 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew Si–O–C composite film anode materials for LIB by electrodeposition
Tetsuya
Osaka
*,
Hiroki
Nara
,
Toshiyuki
Momma
and
Tokihiko
Yokoshima
Faculty of Science and Engineering, Waseda University, 3-4-1, Okubo, Shinjuku-ku, Tokyo, 169-8555, Japan. E-mail: osakatets@waseda.jp; Web: http://www.ec.appchem.waseda.ac.jp/INDEXE.HTM Fax: +81-3-3205-2074; Tel: +81-3-5286-3202
First published on 17th October 2013
Abstract
Silicon is one of the most promising materials for lithium secondary battery anodes. However, silicon anodes have a critical drawback to their practical application, which is capacity degradation due to pulverization of the active material by the large volume change of silicon during charge–discharge cycles. This paper reviews recent studies on silicon-based anodes that have attempted to overcome this poor cycle durability through structural control such as through thin films, porous structures, core–shell structures, and by alloying with other metals, and by application of proper binders. Among them, binder-free Si–O–C composite films prepared by electrodeposition exhibit outstanding cycle durability. The origin of this excellent durability is discussed in depth from the standpoint of chemical and morphological changes. Consequently, the combination of active materials such as Si and Li2Si2O5 and inactive materials such as Li2O, Li2CO3, and organic compounds is suggested to result in outstanding properties as a lithium secondary battery anode.
 Tetsuya Osaka | Tetsuya Osaka is Professor of the Department of Applied Chemistry, Waseda University, Tokyo, Japan. He is President of the Electrochemical Society (ECS) after serving as Vice President of the International Society of Electrochemistry (ISE). His recent studies focus on a new “electrochemical nanotechnology”. His technical contributions have been recognized with many awards including Medal with Purple Ribbon bestowed from the Decoration Bureau of the Cabinet Office, Japan, Pergamon Electrochimica Acta Gold Medal of ISE in 1998, etc. He has been elected as a Fellow of IEEE (2002), ECS (2002), IUPAC (2004) and ISE (2006). |
 Hiroki Nara | Hiroki Nara received his PhD degree in electrochemistry from Waseda University, Tokyo, Japan in 2008. From 2007 to 2009, he worked as a research associate in Waseda University. Currently, he is a junior researcher (assistant professor) in Waseda University. His research interests focused on electrochemical energy devices, especially on the design of the reactive site on anode materials for lithium secondary batteries and impedance analysis for the structured reactive site and diagnosis of electrochemical devices such as the lithium secondary battery and fuel cell. |
 Toshiyuki Momma | Toshiyuki Momma is an associate professor of the Department of Applied Chemistry and Department of Nanoscience and Nanoengineering, Faculty of Science and Engineering, Waseda University. He worked at the University of Minnesota, USA. His research area is in the field of electrochemical energy devices. He is working on materials design of electrodes for rechargeable batteries and fuel cells, including anode and electrolyte for future batteries. He is also interested in the development and improvement of electrochemical impedance spectroscopy. He is currently working on the in situ electrochemical impedance analysis of rechargeable batteries and fuel cells as non-destructive diagnosis of the devices. |
 Tokihiko Yokoshima | Tokihiko Yokoshima is a researcher (associate professor) in the Faculty of Science and Engineering, Waseda University. He worked as a Post-doctoral Research Scientist in the National Institute of Advanced Industrial Science and Technology (AIST). His research area is in the field of materials chemistry for thin film and electrochemical energy devices. He is working on thin film materials design of high functional materials for rechargeable batteries and electronic devices. He is also interested in the development and improvement of the electrochemical impedance spectroscopy for the rechargeable batteries as non-destructive diagnosis of the devices. |
Introduction
The history of secondary batteries, which reversibly store electrical energy as chemical energy, began with the invention of the lead-acid battery in 1859. Lead-acid batteries are still widely used as a power source for vehicle start-up and as backups for commercial power in industry because of their relatively high voltage, ca. 2 V, and moderate price despite their heaviness and toxicity. Fig. 1 shows the history of the secondary battery and future targets. The invention of the nickel–cadmium (NiCd) battery influenced the development of portable electronic devices because of their high power and tolerance against over-discharging and prolonged storage. The development of the nickel-metal-hydride (NiMH) battery was the trigger to making portable electric devices widespread by replacing NiCd batteries because of its high energy density. Commercial production of hybrid electric vehicles (HEV) started in 1997 using the NiMH battery as a power source. Finally, the commercialization of the lithium-ion battery (LIB) has enabled the realization of lap-top computers and high performance smart phones.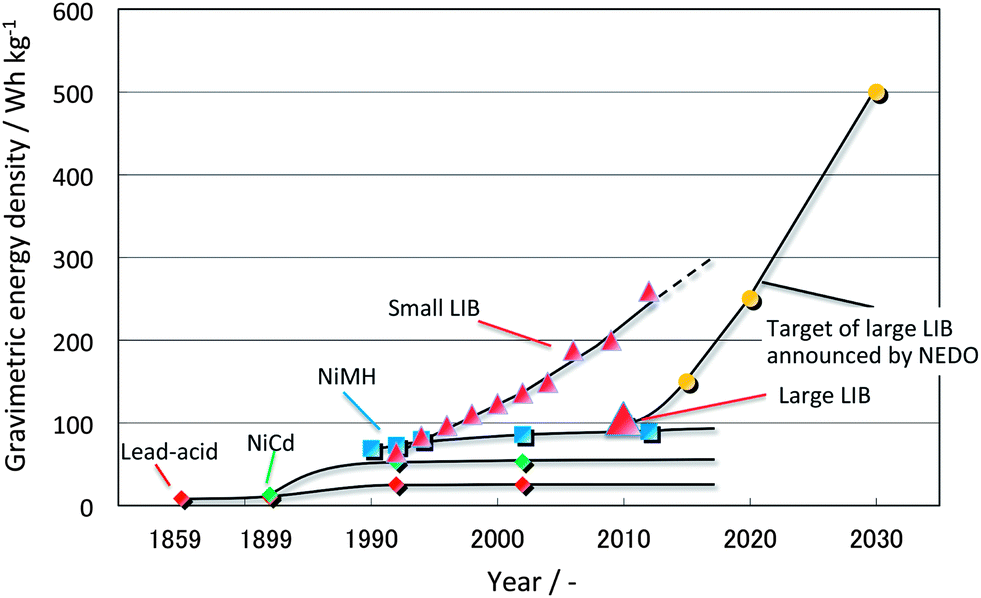 | ||
| Fig. 1 History of secondary batteries and targets for the near future. | ||
Furthermore, progress in LIBs has encouraged the development of electric devices with higher power consumption. The circumstances surrounding energy storage have been changing in recent years. Namely, new applications of LIBs in electric vehicles and stationary grid energy storage have arisen.1 For example, battery electric vehicles (BEV), which use LIBs as their power source, became commercially available in 2010. The development of LIBs is therefore subject to strong demands including increased safety, longer life, lower cost, higher energy density, and higher power. High energy density is especially required for electric vehicles to enable long distance driving. Therefore, enhancement of anode and cathode material capacity is essential for the widespread adoption of electric vehicles. Capacity enhancement must be achieved for both anode and cathode materials; because space is usually limited in a battery, the enhancement of the capacity of one electrode is not enough to enhance the capacity of the overall cell. Fig. 2 shows an estimation of energy density with cathode capacity in a 18650 cylinder-type cell. If the capacity of the anode is fixed to 372 mA h g−1, the over capacity increase is limited even if the capacity of the cathode is increased drastically. A battery that delivers an energy density of 300 W h kg−1 would be difficult to achieve with a graphite anode, and would also be difficult as long as a conventional cathode is used. An increase in the capacities of both anode and cathode would enable the energy density to be increased to over 300 W h kg−1. For example, a battery composed of an anode material with a capacity of 1000 mA h g−1 and a cathode material having with capacity of 800 mA h g−1 would deliver an energy density of ca. 300 W h kg−1.
 | ||
| Fig. 2 Estimation of energy density increase with capacity for various cathodes (LiNixCoyAlzO2, Li2S, O2). Gravimetric energy density values were calculated on the basis of a 18650 cylinder-type cell with graphite anode. Values in brackets in the graph legend are the capacities of the active materials used in the calculations. | ||
Under these circumstances, silicon is one of the most promising anode materials because its theoretical capacity (4200 mA h g−1) is much higher than that of graphite (372 mA h g−1). In addition, because the Clarke number of silicon is 25.8, the cost of the active anode material can be reduced. These advantages are beneficial for both industry and consumers. However, silicon has a critical problem: silicon anodes cannot maintain their structure during charge–discharge cycles because of the large change in the volume of the active material that occurs during the cycles, which results in capacity degradation.2 Recently, Lee et al. provided a deep insight into lithiation of crystalline silicon.3 They used Si nanopillars with circular cross-section in shape with three different axial orientations (〈100〉, 〈110〉, and 〈111〉) as a model system to show cross-sectional dimensional changes during lithiation. Upon lithiation, the cross-sections of nanopillars with 〈100〉, 〈110〉, and 〈111〉 axial orientations expand into cross, ellipse, and hexagonal shapes, respectively. The authors explained this phenomenon by identifying a high-speed lithium ion diffusion channel along the 〈110〉 direction, which causes preferential volume expansion along this direction. Upon further lithiation the amorphous phase begins to form at the surface of the 〈110〉 ion channels where the Li concentration is highest. Interestingly, the 〈111〉 and 〈100〉 nanopillars shrink in height after partial lithiation, while 〈110〉 nanopillars increase in height. The length contraction is considered to be due to the collapse of the {111} planes early in the lithiation process.
Approaches on silicon based anode
To solve this volume change problem, several approaches based on the control of silicon-based anode structure or composition have been carried out. Selected examples are introduced below.Thin silicon films
Graetz et al. reported a thin Si film (100 nm) lithium secondary battery anode prepared using physical vapor deposition (PVD) by vacuum evaporation directly onto the planar current collector.4 The evaporated Si film was amorphous in structure, which avoided the formation of stoichiometric compounds that would have otherwise resulted in considerable suppression of local stress gradients because of electrochemical alloying. The thin Si film anode exhibited an initial discharge (delithiation) capacity of 2500 mA h (g of Si)−1 at 0.25 C rate with a columbic efficiency of 71% on the first cycle, and a stable discharge capacity of 1800 mA h g−1 over 50 cycles. After 20 cycles, the thin film exhibited a mean capacity loss of 8 mA h g−1 per cycle. Ohara et al. reported a thinner Si film anode (50 nm) prepared by PVD using vacuum evaporation from a negative doped Si source.5 The thin Si anode exhibited enhanced specific capacity compared to that achieved with an intrinsic Si source, and a stable discharge capacity of 3800 mA h (g of Si)−1 at 1 C rate over 200 charge–discharge cycles. The thin Si anode prepared by PVD also had an amorphous like structure even after charge–discharge cycles. Thus, an amorphous like structure including a polycrystalline structure can improve the cycle durability of silicon-based anode because amorphous silicon itself can act as a buffer layer during charge–discharge cycles.Such thin film anodes have the advantage of direct deposition on the current collector without any binder or electrically conductive additives. However, thin film anodes have an essential drawback in the fact that it is difficult to improve their energy density by increasing their thickness, because thicker films suffer from more stress because of volume change during charge–discharge cycling. Although studies have been carried out to increase the anode film thickness by roughening the surface of the current collector, the maximum thickness has so far been limited to 3.6 μm.6
Porous silicon powders
Kim et al. reported a three-dimensional and porous Si particle anode7 prepared by thermal annealing of SiO2 and butyl capped Si particles and etching of SiO2. The resulting porous Si particles had bulk sizes of greater than 20 μm with interconnected pores of 200 nm in size and ca. 40 nm wall thickness. The porous structure could accommodate stress during charge–discharge cycling without experiencing pulverization for 100 charge–discharge cycles, and maintained a charge capacity of greater than 2800 mA h (g of Si)−1 at 0.2 C rate. Ge et al. reported porous doped Si nanowire anodes with a pore diameter and wall thickness of ca. 8 nm prepared by direct etching of boron-doped silicon wafers.8 The resulting porous Si nanowire anode exhibited a first discharge capacity of 3038 mA h g−1, and after 250 cycles its discharge capacity remained above 1960 mA h g−1 at 0.5 C rate, indicating good structural stability. Moreover, the porous Si nanowire anode delivered a discharge capacity of above 1000 mA h g−1 even after 2000 cycles.Porous materials are one of the most promising approaches for realization of Si based anodes because a porous structure can shorten the diffusion length in the solid phase and accommodate stress during charge–discharge cycling without pulverization. These phenomena have also been reported for Sn-based anode systems.9 In addition, particle and nanowire powders are favorable for industry because they are suitable for the conventional processes used to prepare graphite anodes, even though the process requires a binder and electron conductive additives. However, such high surface area powder materials often suffer from large irreversible capacity in the initial several charge–discharge cycles because solid electrolyte interphase (SEI) formation, which consumes lithium, occurs in proportion to the surface area.10 This is a serious problem when assembling full cells, in which limited space is available for the anode and cathode materials.
Silicon powders with unique structures
Zhang et al. reported core–shell Si/SiO nanocomposites.11 The core–shell Si/SiO, which was by a sol–gel method and a following heat-treatment process, was ca. 50 nm of the core in average size and in face-centered cubic structure. The core–shell Si/SiO nanocomposite exhibited a discharge capacity of 538 mA h (g of Si)−1 after 20 cycles, which is 65% of the discharge capacity at the 1st cycle. Meanwhile, the anode of the Si nanoparticles showed a rapid capacity fading and retains only 10.3% of the original capacity after 20 cycles. The capacity loss of crystalline silicon nanoparticles could be ascribed to the aggregation of nano-sized active particles caused by electrochemical alloying reaction with lithium and the destruction of crystalline structure accompanied by severe volume expansion.12 On the other, the electroactive Si particles coated by SiO could not combine together because of the existence of the SiO shell. Simultaneously, SiO can also absorb and retain a large quantity of lithium ions. Because the Si–O bond is generally twice as strong as the Si–Si bond, the SiO anode exhibits a good cycling performance. During the lithiation process, SiO can change to form lithium silicates, which expand only half as much as Li–Si alloys, and the Li-silicates serve as a buffer to alleviate volume expansion of the Si nanoparticles, mitigating the destruction of the Si crystalline structure.13 Wu et al. demonstrated double-walled silicon nanotubes (DWSiNTs) consisting of Si nanotubes surrounded by ion-permeable silicon oxide shells.14 The DWSiNT was prepared by using a templating method combined with the electrospinning method. The double-walled structure enables the prevention of the outer surface of the silicon nanotube from expansion by the oxide shell, and the expanding inner surface is not exposed to the electrolyte, resulting in a stable SEI. As a result, the DWSiNT anode exhibited discharge capacities of ca. 2971 mA h (g of Si)−1 and 1780 mA h (g of DWSiNT)−1 at C/5 rate, ca. 940 mA h (g of Si)−1 and 600 mA h (g of DWSiNT)−1 at 12 C rate with 80% capacity retention at room temperature even after 6000 charge–discharge cycles. Furthermore, the DWSiNT anode can operate even at 20 C rate, delivering the discharge capacity of ca. 850 mA h (g of Si)−1. Hwang et al. reported core–shell fibers consisting of Si nanoparticle cores wrapped in carbon shells (SiNPs@C).15 The core–shell fibers, which were prepared by the electrospinning technique, had a one-dimensional fibrous morphology of ca. 1 μm diameter that encapsulated Si particles of ca. 50 nm diameter with compact packing. The core–shell fiber anode exhibited a discharge capacity of 1384 mA h g of (SiNPs@C)−1 at C/10 rate, with almost no capacity loss after 300 charge–discharge cycles. Zhou et al. reported a kind of core–shell structured Si anode in which Si nanoparticles (<200 nm) on graphene sheet were coated by an amorphous carbon shell of 2–3 nm in thickness.16 This core–shell particle anode exhibited an initial discharge capacity of 1328 mA h (g of Si, graphene, and amorphous carbon)−1 at C/3 rate and a discharge capacity of 902 mA h g−1 after 100 charge–discharge cycles. Such modifications by carbonous materials are effective in enhancing electric conductivity,17 reducing electrolyte decomposition,18–21 and buffering volume changes during charge–discharge cycling.22 Okubo et al. reported a Si thin flake anode (4–5 μm in size and 100 nm in thickness) prepared by PVD using vacuum evaporation.23 The Si thin flake anode exhibited an initial discharge capacity of 2651 mA h (g of silicon)−1 at C/6 rate and ca. 2250 mA h g−1 after 50 charge–discharge cycles when vinylene carbonate (VC) was used as an electrolyte additive. A thin flake shape has the advantages of shortening the lithium diffusion length without increasing the surface area, and increasing the tap density compared with nanoparticles. Saito et al. reported the dependence of the thickness for the Si thin flake anodes.24 According to their paper, a Si thin flake of 200 nm in thickness can work as well as a flake of 100 nm in thickness.These silicon powders with unique structures are also favorable for industry, as mentioned above. In addition, carbon-coating treatment can reduce the irreversible capacity attributed to electrolyte decomposition.19–21 Consequently, these materials would be some of the most promising ones for practical realization.
Silicon alloyed with other metals
Anani et al. reported intermetallic compounds such as silicides (M–Si) as anode materials for lithium batteries.25 Among these materials, Choi et al. reported a Fe–Si anode26 and Zhou et al. reported a Ni–Si anode.27 The Fe–Si anode was prepared by radio frequency (RF) magnetron sputtering of a Fe–Si alloy target and was evaluated in an ionic liquid (IL) electrolyte system. The anode exhibited an initial discharge capacity of 1073 mA h (g of FeSi2.7)−1 and 756 mA h g−1 at 0.3 C rate in conventional liquid electrolyte and IL electrolyte, respectively. The discharge capacity declined to 783 mA h g−1 and 694 mA h g−1 after 100 charge–discharge cycles in the conventional and IL electrolytes, respectively. A.C. impedance analysis revealed that the difference was due to the stability of the interface between the electrolyte and the electrode. The Ni–Si anode was prepared by pulse laser deposition from a target made from Ni and Si powders. The anode exhibited an initial discharge capacity of ca. 1120 mA h (g of NiSi1.2)−1 at ca. 0.013 C rate, estimated from the theoretical capacity of Si. The discharge capacity after 100 charge–discharge cycles was ca. 1000 mA h g−1.The alloying of silicon with other metals is effective in increasing the electric conductivity of the material and suppressing the pulverization of the active material. This effect has also been confirmed in Sn-based anode systems.28,29
Approaches on polymer binder
Much research has been devoted to the design of active materials to moderate the stress due to the volume change of the active materials. Meanwhile, the other approaches to inhibit the mechanical fracture of silicon-based anode, namely an improvement of electrically inactive materials such as binders, have been also reported. Carboxymethyl cellulose (CMC) and polyacrylic acid (PAA) binders are well known to perform better than polyvinylidene fluoride (PVdF) for silicon-based anodes.30,31 Unlike PVdF is attached to Si particles via weak van der Waals forces only, CMC and PAA are bonded to the surface of silicon particles via a covalent ester bonding between the free carboxylic acid groups of CMC and PAA and the OH groups on silicon particles.31,32 The stronger interactions enable the prevention of destruction of the electric network under the large volume change during charge–discharge cycles. Furthermore, these binders, which bond with silicon particles by hydrogen bonding between the carboxyl groups of the binder and the silicon oxides on the silicon particles, were proposed to exhibit a self-healing effect and reform if locally broken.33 Kovalenko et al. reported alginate, which is a high-modulus natural polysaccharide extracted from blown algae, as a binder to stabilize an operation of the silicon powder anode.34 The silicon powder anode with the alginate binder exhibited a reversible capacity of ca. 1700 mA h (g of Si)−1 after 100 cycles at 1 C rate at room temperature. Alginate macromolecules, which are more polar than the CMC polymer, are attributed to the better cycle durability. Koo et al. reported a cross-linked polymeric binder, namely a three-dimensional interconnected network of PAA and Na–CMC. The nanosized silicon powder formed by the binder exhibited a high reversible capacity of over 2000 mA h (g of Si)−1 after 100 cycles at 0.1 C rate at 30 °C and maintains a superior capacity of 1500 mA h g−1 at 10 C rate at 60 °C.32Electrodeposited Si–O–C composite film
As mentioned above, there have been many studies on the Si-based anode and binder. Among them, we have successfully developed a binder-free Si–O–C composite film for lithium secondary battery anodes by means of electrodeposition.35–38 The Si–O–C composite film was electrodeposited from a bath containing silicon tetrachloride as a silicon source, tetrabutylammonium perchlorate as a supporting electrolyte, and propylene carbonate solvent directly onto a copper substrate. The electrodeposition of silicon is considered to occur with accompanying decomposition of the organic solvent, leading to the formation of a Si–O–C composite film.Charge–discharge property of the Li–Si–O–C composite anode
The electrochemical performance of a Li–Si–O–C composite anode, which was firstly electrochemically lithiated, was examined through a constant current (CC) – constant voltage (CV) charge (lithiation) – CC discharge (delithiation) test. Fig. 3 shows the charge and discharge capacity values of the Li–Si–O–C composite anode under a loading current of 250 μA cm−2, which corresponds to a value of 1.0 in C-rate. At the initial stage, the Li–Si–O–C composite anode exhibited a high discharge capacity of above 1200 mA h (g of Si)−1. Then, the discharge capacity decreased to 900 mA h (g of Si)−1 until the 20th cycle. After the 20th cycle, the discharge capacity increased beyond 1100 mA h (g of Si)−1 and then gradually decreased, maintaining a value higher than 1000 mA h (g of Si)−1 for more than 2000 cycles. The columbic efficiency during charge–discharge cycling was found to be higher than 98%, except for a few initial cycles. The low efficiency in the first cycle was attributed to the consumption of reducing charges by Si oxide39,40 and SEI formation21,41 as discussed with EELS and XPS data below. | ||
| Fig. 3 Plots of charge–discharge capacity of the Si–O–C composite anode against the number of cycles (upper) and potential profiles during charge–discharge cycling at various cycle numbers (bottom). The inset in the bottom shows potential profiles during the first lithiation and delithiation. The capacity values were normalized with respect to the weight of the initial deposit or the calculated amount of Si in the deposit. The applied current density was 250 μA cm−2 (1.0 C-rate). Reproduced from data published in ref. 36. | ||
The potential profiles measured during charge–discharge cycling (bottom, Fig. 3) give us a deeper insight. A negative potential overshoot was observed during the initial lithiation process on the Si–O–C composite film because of the nucleation of a new phase, namely the Li2O phase.40 The capacity for the first charge (lithiation) was 4731 mA h (g of Si)−1, while the capacity for the first discharge (delithiation) was 1403 mA h (g of Si)−1. This large irreversible capacity is attributed mainly to the conversion of SiOx to SiLiy and Li2O,39,40 and partly to SEI formation,21,41 indicated by the potential plateau above 0.4 V vs. Li/Li+ in Fig. 3 (bottom) and confirmed by cyclic voltammograms (see ESI of ref. 36). The potential plateau for delithiation below 0.6 V vs. Li/Li+ during the first discharge was confirmed, and corresponded to the anodic peaks repeated at 0.32 and 0.46 V vs. Li/Li+ in the cyclic voltammogram (see ESI of ref. 36), the results of which accord with the delithiation of silicon.42 After the first cycle, the Li–Si–O–C composite anode was charged at a potential below 0.3 V vs. Li/Li+, and discharged at a potential plateau below 0.6 V vs. Li/Li+. For 300 cycles, the electrode potential dropped to about 0.2 V vs. Li/Li+ at the initial stage of charging in each cycle and then decreased while the current was being passed. The initial potential shifted towards 0.4 V vs. Li/Li+ with increasing cycle number. The potential profile observed during charging did not change significantly after the 300th cycle. The potential profiles during discharge were identical at all stages except for the amount of discharge capacity, which was found to be above 1000 mA h (g of Si)−1 or above 630 mA h (g of deposit)−1 even after 2000 cycles. Moreover, the Li–Si–O–C composite anode delivered a discharge capacity of 831 mA h (g of Si)−1 or 512 mA h (g of deposit)−1 even after 7000 cycles.
Analysis of the Si–O–C composite film and Li–Si–O–C composite anode with charge–discharge cycle
To reveal the reason why the Li–Si–O–C composite film exhibited outstanding durability for more than 7000 cycles, the Si–O–C composite film and Li–Si–O–C composite anode were analysed from the standpoints of changes in morphology and chemical composition.Change of the Si–O–C composite film and Li–Si–O–C composite anode morphology with charge–discharge cycle
FESEM images of the Si–O–C composite film prepared by the electrodeposition of silicon are shown in Fig. 4. The images of the as electrodeposited Si–O–C composite film show a macroscopically homogeneous deposit. The enlarged FESEM image indicates that the deposit consisted of sub-micrometer clusters with voids that covered the Cu substrate. Plane view FESEM images of the Li–Si–O–C composite anode after the first lithiation are shown in Fig. 4. After the first lithiation, the voids that were observed in the as electrodeposited Si–O–C composite film before the first lithiation decreased in size. This is attributed to the expansion of silicon and the formation of Li2O by the lithiation of SiOx leading to a reduction of the space in the voids and also to SEI formation,21,41 leading to the covering of the film. Although the SEI was formed on the Li–Si–O–C composite anode, the particles which were observed on the Si–O–C composite film were only slightly visible on the Li–Si–O–C composite anode. FESEM images of the Li–Si–O–C composite anodes after the 10th and 300th cycles are also shown in Fig. 4. The particles observed on the Li–Si–O–C composite anode surface after the first lithiation were absent after the 10th cycle. This phenomenon must be due to the progress of SEI formation, which is evidenced by the lower columbic efficiency observed during the initial cycles (Fig. 3). Cracking of the Li–Si–O–C composite anode caused by a large volume change of silicon, which is a problem associated with the Si anode system,18,20,43 was not observed. This was attributed to a uniform reaction of the well-dispersed SiOx at the nanometer scale as confirmed by STEM-EDX (Fig. 6), and to the buffering effect of surrounding decomposition products of the organic solvent. Although cracking was observed to a slight extent after the 100th cycle, the surface morphology was almost unchanged even after the 300th cycle. The results of the FESEM observation imply that the suppression of crack formation was the reason for the excellent cycle durability exhibited by the Li–Si–O–C composite anode. Fig. 5 shows the variation in thickness of the as deposited Si–O–C composite film and of the Li–Si–O–C composite anodes after the 1st charge, 1st discharge, 2nd charge, 2nd discharge, 5th charge, 5th discharge, 10th charge, 10th discharge, 100th charge, and 100th discharge, as measured from SEM images of fracture cross-sections of a number of samples after electrodeposition. The thickness of the as electrodeposited Si–O–C composite film varied from ca. 1 to 5 μm because of the roughness of the deposit, as seen in Fig. 4. After the 1st charge, the overall film thickness of the Li–Si–O–C composite anode showed a tendency to increase, caused by the alloying of silicon with lithium. Increased film thickness would also be due to SEI formation.41,44 After the 1st discharge, the distribution of the film thickness became more uniform. Especially, the thicker parts of the Li–Si–O–C composite anode showed a tendency to become thinner after the 1st charge, which should be due to the dealloying of silicon from lithium. This non-uniform variation may be due to SEI formation during the 1st charge and the initial structure of the Si–O–C composite film. The projecting parts of the film delithiated preferentially during the 1st discharge. The film thickness distribution tended to become uniform up to the 100th cycle; in fact, the projecting part of the deposit reorganized into the void space during charge–discharge cycles. Furthermore, the variation of the film thickness between charged and discharged states showed a tendency to become smaller. Thus, after several charge–discharge cycles the Li–Si–O–C composite anode, which buffers against volume change during charge–discharge cycles, is likely to be formed with not only SEIs such as Li2O, Li2CO3, and organic compounds, but also inorganic phases such as lithium silicates as discussed below. The smaller variation of the film thickness between charged and discharged states suggests that the Li–Si–O–C composite anode varied correspondingly between dense and non-dense states, respectively. | ||
| Fig. 4 Plane view FESEM images of as the electrodeposited Si–O–C composite film and Li–Si–O–C composite anodes after the 1st charge, the 10th cycle, and after the 300th cycle. Reproduced from data published in ref. 36. | ||
 | ||
| Fig. 5 Film thickness of the as electrodeposited Si–O–C composite film and of the Li–Si–O–C composite anode after the 1st charge, 1st discharge, 2nd charge, 2nd discharge, 5th charge, 5th discharge, 10th charge, 10th discharge, 100th charge, and 100th discharge. The film thickness was measured at fractured cross-sections for a number of samples. Reproduced from the data published in ref. 37. | ||
Change in composition of the Si–O–C composite film and Li–Si–O–C composite anode with charge–discharge cycle
Fig. 6 shows STEM-EDX mapping of Si, C, O, Cl, and Cu elements in the Si–O–C composite film and the Li–Si–O–C composite anodes after the 1st charge and the 100th discharge. The EDX mapping shown in Fig. 6(a) reveals uniform dispersion of Si, O, and C at the nano-scale level and incorporation of Cl derived from SiCl4, which was the source of silicon for the electrodeposition. Fig. 6(b) also indicates homogeneous dispersion of Si, O, and C at the nano-scale, and Fig. 6(c) indicates that these elements were still homogenously dispersed after the 100th discharge. | ||
| Fig. 6 Cross-sectional Z-contrast images and micro EDX mapping of Si, C, O, Cl, and Cu elements of the Si–O–C composite film (a), Li–Si–O–C composite anode after the 1st charge, (b), and after the 100th discharge (c). The contrast in the Z-contrast image depends on the average atomic number. Reproduced from data published in ref. 37. | ||
Fig. 7 shows cross-sectional Z-contrast (ZC) images, which were obtained by high-angle annular dark-field STEM (HAADF-STEM), and bright field (BF) images, which were obtained by annular bright-field STEM (ABF-STEM), of the Si–O–C composite film and the Li–Si–O–C composite anodes after the 1st charge and the 100th discharge. Fig. 7(a) and (d) indicate that the Si–O–C composite film was uniformly deposited and had pores of 10–100 nm in size. The cross-sectional images are considered to have been observed at the thinnest part because the surface of the Si–O–C composite film was rough as confirmed by the thickness variation shown in Fig. 5 and by the SEM images shown in Fig. 4. The crystallinity of the Si–O–C composite anode was confirmed to be completely amorphous from the transmission electron diffraction (TED) pattern obtained from the STEM (see the ref. 37). Pores measuring ca. 500 nm in size, which is larger than the pores observed in the Si–O–C composite film, were observed on the Li–Si–O–C composite anode after the 1st charge (Fig. 7(b) and (e)). These pores would have been formed during the charge process, in which inhomogeneous expansion of silicon and Li2O formation take place because of the reaction with lithium. The contrasts in the HAADF-STEM and BF images changed from homogeneous to the inhomogeneous contrasts observed for the Si–O–C composite film, which means that the Li–Si–O–C composite anode was composed of multiple phases after the 1st charge. The pores that were observed in the Si–O–C composite film and the Li–Si–O–C composite anode after the 1st charge clearly decreased in number after the 100th discharge (Fig. 7(c) and (f)). This implies that decomposition of the electrolyte, such as by SEI formation, takes place even in the Li–Si–O–C composite anode as the number of charge–discharge cycles increases, and that the repeated expansion and contraction of silicon accelerates the uniform dispersion of Li, Si, O, and C elements. The inhomogeneous contrasts in the HAADF-STEM and BF images also mean that the microstructure of the Li–Si–O–C composite anode had at least two phases after the 100th discharge, as in the microstructure observed after the 1st charge.
 | ||
| Fig. 7 Cross-sectional Z-contrast images and BF images of the Si–O–C composite film (a) and (d), and the Li–Si–O–C composite anodes after the 1st charge (b) and (e), and 100th discharge (c) and (f), respectively. Samples were prepared by focused ion beam. The contrast in the Z-contrast image depends on the average atomic number. Reproduced from data published in ref. 37. | ||
The chemical states of Li, Si, C, and O on the Si–O–C composite film and the Li–Si–O–C composite anode after the 1st charge and after 100th discharge were evaluated by EELS. Fig. 8 shows the EELS spectra of Si, C, and O on the Si–O–C composite film obtained at different depths. All peak positions were identified in all spectra, revealing that the Si–O–C composite film was chemically uniform in the depth direction. Fig. 9 shows the EELS spectra of Si, C, and O on the Si–O–C composite film obtained at different points and the Si spectra of standard SiO2 and Si samples. Each peak position in the EELS spectra of Si, C, and O was similar even at different points on the sample, which suggests that the composition of the Si–O–C composite film was chemically uniform as well as has good elemental dispersion. The state of silicon in the Si–O–C composite film was investigated through comparison with standard SiO2 and Si samples. The EELS spectra of Si in the Si–O–C composite exhibited a peak attributed to Si and peaks attributed to SiO2. Therefore, the silicon in the Si–O–C composite film was suggested to exist as SiOx (0 < x < 2).
 | ||
| Fig. 8 Cross-sectional Z-contrast image of the Si–O–C composite film and EELS spectra of Si on the Si–O–C composite film obtained at the colored points, the colors of which correspond to those of the EELS spectra. The baselines in each graph are displaced for clarity. | ||
 | ||
| Fig. 9 EELS spectra of Si (a), C (b), and O (c) on the Si–O–C composite film obtained at different locations. The spectra of SiO2 and Si standard samples are included for comparison. The baselines in each graph are displaced for clarity. Reproduced from data published in ref. 37. | ||
Fig. 10 shows the EELS spectra of Li, Si, O, and C in the Li–Si–O–C composite anode after the 1st charge at points 1 and 2 as indicated in Fig. 7(b). The points were selected on the basis of the contrast difference observed in the HAADF-STEM image (Fig. 7(b)). Although the EELS spectra were obtained at several points in the Li–Si–O–C composite anode after the 1st charge, the spectra were classified as either point 1 or point 2, corresponding to the dark area or bright area, respectively, in the HAADE-STEM image. Although the intensities of the Li, Si, O, and C spectra were different, the peak positions were similar to each other. These results suggested that the Li–Si–O–C composite anode had at least two phases after the 1st charge; a Li rich phase and a Si–O rich phase, as confirmed from Fig. 10(a). The spectra of Li2CO3 and Li2O standard samples, which are well known to be SEI components,41 are shown in Fig. 10(a). The comparison suggests the possibility of formation of Li2O and Li2CO3 in the Li–Si–O–C composite anode after the 1st charge.
 | ||
| Fig. 10 EELS spectra of Li (a), Si (b), C (c), and O (d) obtained at different locations on the Li–Si–O–C composite anode after the 1st charge. The spectra of SiO2, Si, Li2O, and Li2CO3 standard samples are also included. The baselines of each graph have been displaced for clarity. Reproduced from data published in ref. 37. | ||
Fig. 11 shows the EELS spectra of Li, Si, O, and C in the Li–Si–O–C composite anode after the 100th discharge at points 3–6 indicated in Fig. 7(c). Although the intensities observed in the Li spectra were different, the existence of Li was confirmed at all points even in the discharged state. These results suggest the existence of irreversible lithium in the form of Li2O, Li2CO3, and lithium silicates as suggested from the XPS analysis below. The Si spectra measured at points 3 and 5 indicate the possibility of the existence of SiO2. While the states of Li, Si, O, and C were difficult to identify from the EELS spectra, the EELS spectra in Fig. 11 indicate that the Li–Si–O–C composite anode had several phases including Li rich and Si–O rich phases after the 100th discharge. In addition, organic phases resulting from the electrolyte decomposition, which cannot be identified by EELS, should exist. Thus, the composite, which contained several phases which do not react with lithium ions such as Li2O, Li2CO3, and flexible organic phases, would have buffered the volume change caused by the lithiation and delithiation of silicon.
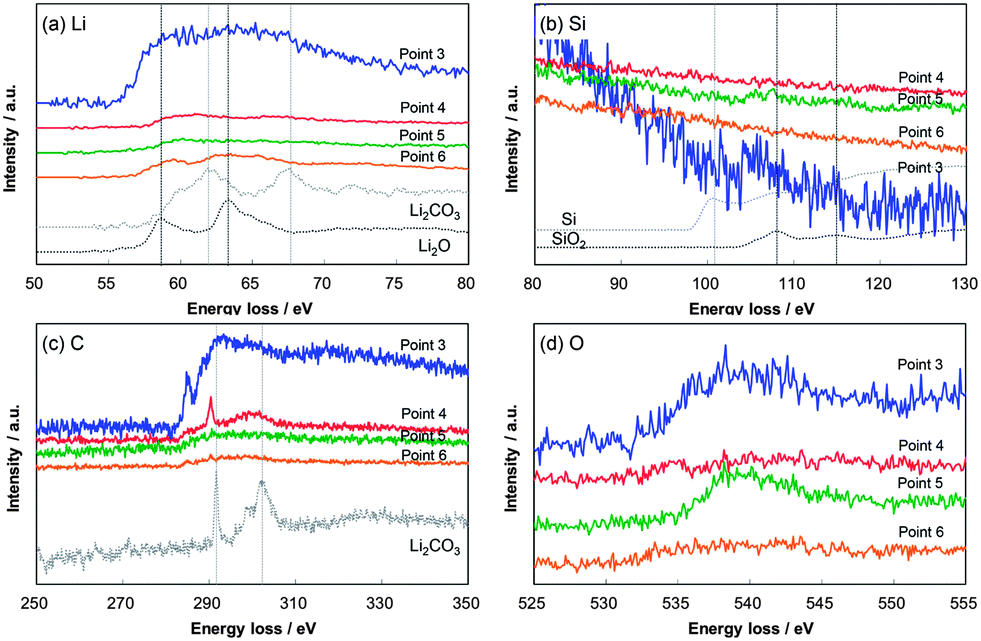 | ||
| Fig. 11 EELS spectra of Li (a), Si (b), C (c), and O (d) obtained at different locations on the Li–Si–O–C composite anode after the 100th discharge. The spectra of SiO2, Si, Li2O, and Li2CO3 standard samples are included for comparison. The baselines of each graph are displaced for clarity. Reproduced from data published in ref. 37. | ||
Fig. 12(a)–(d) shows the XPS spectra of C1s, Li1s, Si2p3/2, and O1s, respectively. The spectra of C1s, shown in Fig. 12(a), suggest an increase of Li2CO3, (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak at ca. 289 eV (ref. 45)) with charge–discharge cycle. The formation of Li2CO3 was also suggested by the EELS spectra shown in Fig. 9 and 10. The peaks at 284–286 eV, which were observed in all spectra, are attributed to the overlapping of several C–C and C–O bonds.
O peak at ca. 289 eV (ref. 45)) with charge–discharge cycle. The formation of Li2CO3 was also suggested by the EELS spectra shown in Fig. 9 and 10. The peaks at 284–286 eV, which were observed in all spectra, are attributed to the overlapping of several C–C and C–O bonds.
 | ||
| Fig. 12 XPS spectra of C1s (a), Li1s (b), Si2p3/2 (c), and O1s (d) on the Si–O–C composite film and Li–Si–O–C composite anodes after the 1st charge, 1st discharge, 2nd charge, 2nd discharge, 5th charge, 5th discharge, 10th charge, 10th discharge, 100th charge, and 100th discharge. All spectra were corrected based on the C–C bond at 284 eV after peak separation of the C1s spectra into C–C, C–O, and CO peaks. The baselines of each graph are displaced for clarity. Reproduced from data published in ref. 37. | ||
The spectra of Li1s shown in Fig. 12(b), suggest the appearance of Li2CO3, Li2O, and Li alloyed with silicon after the 1st charge. By comparing the spectra of charged and discharged samples at the same cycle number, the peak attributed to Li could be seen to increase after charging. Although the absolute intensities were incommensurable, the decrease in Li/Li2O in each cycle was confirmed. These results imply that the lithiation and delithiation of silicon occurred during charge and discharge, respectively. The peak shift, which was attributed to the increase in the Li2CO3 content of the Li–Si–O–C composite anodes, was confirmed to occur with increasing number of cycles, as also suggested by EELS analysis. In addition, the phases of Li2CO3 and Li2O were suggested to have remained unchanged during the charge–discharge cycles. The unchanged phases should act as a buffer against volume change during charge–discharge cycles, resulting in such a highly durable silicon based anode suitable for lithium secondary batteries.
The Si2p3/2 spectra shown in Fig. 12(c) suggest that the silicon in the Si–O–C composite film existed in the form of Si, SiO2 and intermediate oxides of silicon. The results correspond to those of the EELS analysis. Taking into consideration that the discharge capacity was delivered and that the existence of lithium was confirmed by the EELS analysis, the silicon in the Si–O–C composite film should react with lithium ions during the 1st charge process. However, although the shift of peak position to a lower binding energy due to silicon lithiation was confirmed, the peaks remained located mostly at ca. 102 eV. This result suggests that lithium silicates such as Li4SiO4 were formed instead of LixSi, as has been reported by several groups.13,46 Comparing the charged state with the discharged state in each cycle shows that the intensity of the peaks at a lower binding energy increased after discharge. This difference is attributable to the formation of LixSi due to the lithiation of SiOx. Therefore, it is suggested that LixSi and lithium silicates are formed in the charged state of Li–Si–O–C composite anodes.
The broad peak in the O1s spectra shown in Fig. 12(d) is likely to be composed of several peaks attributed to SiO2 as the main phase and to Si–O and C–O bonding. Peaks attributed to Li2O and Li2CO3 were also observed. The broad peak shifted toward lower binding energies after the 1st charge, which was apparently caused by the reduction of SiO2 by lithiation. The peak at ca. 532 eV was attributed to Li4SiO4 as reported in a previously published paper.46 Additional peaks appeared at 530–531 eV after the 1st charge. These peaks were observable even after the 1st discharge and subsequent cycles. Because the magnitude of the peaks decreased after each discharge, the peaks appear to be related to lithiation. This phenomenon corresponds to the reversible conversion reaction of SiO2 into Li2Si2O5, as reported by Sun et al.47 The peaks of Li2Si2O5 are reported to be at 530.7 and 532.5 eV.13,48 While these peaks would be expected to decrease after discharge, the observed peak remained at 530–531 eV. This result therefore implies the existence of irreversible lithium silicates. The peaks of Li2SiO3 and Li2Si2O3 are also reported to appear at 530.3 eV (ref. 46) and 530.8, 532.4 eV,13,48 respectively. Therefore, the Li–Si–O–C composite anode is suggested to contain several different lithium silicate states. Because the peak at 532 eV was observed independently of the charge state and the cycle number, Li4SiO4 consistently appears to exist in the Li–Si–O–C composite anode. The peaks shifted to lower binding energies with increasing cycle number, which implies the possibility of a transformation of Li4SiO4 to Li2SiO3.
Mechanism of Si–O–C composite film formation
From the EDX, EELS, and XPS analyses, the Si–O–C composite film was suggested to be uniformly composed of SiOx and organic compounds. Such a uniform Si–O–C composite film can be achieved by the simultaneous deposition of silicon and organic compounds as mentioned above. This was also supported by linear sweep voltammogram (LSV) of the electrodeposition bath composed of propylene carbonate (PC) and TBAClO4 with SiCl4 and glow discharge-optical emission spectroscopy (GDOES). The linear sweep voltammogram in Fig. 13 indicates that the deposition of SiCl4 begins at ca. 1.3 V vs. Li/Li+. In contrast, the decomposition of PC or TBAClO4 begins at ca. 0.45 V vs. Li/Li+. PC and TBAClO4 may be considered to be stable against reduction above a potential of ca. 0.45 V vs. Li/Li+. However, the decomposition of the electrolyte must have occurred due the fact that a small cathodic current was confirmed over the entire range and that the amount of silicon in the Si–O–C composite film, which was electrodeposited at a constant current of 0.7 mA cm−2 and a potential of ca. 1.3 V vs. Li/Li+, was 60 wt% from the GDOES data (not shown). So far, it is difficult to conclude that the SiCl4 accelerated the decomposition of the electrolyte or that impurities in the SiCl4 decomposed. However, the facts suggest that the coinstantaneous deposition of silicon and organic compounds is possible at a potential of ca. 1.3 V vs. Li/Li+ in the present case.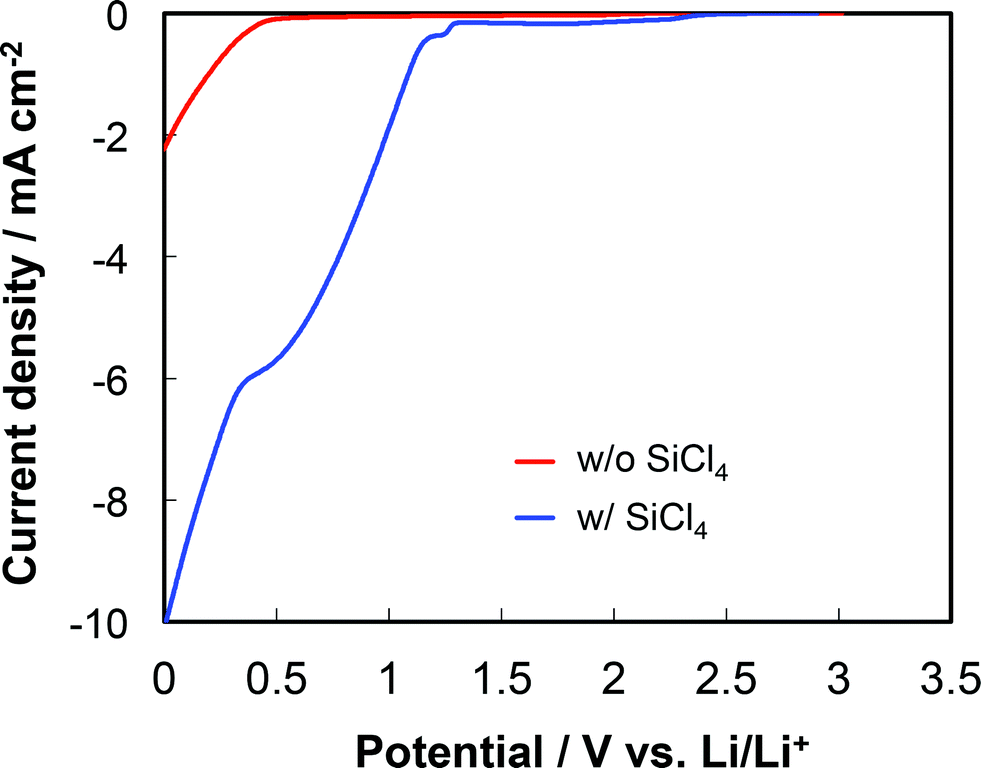 | ||
| Fig. 13 Linear sweep voltammogram of electrodeposition bath containing PC and TBAClO4 with or without SiCl4. Scan rate is 1.0 mV s−1 in the cathodic direction. Reproduced from the data published in ref. 36. | ||
Fig. 14 schematically illustrates the electrodeposition of the Si–O–C composite film summarized from the analytical data. SiCl4 and PC are decomposed simultaneously by electrons that are conducted through the deposit, resulting in the formation of the Si–O–C composite film. The composite film is composed of SiOx, which is a mixture of Si and SiO2 as shown by EELS analysis, and organic compounds. After the 1st lithiation, SiOx is converted to LixSi, Li2O, and lithium silicates and SEI, as typified by Li2CO3, are formed by the decomposition of the electrolyte. The mixture of active materials such as Si and Li2Si2O5 and inactive materials such as Li2O, Li2CO3, and organic compounds results in outstanding properties for a lithium secondary battery anode.
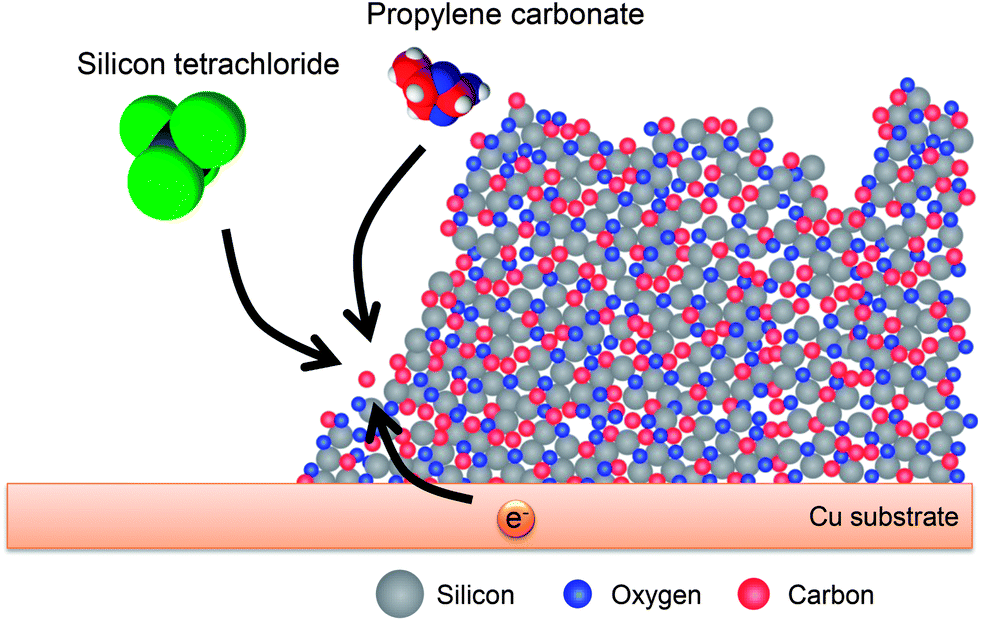 | ||
| Fig. 14 Schematic of the electrodeposition of the Si–O–C composite film. Reproduced from data published in ref. 36. | ||
Summary
The increase of energy density on energy storage is essential to make our sustainable life with a future come true. The reasons to increase the energy density of batteries are to increase the capacity of both anode and cathode, to expand the potential difference between anode and cathode, and to balance the capacities between anode and cathode: of course, decrease of weight and volume of other components in batteries is also important. For the purpose, the active materials of anode and cathode have been shifting to alloying materials (including conversion materials) from insertion materials. However, the alloying materials have some problems which should be solved for their practical application, namely, volume change during charge–discharge cycles, and low electric conductivity and low ionic conductivity in the materials. Especially, the volume change during charge–discharge cycles is a crucial problem for anode materials such as Si and Sn, resulting in capacity decay caused by pulverization of the active material. To solve those problems, materials design such as reduction in size down to nanometric scale and two-dimensionalization of the materials, and combination with carbon materials have been proposed. Firstly, this paper reviewed recent work on silicon-based anodes to overcome the poor cycle durability from the standpoints of structural control such as with thin films,4–6 porous structures,7,8,49 core–shell structures,14–16,50 nano structures,23,24,51 and by alloying with other metals.25–27,29 All these efforts have been focused on the reduction of the stress experienced during charge–discharge cycling by nano-structuring the material and/or surrounding materials to buffer the stress. The approaches used can be divided into two types. The first is nano-structured powder, which is suitable for conventional processes used to assemble batteries and can increase the capacity of the electrode by increasing its thickness. Cui's group has kept the heat on the development of nano-structured silicon materials.14,49–52 Among them, the work on the DWSiNT anode demonstrated high durability for charge–discharge cycles i.e. 88% capacity retention even after 6000 cycles.14 Coinstantaneously, good binders that suppress the degradation of silicon-based anodes have also been developed.32,34,53 The other is a nano-structured film deposited directly onto the substrate that does not require any other additives such as electron conductor and binder. The applications of these two types will therefore be different; nano-structured powder will be preferentially applied in high-energy-oriented batteries because it can be made thick, and the nano-structured film will be preferentially applied in energy density-oriented batteries because other additives are not required. The Si–O–C composite film has the potential to achieve both properties, leading to much higher energy density, because the thickness of the Si–O–C composite film can be increased without any additives by increasing the columbic amount for electrodeposition.38 Moreover, the technique of alloy anode preparation by electrodeposition is applicable to other materials that can alloy with lithium.54 This paper focused especially on a binder-free Si–O–C composite film for lithium secondary battery anodes that delivered a discharge capacity of 1125 mA h (g of Si)−1 or 693 mA h (g of deposit)−1 at the 500th cycle and can operate even over 7000 charge–discharge cycles with a discharge capacity of 831 mA h (g of Si)−1 or 512 mA h (g of deposit)−1. The mechanism of the Si–O–C composite film formation was discussed using STEM, EELS, and XPS data. The results suggested that the mixture of active materials such as Si and Li2Si2O5 and inactive materials such as Li2O, Li2CO3, and organic compounds results in outstanding properties for a lithium secondary battery anode.Silicon anode-based lithium ion batteries will become standard batteries like the lead-acid battery, which has been used for over 150 years in various applications, in future because of its high theoretical capacity. Therefore, a cathode should be also developed for the realization of silicon anode-based lithium ion batteries. The replacement of the graphite anode with a silicon-based anode in a conventional LIB system can hardly ask for drastic increase in capacity in the same manner mentioned in Fig. 2. Fortunately, numerous efforts to increase the capacity of cathode have been made. For example, sulfur cathodes, which exhibit high capacity, have been investigated.55 In addition, some studies that combine conversion anodes with sulfur cathodes have been reported.56 Thus, the lithium ion battery composed of the silicon-based anode and sulfur-based cathode will be realized in the near future. A battery with air cathode has been also investigated as an ultimate battery with high energy density.57 Even in the battery system for the future, silicon-based anode can be one of the choices for safety issue due to the reactivity of lithium metal.58 Therefore, the silicon-based anode will be one of the critical technologies for future energy storage.
Acknowledgements
We acknowledge the support by the Grant-in-Aid for Specially Promoted Research “Establishment of Electrochemical Device Engineering” from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We also acknowledge the support in part by the “Research & Development Initiative for Scientific Innovation of New Generation Batteries” of the New Energy and Industrial Technology Development Organization of Japan.Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed; P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef PubMed; B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef PubMed.
- R. A. Huggins and W. D. Nix, Ionics, 2000, 6, 57–63 CrossRef CAS; T. D. Hatchard and J. R. Dahn, J. Electrochem. Soc., 2004, 151, A838–A842 CrossRef; P. Limthongkul, Y. I. Jang, N. J. Dudney and Y. M. Chiang, Acta Mater., 2003, 51, 1103–1113 CrossRef; M. N. Obrovac and L. Christensen, Electrochem. Solid-State Lett., 2004, 7, A93–A96 CrossRef; J. H. Ryu, J. W. Kim, Y. E. Sung and S. M. Oh, Electrochem. Solid-State Lett., 2004, 7, A306–A309 CrossRef; J. Li and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A156–A161 CrossRef.
- S. W. Lee, M. T. McDowell, J. W. Choi and Y. Cui, Nano Lett., 2011, 11, 3034–3039 CrossRef CAS PubMed.
- J. Graetz, C. C. Ahn, R. Yazami and B. Fultz, Electrochem. Solid-State Lett., 2003, 6, A194–A197 CrossRef CAS.
- S. Ohara, J. Suzuki, K. Sekine and T. Takamura, J. Power Sources, 2004, 136, 303–306 CrossRef CAS.
- T. Takamura, M. Uehara, J. Suzuki, K. Sekine and K. Tamura, J. Power Sources, 2006, 158, 1401–1404 CrossRef CAS.
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 47, 10151–10154 CrossRef CAS PubMed.
- M. Y. Ge, J. P. Rong, X. Fang and C. W. Zhou, Nano Lett., 2012, 12, 2318–2323 CrossRef CAS PubMed.
- H. Nara, Y. Fukuhara, A. Takai, M. Komatsu, H. Mukaibo, Y. Yamauchi, T. Momma, K. Kuroda and T. Osaka, Chem. Lett., 2008, 37, 142–143 CrossRef CAS.
- F. Beguin, F. Chevallier, C. Vix-Guterl, S. Saadallah, V. Bertagna, J. N. Rouzaud and E. Frackowiak, Carbon, 2005, 43, 2160–2167 CrossRef CAS; C. K. Chan, R. Ruffo, S. S. Hong and Y. Cui, J. Power Sources, 2009, 189, 1132–1140 CrossRef; S. P. V. Nadimpalli, V. A. Sethuraman, S. Dalavi, B. Lucht, M. J. Chon, V. B. Shenoy and P. R. Guduru, J. Power Sources, 2012, 215, 145–151 CrossRef.
- T. Zhang, J. Gao, H. P. Zhang, L. C. Yang, Y. P. Wu and H. Q. Wu, Electrochem. Commun., 2007, 9, 886–890 CrossRef CAS.
- J. O. Besenhard, J. Yang and M. Winter, J. Power Sources, 1997, 68, 87–90 CrossRef CAS; H. Li, X. Huang, L. Chen, G. Zhou, Z. Zhang, D. Yu, Y. Jun Mo and N. Pei, Solid State Ionics, 2000, 135, 181–191 CrossRef.
- M. Miyachi, H. Yamamoto, H. Kawai, T. Ohta and M. Shirakata, J. Electrochem. Soc., 2005, 152, A2089–A2091 CrossRef CAS.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS PubMed.
- T. H. Hwang, Y. M. Lee, B. S. Kong, J. S. Seo and J. W. Choi, Nano Lett., 2012, 12, 802–807 CrossRef CAS PubMed.
- M. Zhou, T. W. Cai, F. Pu, H. Chen, Z. Wang, H. Y. Zhang and S. Y. Guan, ACS Appl. Mater. Interfaces, 2013, 5, 3449–3455 CAS.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed; Z. P. Guo, E. Milin, J. Z. Wang, J. Chen and H. K. Liu, J. Electrochem. Soc., 2005, 152, A2211–A2216 CrossRef; Y. Liu, T. Matsumura, N. Imanishi, A. Hirano, T. Ichikawa and Y. Takeda, Electrochem. Solid-State Lett., 2005, 8, A599–A602 CrossRef.
- H. Li, X. Huang, L. Chen, Z. Wu and Y. Liang, Electrochem. Solid-State Lett., 1999, 2, 547–549 CrossRef CAS.
- J. Yang, B. F. Wang, K. Wang, Y. Liu, J. Y. Xie and Z. S. Wen, Electrochem. Solid-State Lett., 2003, 6, A154–A156 CrossRef CAS.
- U. Kasavajjula, C. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003–1039 CrossRef CAS.
- Y.-C. Yen, S.-C. Chao, H.-C. Wu and N.-L. Wu, J. Electrochem. Soc., 2009, 156, A95–A102 CrossRef CAS.
- T. Kim, Y. H. Mo, K. S. Nahm and S. M. Oh, J. Power Sources, 2006, 162, 1275–1281 CrossRef CAS; Y. Xu, G. Yin, Y. Ma, P. Zuo and X. Cheng, J. Mater. Chem., 2010, 20, 3216–3220 RSC.
- T. Okubo, T. Yamada, M. Saito, C. Yodoya, A. Kamei, M. Hirota, T. Takenaka, A. Tasaka and M. Inaba, Electrochemistry, 2012, 80, 720–724 CrossRef CAS.
- M. Saito, T. Yamada, C. Yodoya, A. Kamei, M. Hirota, T. Takenaka, A. Tasaka and M. Inaba, Solid State Ionics, 2012, 225, 506–509 CrossRef CAS.
- A. Anani and R. A. Huggins, J. Power Sources, 1992, 38, 351–362 CrossRef CAS.
- J. A. Choi, D. W. Kim, Y. S. Bae, S. W. Song, S. H. Hong and S. M. Lee, Electrochim. Acta, 2011, 56, 9818–9823 CrossRef CAS.
- Y. N. Zhou, W. J. Li, H. J. Chen, C. Liu, L. Zhang and Z. Fu, Electrochem. Commun., 2011, 13, 546–549 CrossRef CAS.
- H. Mukaibo, T. Momma and T. Osaka, J. Power Sources, 2005, 146, 457–463 CrossRef CAS.
- J. Chen, S. J. Bull, S. Roy, A. Kapoor, H. Mukaibo, H. Nara, T. Momma, T. Osaka and Y. Shacham-Diamand, Tribol. Int., 2009, 42, 779–791 CrossRef CAS.
- H. Buqa, M. Holzapfel, F. Krumeich, C. Veit and P. Novák, J. Power Sources, 2006, 161, 617–622 CrossRef CAS; B. Lestriez, S. Bahri, I. Sandu, L. Roué and D. Guyomard, Electrochem. Commun., 2007, 9, 2801–2806 CrossRef; N. S. Hochgatterer, M. R. Schweiger, S. Koller, P. R. Raimann, T. Wöhrle, C. Wurm and M. Winter, Electrochem. Solid-State Lett., 2008, 11, A76–A80 CrossRef.
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004–3010 CAS.
- B. Koo, H. Kim, Y. Cho, K. T. Lee, N.-S. Choi and J. Cho, Angew. Chem., Int. Ed., 2012, 51, 8762–8767 CrossRef CAS PubMed.
- J. S. Bridel, T. Azaïs, M. Morcrette, J. M. Tarascon and D. Larcher, Chem. Mater., 2009, 22, 1229–1241 CrossRef.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- T. Momma, S. Aoki, H. Nara, T. Yokoshima and T. Osaka, Electrochem. Commun., 2011, 13, 969–972 CrossRef CAS.
- H. Nara, T. Yokoshima, T. Momma and T. Osaka, Energy Environ. Sci., 2012, 5, 6500–6505 CAS.
- H. Nara, T. Yokoshima, M. Otaki, T. Momma and T. Osaka, Electrochim. Acta, 2013 DOI:10.1016/j.electacta.2013.07.143.
- T. Hang, H. Nara, T. Yokoshima, T. Momma and T. Osaka, J. Power Sources, 2013, 222, 503–509 CrossRef CAS.
- A. H. Robert, Solid State Ionics, 1998, 113–115, 57–67 Search PubMed.
- A. Netz, R. A. Huggins and W. Weppner, J. Power Sources, 2003, 119, 95–100 CrossRef.
- K. Xu, Energies, 2010, 3, 135–154 CrossRef CAS; J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 Search PubMed.
- T. Jiang, S. Zhang, X. Qiu, W. Zhu and L. Chen, Electrochem. Commun., 2007, 9, 930–934 CrossRef CAS.
- S.-J. Lee, J.-K. Lee, S.-H. Chung, H.-Y. Lee, S.-M. Lee and H.-K. Baik, J. Power Sources, 2001, 97–98, 191–193 CrossRef CAS.
- M. Winter, W. K. Appel, B. Evers, T. Hodal, K.-C. Möller, I. Schneider, M. Wachtler, M. R. Wagner, G. H. Wrodnigg and J. O. Besenhard, Monatshefte für Chemie/Chemical Monthly, 2001, 132, 473–486 CrossRef CAS; P. Verma, P. Maire and P. Novak, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef.
- J. P. Contour, A. Salesse, M. Froment, M. Garreau, J. Thevenin and D. Warin, J. Microsc. Spectrosc. Electron., 1979, 4, 483–491 Search PubMed; S. Contarini and J. W. Rabalais, J. Electron Spectrosc. Relat. Phenom., 1985, 35, 191–201 CrossRef CAS.
- X. Liu, M. C. Zheng and K. Xie, J. Power Sources, 2011, 196, 10667–10672 CrossRef CAS.
- Q. Sun, B. Zhang and Z. W. Fu, Appl. Surf. Sci., 2008, 254, 3774–3779 CrossRef CAS.
- B. Dejong, D. Ellerbroek and A. L. Spek, Acta Crystallogr., Sect. B: Struct. Sci., 1994, 50, 511–518 Search PubMed.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed.
- L.-F. Cui, R. Ruffo, C. K. Chan, H. Peng and Y. Cui, Nano Lett., 2008, 9, 491–495 CrossRef CAS PubMed; L.-F. Cui, Y. Yang, C.-M. Hsu and Y. Cui, Nano Lett., 2009, 9, 3370–3374 CrossRef PubMed.
- C. K. Chan, R. N. Patel, M. J. O'Connell, B. A. Korgel and Y. Cui, ACS Nano, 2010, 4, 1443–1450 CrossRef CAS PubMed; C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef PubMed; M.-H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef PubMed.
- L. Hu, H. Wu, Y. Gao, A. Cao, H. Li, J. McDough, X. Xie, M. Zhou and Y. Cui, Adv. Energy Mater., 2011, 1, 523–527 CrossRef CAS; H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef.
- J. Li, R. B. Lewis and J. R. Dahn, Electrochem. Solid-State Lett., 2007, 10, A17–A20 CrossRef CAS; M. N. Obrovac and L. J. Krause, J. Electrochem. Soc., 2007, 154, A103–A108 CrossRef; S. D. Beattie, D. Larcher, M. Morcrette, B. Simon and J. M. Tarascon, J. Electrochem. Soc., 2008, 155, A158–A163 CrossRef; S. Komaba, K. Shimomura, N. Yabuuchi, T. Ozeki, H. Yui and K. Konno, J. Phys. Chem. C, 2011, 115, 13487–13495 Search PubMed.
- T. Momma, M. Jeong, T. Yokoshima, H. Nara, A. Toyoda and T. Osaka, J. Power Sources, 2013, 242, 527–532 CrossRef CAS.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed; S. Evers and L. F. Nazar, Acc. Chem. Res., 2012, 46, 1135–1143 CrossRef PubMed; J. Kim, D.-J. Lee, H.-G. Jung, Y.-K. Sun, J. Hassoun and B. Scrosati, Adv. Funct. Mater., 2013, 23, 1076–1080 CrossRef; S. Zhi Wei, L. Weiyang, J. C. Judy, Z. Guangyuan, Y. Yuan, T. M. Matthew, H. Po-Chun and C. Yi, Nat. Commun., 2013, 4, 1331 CrossRef PubMed; G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef PubMed; W. Li, G. Zheng, Y. Yang, Z. W. Seh, N. Liu and Y. Cui, Proc. Natl. Acad. Sci., 2013, 110, 7148–7153 CrossRef PubMed; Y. Yang, G. Zheng, S. Misra, J. Nelson, M. F. Toney and Y. Cui, J. Am. Chem. Soc., 2012, 134, 15387–15394 CrossRef PubMed.
- J. Hassoun, J. Kim, D.-J. Lee, H.-G. Jung, S.-M. Lee, Y.-K. Sun and B. Scrosati, J. Power Sources, 2012, 202, 308–313 CrossRef CAS; J. Hassoun, Y.-K. Sun and B. Scrosati, J. Power Sources, 2011, 196, 343–348 CrossRef; Y. Yang, M. T. McDowell, A. Jackson, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2010, 10, 1486–1491 CrossRef PubMed; R. Elazari, G. Salitra, G. Gershinsky, A. Garsuch, A. Panchenko and D. Aurbach, Electrochem. Commun., 2012, 14, 21–24 CrossRef.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS; P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef PubMed; J. Christensen, P. Albertus, R. S. Sanchez-Carrera, T. Lohmann, B. Kozinsky, R. Liedtke, J. Ahmed and A. Kojic, J. Electrochem. Soc., 2011, 159, R1–R30 CrossRef; J.-S. Lee, S. Tai Kim, R. Cao, N.-S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef.
- J. Hassoun, H.-G. Jung, D.-J. Lee, J.-B. Park, K. Amine, Y.-K. Sun and B. Scrosati, Nano Lett., 2012, 12, 5775–5779 CrossRef CAS PubMed; P. Jakes, G. Cohn, Y. Ein-Eli, F. Scheiba, H. Ehrenberg and R.-A. Eichel, ChemSusChem, 2012, 5, 2278–2285 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |