electronic structure package.32 Unless noted otherwise, calculations used density functional theory with the M06 exchange–correlation functional;33 the LANL08 basis set and relativistic effective core potential on Ni, Pd, and Pt;34,35 and the 6-31+G(d,p) basis set on all other atoms.36,37 Geometries and free energy corrections are evaluated with the PBE0 hybrid exchange-correlation functional and the LANL2DZ basis set and core potential. M06 self-consistent field calculations use Gaussian keyword “SCF = Tight”, and use a numerical integration grid with 99 radial and 590 angular points per atom (Gaussian keyword “Integral(Grid = UltraFine)”).Results and discussion
A magnesium halogen exchange reaction between DBHT and t-butylmagnesium chloride generated regioisomers (2) and (3) (Scheme 1) in a ratio of 80 to 20, as determined by GC-MS analysis. Initially, Ni(dppe)Cl2 was used as a catalyst and the kinetics of polymerization was monitored. GC-MS analysis of the quenched reaction mixture, after the addition of Ni(dppe)Cl2 indicated that only monomer (2) was consumed during polymerization. Isomer (3) was not consumed during the nickel-mediated polymerization. The monomer conversion for the Ni(dppe)Cl2 catalyzed reaction was thus calculated from the consumption of isomer (2). The conversion vs. time plot (Fig. 1B, top) shows that the polymerization was fast, reaching 98% conversion in 10 minutes. The molecular weight vs. conversion plot (Fig. 1B, bottom) shows a linear increase of molecular weight with conversion. This is consistent with previous reported data which demonstrated the quasi-“living” nature of GRIM polymerization of DBHT with Ni(dppp)Cl2 initiator.14 The polydispersity index of the collected polymer samples has a value of ∼1.23 (the polydispersity index vs. conversion plots is shown in the ESI†).![Conversion vs. time (top) and number average molecular weight vs. conversion (bottom) plots for GRIM polymerization of 2,5-dibromo-3-hexylthiophene. (A) [DBHT]0 = 0.1 mol L−1; [Pd(dppe)Cl2]0 = 0.0015 mol L−1; temp = 45 °C. (B) [DBHT]0 = 0.1 mol L−1; [Ni(dppe)Cl2]0 = 0.0015 mol L−1; temp = 23 °C.](/image/article/2013/TA/c3ta13258g/c3ta13258g-f1.gif) | ||
| Fig. 1 Conversion vs. time (top) and number average molecular weight vs. conversion (bottom) plots for GRIM polymerization of 2,5-dibromo-3-hexylthiophene. (A) [DBHT]0 = 0.1 mol L−1; [Pd(dppe)Cl2]0 = 0.0015 mol L−1; temp = 45 °C. (B) [DBHT]0 = 0.1 mol L−1; [Ni(dppe)Cl2]0 = 0.0015 mol L−1; temp = 23 °C. | ||
GRIM polymerization of DBHT was also performed with Pd(dppe)Cl2 catalyst. Polymerization under the above conditions was very slow, reaching less than 30% conversion in 24 h. To increase the rate of polymerization, the reaction with Pd(dppe)Cl2 catalyst was performed at 45 °C. The conversion vs. time plot (Fig. 1A, top) showed that, even at this elevated temperature, 98% monomer conversion required 12 hours of polymerization time. In a remarkable contrast to the nickel-mediated reaction, polymerization with Pd(dppe)Cl2 catalyst consumed both isomers (2) and (3). (Conversion vs. time plots for isomers (2) and (3) are shown in ESI†).
The observed non-linear dependence of the molecular weight as a function of conversion resembles the behavior of a step-growth polymerization (Fig. 1A, bottom). GRIM polymerization of DBHT was also performed with Pt(dppe)Cl2 catalyst. This polymerization was very slow, reaching only 80% conversion in 48 h at 45 °C and producing only dimer and small oligomers (Mn = 800 g mol−1). GC-MS analysis of the reaction mixture indicated the presence of the 5,5′-dibromo-4,4′-dihexyl-2,2′-bithiophene dimer intermediate (6). This is consistent with the fact that, to our knowledge, platinum catalysts have not been successfully used for Kumada cross-coupling.38–45
The polymerization experiments were repeated with IBHT monomer. This monomer is known to form only one regioisomer (2), which was expected to lead to the formation of regioregular P3HT. The observed reaction rates are comparable to those for DBHT (Fig. 2, top). Molecular weight vs. conversion plots also show the same trends as observed for DBHT polymerization, with the Ni(dppe)Cl2 and Pd(dppe)Cl2 catalysts respectively displaying linear and non-linear dependence of molecular weight with conversion (Fig. 2, bottom). As for DBHT, the Pt(dppe)Cl2 catalyst gave very slow polymerization yielding only dimer and small oligomers. The polydispersity index vs. conversion plots for IBHT polymerizations are included in the ESI.†
![Conversion vs. time (top) and number average molecular weight vs. conversion (bottom) plots for GRIM polymerization of 2-bromo-5-iodo-3-hexylthiophene. (A) [IBHT]0 = 0.1 mol L−1; [Pd(dppe)Cl2]0 = 0.0015 mol L−1; temp = 45 °C. (B) [IBHT]0 = 0.1 mol L−1; [Ni(dppe)Cl2]0 = 0.0015 mol L−1; temp = 23 °C.](/image/article/2013/TA/c3ta13258g/c3ta13258g-f2.gif) | ||
| Fig. 2 Conversion vs. time (top) and number average molecular weight vs. conversion (bottom) plots for GRIM polymerization of 2-bromo-5-iodo-3-hexylthiophene. (A) [IBHT]0 = 0.1 mol L−1; [Pd(dppe)Cl2]0 = 0.0015 mol L−1; temp = 45 °C. (B) [IBHT]0 = 0.1 mol L−1; [Ni(dppe)Cl2]0 = 0.0015 mol L−1; temp = 23 °C. | ||
Computational investigations
The previously reported paper by McCullough suggested that the “associated pair” (9) is critical to the chain-growth mechanism of GRIM.14Based on this prior report, we hypothesized that the high polydispersity and regioirregularity observed for Pd(dppe) catalyst was due to side reactions of free Pd(0), which could be produced by dissociation of the associated pair. (Note that the accepted mechanism of Scheme 1 does not contain free zero-valent metal.) We further speculate that the very slow Pt-catalyzed polymerization is due to “poisoning” by thiophene.
First, we examined the potential energy surface for the M(0) [1,2-bis(dimethylphosphino) ethane (dmpe)]-thiophene associated pair to undergo either oxidative insertion into the chain end, or dissociation off of the growing chain. Fig. 3 shows the calculated potential energy surfaces for M = (Ni, Pd, Pt), with the zero of free energy set to the associated pair. The three metals give roughly comparable free energy differences and barriers for oxidative addition. However, M = Pd had a much smaller free energy of dissociation. Counterpoise corrected calculations give similar trends, with dissociation free energies of 32.9 kcal mol−1 for Ni, 8.3 kcal mol−1 for Pd, and 22.7 kcal mol−1 for Pt. These calculations are unlikely to reproduce the experimental binding free energies, due to limitations of the solvent model, an ideal gas model for translational and rotational free energies, and so on. However, the trends for different metals may reasonably be expected to be qualitatively correct. This suggested that the Pd associated pair is much less stable than Ni or Pt. The calculated total energy for dissociation of the associated pair between Ni(dmpe) and 2-bromo-3-methyl-5-(3-methylthiophenyl) thiophene is 48.8 kcal mol−1, vs. 47.2 kcal mol−1 for the system in Fig. 3. The calculated oxidative addition barrier for this dithiophene model is 12.3 kcal mol−1, vs. 11.8 kcal mol−1 for the system in Fig. 3. It is worth noting that dissociation of the associated pair (9) would yield a di-bromo-terminated thiophene chain. This is in line with the experimental studies, showing that “living” Grignard metathesis yields a high proportion of (H/Br)-terminated chains.14
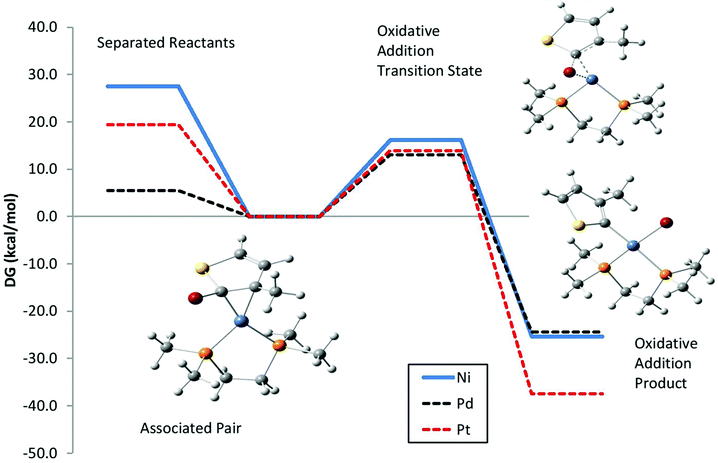 | ||
| Fig. 3 Calculated free energy surface for the associated pair between M(dmpe) and a 2-bromo-3-methylthiophene model for the growing chain to undergo dissociation (left) or oxidative addition (right). Calculated geometries for M = Ni are shown for each species. | ||
Next, we explored one possible mechanism for step-growth kinetics, oxidative addition of free Pd(0)(dmpe) to monomer. Fig. 4 compares the PBE0/LANL2DZ free energy surfaces for Pd(0)(dmpe) oxidative addition to the growing chain (modeled as in Fig. 3 as 2-bromo-3-methylthiophene) vs. Monomer 2 (modeled as 2-bromo-3-methyl-5-magnesiochloro thiophene). The potential energy surfaces are almost identical, indicating that free Pd(0) is as likely to react with monomer as with the growing chain. M06 calculations give qualitatively similar results, with oxidative addition barriers of 13.1 kcal mol−1 for the growing chain and 12.9 kcal mol−1 for the monomer. Transmetallation of this oxidative addition product could yield multiple thiophene chains per Pd and subsequently step-growth kinetics.
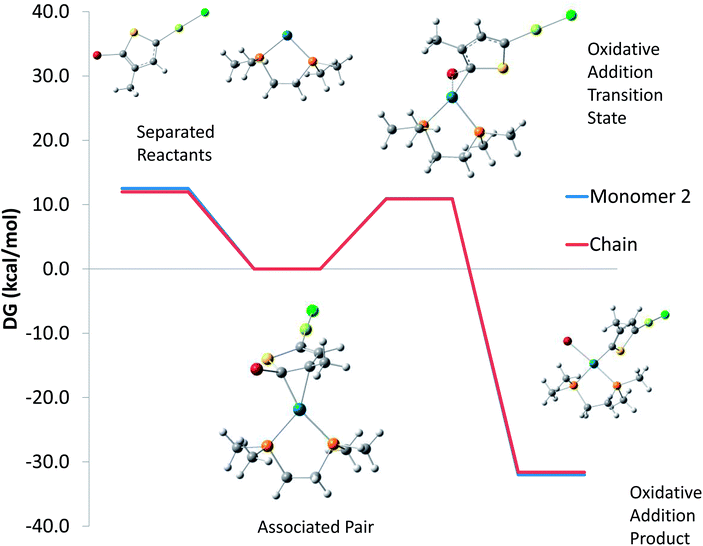 | ||
| Fig. 4 Calculated free energy surface for the associated pair between Pd(dmpe) and either 2-bromo-3-methyl-5-magnesiochloro thiophene monomer or a 2-bromo-3-methylthiophene model for the growing chain to undergo dissociation (left) or oxidative addition (right). Calculated geometries for the monomer case are shown for each species. | ||
Similar results were found for Ni(0)(dppe). Relative to the associated pair, the PBE0/LANL2DZ total energy barriers to Ni(0) oxidative addition are 11.8 kcal mol−1 for 2-bromo-3-methylthiophene, 12.3 kcal mol−1 for 2-bromo-3-methyl-5-(3-methylthiophenyl) thiophene, 11.6 kcal mol−1 for 2-bromo-3-methyl-5-magnesiochloro thiophene (equivalent to monomer 2 in Scheme 1), and only 10.0 kcal mol−1 for 2-bromo-4-methyl-5-magnesiochloro thiophene (equivalent to monomer 3 in Scheme 1). Next, we explored whether free Pd(0)(dmpe) could produce regioregular polymer.
Here it is critical to recall that Pd(dppeCl2) catalyzed polymerization of IBHT, which only used monomer [2], still yielded regioirregular polymer. We suggest one possible mechanism for this in Scheme 2. In this mechanism, the free Pd(0)(dppe) discussed above inserts into the middle of regioregular polymer chain [10] (Scheme 2), which could be considered the equivalent of the reverse of reaction 8 → 9 from Scheme 1. The product undergoes transmetalation with free MgBr2 (Scheme 2) which is the equivalent of the reverse of reaction 7 → 8 from Scheme 1. This transmetalation could produce two possible products depending on which of the two chains is displaced from Pd. Products [13] and [14] (Scheme 2) would produce regioregular polymer, though the kinetics would likely not be chain-growth.
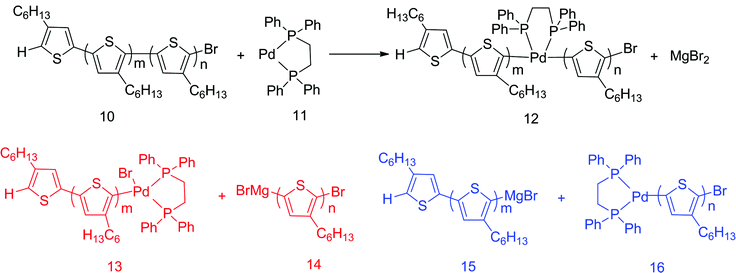 | ||
| Scheme 2 Proposed chain scissoring mechanism in Pd(dppe)Cl2 catalyzed polymerization of 3-hexylthiophene. | ||
In contrast, transmetalation of product [16] (Scheme 2) with monomer [2] (Scheme 1) would generate a second TT defect, and transmetalation of product [15] (Scheme 2) with growing chain [7] (Scheme 1) would introduce a HH defect.
To test whether the back-reaction of Scheme 2 is feasible, we calculated the free energy surface for transmetalation of [7] (which is the equivalent of the oxidative addition product of Fig. 2) with monomer [2] (Scheme 1), and subsequent reductive elimination to the associated pair [9] (Scheme 1). These calculations use Ni(dmpe), preliminary results suggest comparable values for Pd(dmpe). Fig. 5 shows the PBE0/LANL2DZ free energy surface. M06 calculations give qualitatively similar results. The reactant [7 + 2] and product [9] + MgClBr have comparable free energies. The largest free energy barrier was found for the forward direction, and encompasses the loss of MgClBr from the transmetalation product and subsequent reductive elimination.
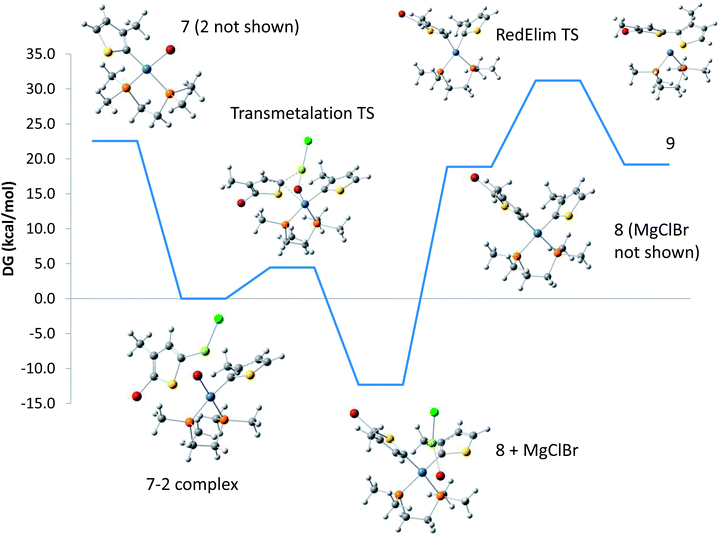 | ||
| Fig. 5 Calculated free energy surface for reaction 7 → 8 → 9 of Scheme 1, catalyzed by Ni(dmpe). | ||
This suggests that the reactions 7 → 8 → 9 and 9 → 8 → 7 are comparably favorable.
To test whether the proposed chain scissoring for the Pd catalyzed polymerization was possible we performed an experiment using a regioregular P3HT polymer of a known molecular weight (Mn = 38000 g mol−1, PDI = 1.36). The P3HT polymer used in this study was synthesized by GRIM method using Ni(dppp)Cl2 as a catalyst. The 1HNMR spectrum of the P3HT showed 66% of H/Br and 40% of H/H end groups before the chain scissoring experiment. The polymer was dissolved in THF and reacted with Pd(0) for 12 hours. The Pd(0) complex was generated from the reduction of Pd(dppe)Cl2 with sodium metal. The colour change from brown to green yellow was used as an indicative for the formation of Pd(0) complex. MgBr2 was added to the reaction mixture containing P3HT and Pd(0) complex to facilitate the transmetallation as proposed in the mechanism shown in Scheme 2. The resulting polymer exhibited a lower molecular weight and a broader PDI as compared to the starting P3HT polymer (ESI†). The molecular weight of the resulting polymer was also reduced to 25000 g mol−1 from 38000 g mol−1 while the PDI broadened from 1.36 to 1.60. While it is difficult to predict at which position the scissoring is taking place, the decrease in the molecular weight of P3HT and the broadening of PDI can be reasonably considered as an indicative of the chain scissoring. Moreover, the 1H NMR analysis indicated that the final polymer obtained after the chain scissoring experiment had only H/H end groups (see the 1H NMR spectra in ESI†) which show the absence of a triplet at 2. 6 ppm which is due to the hexyl methylene protons adjacent to the bromine end group.
Conclusions
In conclusion, we studied the mechanistic differences of group 10 transition metal-mediated GRIM polymerization for the synthesis of P3HT. The presented experimental evidence and the proposed mechanisms bring additional support particularly for the presence of the associated pair of 5,5′-dibromo-4,4′-dihexyl-2,2′-bithiophene dimer and Ni(0) in the mechanism of Grignard metathesis (GRIM) polymerization.Acknowledgements
Financial support for this project from NSF (Career DMR-0956116) and Welch Foundation (AT-1740) is gratefully acknowledged. We gratefully acknowledge the NSF-MRI grant (CHE-1126177) used to purchase the Bruker Avance III 500 NMR instrument.Notes and references
- T. A. Skotheim and J. Reynolds, Handbook of Conducting Polymers, CRC Press, LLC, Boca Raton, 2006 Search PubMed.
- Handbook of Thiophene-Based Materials: Applications in Organic Electronics and Photonics, Volume One: Synthesis and Theory, ed. I. F. Perepichka and D. F. Perepichka, 2009 Search PubMed.
- R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- R. D. McCullough and R. D. Lowe, J. Chem. Soc., Chem. Commun., 1992, 70–72 RSC.
- R. D. McCullough, R. D. Lowe, M. Jayaraman, P. C. Ewbank, D. L. Anderson and S. Tristram-Nagle, Synth. Met., 1993, 55, 1198–1203 CrossRef CAS.
- R. D. McCullough, R. D. Lowe, M. Jayaraman and D. L. Anderson, J. Org. Chem., 1993, 904–912 CrossRef CAS.
- E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526–3528 CrossRef CAS.
- S. A. Chen and C. S. Liao, Macromolecules, 1993, 26, 2810–2816 CrossRef CAS.
- T. A. Chen and R. D. Rieke, J. Am. Chem. Soc., 1992, 114, 10087–10088 CrossRef CAS.
- T.-A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
- R. S. Loewe, P. C. Ewbank, J. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS.
- R. S. Loewe, S. M. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250–253 CrossRef CAS.
- A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169–1171 CrossRef CAS.
- M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649–8656 CrossRef CAS.
- M. Jeffries-El, G. Sauve and R. D. McCullough, Adv. Mater., 2004, 1017–1019 CrossRef CAS.
- M. Jeffries-El, G. Sauve and R. D. McCullough, Macromolecules, 2005, 38, 10346–10352 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, Macromol. Rapid Commun., 2004, 25, 1663–1666 CrossRef CAS.
- T. Beryozkina, V. Senkovskyy, E. Kaul and A. Kiriy, Macromolecules, 2008, 41, 7817–7823 CrossRef CAS.
- N. Doubina, M. Stoddard, H. A. Bronstein, A. K. Y. Jen and C. K. Luscombe, Macromol. Chem. Phys., 2009, 210, 1966–1972 CrossRef CAS.
- N. Doubina, A. Ho, A. K. Y. Jen and C. K. Luscombe, Macromolecules, 2009, 42, 7670–7677 CrossRef CAS.
- N. Doubina, S. A. Paniagua, A. V. Soldatova, A. K. Y. Jen, S. R. Marder and C. K. Luscombe, Macromolecules, 2011, 44, 512–520 CrossRef CAS.
- H. A. Bronstein and C. K. Luscombe, J. Am. Chem. Soc., 2009, 131, 12894–12895 CrossRef CAS PubMed.
- N. Marshall, S. K. Sontag and J. Locklin, Chem. Commun., 2011, 47, 5681–5689 RSC.
- V. Senkovskyy, N. Khanduyeva, H. Komber, U. Oertel, M. Stamm, D. Kuckling and A. Kiriy, J. Am. Chem. Soc., 2007, 129, 6626–6632 CrossRef CAS PubMed.
- V. Senkovskyy, M. Sommer, R. Tkachov, H. Komber, W. T. S. Huck and A. Kiriy, Macromolecules, 2010, 43, 10157–10161 CrossRef CAS.
- R. Tkachov, V. Senkovskyy, H. Komber and A. Kiriy, Macromolecules, 2011, 44, 2006–2015 CrossRef CAS.
- R. Tkachov, V. Senkovskyy, H. Komber, J.-U. Sommer and A. Kiriy, J. Am. Chem. Soc., 2010, 132, 7803–7810 CrossRef CAS PubMed.
- E. L. Lanni and A. J. McNeil, J. Am. Chem. Soc., 2009, 131, 16573–16579 CrossRef CAS PubMed.
- E. L. Lanni and A. J. McNeil, Macromolecules, 2010, 43, 8039–8044 CrossRef CAS.
- J. R. Locke and A. J. McNeil, Macromolecules, 2010, 43, 8709–8710 CrossRef CAS.
- R. Saha, M. A. Qaium, D. Debnath, M. Younus, N. Chawdhury, N. Sultana, G. Kociok-Koehn, L.-l. Ooi, P. R. Raithby and M. Kijima, Dalton Trans., 2005, 2760–2765 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, J. L. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. A. Montgomery,J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, in Gaussian 09, Revision B.01, Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- Y. Zhao, B. J. Lynch and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 2715–2719 CrossRef CAS.
- K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374–4376 CrossRef CAS.
- R. J. P. Corriu and J. P. Masse, J. Chem. Soc., Chem. Commun., 1972, 144 RSC.
- M. Kumada, Pure Appl. Chem., 1980, 52, 669–679 CrossRef CAS.
- E. Negishi, Acc. Chem. Res., 1982, 15, 340–348 CrossRef CAS.
- E. Negishi, T. Takahashi, S. Baba, H. D. E. Van and N. Okukado, J. Am. Chem. Soc., 1987, 109, 2393–2401 CrossRef CAS.
- F. Ozawa, T. Hidaka, T. Yamamoto and A. Yamamoto, J. Organomet. Chem., 1987, 330, 253–263 CrossRef CAS.
- A. Yamamoto, T. Yamamoto and F. Ozawa, Pure Appl. Chem., 1985, 57, 1799–1808 CrossRef CAS.
- T. Yamamoto, S. Wakabayashi and K. Osakada, J. Organomet. Chem., 1992, 428, 223–237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, kinetic plots, and SEC traces. See DOI: 10.1039/c3ta13258g |
| This journal is © The Royal Society of Chemistry 2013 |