 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow fractions of ionic liquid or poly(ionic liquid) can activate polysaccharide biomass into shaped, flexible and fire-retardant porous carbons†
Yongjun Mena,
Miriam Siebenbürgerb,
Xunlin Qiuc,
Markus Antoniettia and
Jiayin Yuan*a
aDepartment of Colloid Chemistry, Max-Planck-Institute of Colloids and Interfaces, D-14476 Potsdam, Germany. E-mail: jiayin.yuan@mpikg.mpg.de; Fax: +49-331-5679502; Tel: +49-331-5679552
bSoft Matter and Functional Materials, Helmholtz-Zentrum Berlin für Materialien und Energie, D-14109 Berlin, Germany
cDepartment of Physics and Astronomy, University of Potsdam, D-14476 Potsdam, Germany
First published on 1st August 2013
Abstract
Sugar-based molecules and polysaccharide biomass can be turned into porous functional carbonaceous products at comparably low temperatures of 400 °C under a nitrogen atmosphere in the presence of an ionic liquid (IL) or a poly(ionic liquid) (PIL). The IL and PIL act as “activation agents” with own structural contribution, and effectively promote the conversion and pore generation in the biomaterials even at a rather low doping ratio (7 wt%). In addition, this “induced carbonization” and pore forming phenomenon enables the preservation of the biotemplate shape to the highest extent and was employed to fabricate shaped porous carbonaceous materials from carbohydrate-based biotemplates, exemplified here with cellulose filter membranes, coffee filter paper and natural cotton. These carbonized hybrids exhibit comparably good mechanical properties, such as bendability of membranes or shape recovery of foams. Moreover, the nitrogen atoms incorporated in the final products from the IL/PIL precursors further improve the oxidation stability in the fire-retardant tests.
Introduction
Bio-based materials derived from plants and lower organisms are an important class of renewable and abundant resources that regain rapidly increasing interest in both the scientific community and industrial production, due to sustainability issues.1–6 The capability of making functional materials entirely or partially from biomass is a part of humankind's ancient history and is how technology started. Excitingly, this concept is returning into the focus of modern material design and has already made a prominent impact on current research.7–14 In particular, most natural materials are able to provide unique and sophisticated hierarchical structures and morphologies that are by far superior to most man-made constructions. Such refined structures can be immediately adopted as biotemplates or construction units to fabricate products with superior performance, as recently illustrated by Mann and other groups.15–18Activated carbon is a class of widely used absorbents in a multitude of environmental applications for wastewater treatment, gas separation and purification, and more.19 It features large surface area, good chemical stability, oxygen-rich surface functionalities, and open pore structures ideal for the adsorption of large molecules. Typically, activated carbon can be produced from carbon-rich precursors, such as coal, lignite and agricultural waste via physical and chemical activation.20,21 The former method deals with the pyrolysis of the precursors and gasification of the resulting char in steam or carbon dioxide.22 In the latter method, carbon precursors are impregnated with excessive chemical reagents, such as phosphoric acid, zinc chloride, and alkaline hydroxides, and pyrolysed under an inert gas atmosphere at temperatures often between 600 and 800 °C.23 In addition, a purification process is required to remove the residue activation chemicals. It is therefore highly desired to develop new activation agents, which work at low doping ratios and low temperatures, and can ideally be incorporated into the final carbon products to avoid the post-synthesis purification.
In this article, we report on the facile preparation of porous carbonaceous architectures with well-defined shape and complex structure by lowering the activation temperature of bio(macro)molecules and plant biostructures down to the point of structural preservation with the assistance of imidazolium-based ionic liquids (ILs) or poly(ionic liquid)s (PILs). Surprisingly it was found that even at rather low added amounts, ILs or PILs can efficiently promote the carbonization and micropore generation in the bio-entities without destroying the structural skeleton pre-existing in the biotemplates down to very fine structural details. Finally following the biomass-activation procedure the ILs or PILs favorably ended up as porous nitrogen-doped carbon in the final porous product, i.e. no need to remove any residue activation agent, and it additionally improved the fire-retardancy.
Experimental section
Materials
1-Vinylimidazole (99%), bromoacetonitrile (97%), 1,3-bis(cyanomethyl)imidazolium bis(trifluoromethylsulfonyl)imide (≥94%), lithium bis(trifluoromethanesulfonyl)imide (99.95%), α- and β-cyclodextrin (≥97%), D-(+)-glucose (99.5%), starch (puriss. p.a., from potato), cellulose (fibers, particle size: medium), poly(ethylene oxide) (Mv ∼ 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), poly(vinyl alcohol) (Mw ∼ 89000–98000 g mol−1), polystyrene (Mw ∼ 192000 g mol−1) and poly(acrylic acid) (Mv ∼ 450000 g mol−1), crystal violet (dye content ≥90%), and methylene blue were obtained from Sigma-Aldrich and used without further purification. 2,2′-Azobis(2-methylpropionitrile) (98%, Aldrich) was recrystallized from methanol. An ionic liquid monomer, 3-cyanomethyl-1-vinyl imidazolium bromide (CMVImBr) was prepared via quaternization of 1-vinylimidazole with bromoacetonitrile, according to our previous paper.24 The solvents and other materials were used as received.
000 g mol−1), poly(vinyl alcohol) (Mw ∼ 89000–98000 g mol−1), polystyrene (Mw ∼ 192000 g mol−1) and poly(acrylic acid) (Mv ∼ 450000 g mol−1), crystal violet (dye content ≥90%), and methylene blue were obtained from Sigma-Aldrich and used without further purification. 2,2′-Azobis(2-methylpropionitrile) (98%, Aldrich) was recrystallized from methanol. An ionic liquid monomer, 3-cyanomethyl-1-vinyl imidazolium bromide (CMVImBr) was prepared via quaternization of 1-vinylimidazole with bromoacetonitrile, according to our previous paper.24 The solvents and other materials were used as received.
Polymerization of the CMVImBr monomer
Monomer 3-cyanomethyl-1-vinyl imidazolium bromide (16.2 g) and AIBN (320 mg) were dissolved in 160 mL of DMSO in a 500 mL Schlenk flask equipped with a magnetic stirring bar. After complete dissolution of the monomer, the solution was sealed, degassed by vacuum and washed with argon several times. The polymerization was conducted in argon at 90 °C under magnetic stirring for 24 hours. After cooling down, the reaction mixture was purified by precipitation twice into acetone. A yellowish powder was obtained and dried at 60 °C under vacuum to constant weight. Yield: 80%. 1H-NMR (DMSO-d6, δ, ppm): 8.9–9.6 (1H), 7.0–8.4 (2H), 5.2–5.7 (2H), 3.5–4.6 (1H), 1.9–2.7 (2H).Anion exchange of the PCMVImTf2N polymer
6 g of poly(3-cyanomethyl-1-vinyl imidazolium bromide) was added to 800 mL of water under gentle heating. After complete dissolution and cooling down, 200 mL of an aqueous solution containing 1.05 molar equivalents of lithium bis(trifluoromethanesulfonyl)imide salt was added under vigorous stirring. The solution became turbid and precipitation occurred. The precipitate was filtered, washed with pure water several times and dried at 60 °C to constant weight under high vacuum (1 × 10−2 mbar). Yield: 70%.Loading of an IL/PIL onto various molecular substrates
To load an IL/PIL onto β-CD, a mixture of IL or PIL (0.50 g) and different amounts of β-CD powder at designed weight ratios (entries 3–8, 17 and 18 in Table 1) was finely ground and mixed in an alumina crucible. The same procedure was applied also to glucose, starch, cellulose, and synthetic polymers (PAA, PEG, and PS), in which the IL occupied 43 wt% in the mixture (entries 9–14 in Table 1).| Entry | Component | IL or PIL fraction (wt%) | SBET (m2 g−1) | Yield (%) | N (%) | H (%) |
|---|---|---|---|---|---|---|
| 1 | IL | 0 | 570 | 39.1 | 31.5 | 2.5 |
| 2 | β-CD | 100 | 0 | 26.0 | 0.4 | 4.0 |
| 3 | IL + β-CD | 21 | 724 | 38.4 | 22.9 | 2.5 |
| 4 | IL + α-CD | 21 | 735 | 35.9 | 23.0 | 2.3 |
| 5 | IL + β-CD | 57 | 503 | 39.8 | 12.9 | 2.9 |
| 6 | IL + β-CD | 73 | 486 | 38.9 | 9.3 | 2.3 |
| 7 | IL + β-CD | 84 | 266 | 37.6 | 5.4 | 3.3 |
| 8 | IL + β-CD | 93 | 116 | 35.8 | 3.0 | 3.6 |
| 9 | IL + glucose | 57 | 586 | 36.9 | 15.9 | 2.1 |
| 10 | IL + starch | 57 | 605 | 37.6 | 12.9 | 2.7 |
| 11 | IL + cellulose | 57 | 623 | 35.9 | 13.3 | 2.6 |
| 12 | IL + PEG | 57 | 22 | 22.8 | 14.4 | 2.7 |
| 13 | IL + PAA | 57 | 40 | 25.6 | 10.3 | 2.7 |
| 14 | IL + PS | 57 | 57 | 19.7 | 23.8 | 2.0 |
| 15 | IL + filter membrane | 57 | 680 | 36.9 | 13.4 | 3.0 |
| 16 | IL + coffee paper | 57 | 720 | 37.5 | 15.5 | 3.3 |
| 17 | PIL + β-CD | 57 | 530 | 32.2 | 9.3 | 2.9 |
| 18 | PIL + β-CD(500 °C) | 73 | 534 | 31.5 | 7.2 | 2.7 |
| 19 | PIL + filter membrane | 88 | 400 | 35.6 | 2.1 | 2.7 |
| 20 | PIL + cotton | 88 | 350 | 35.4 | 2.2 | 2.9 |
Loading of an IL/PIL onto the filter membrane and coffee paper
To load an IL onto the biotemplates (entries 15 and 16), an IL (1 g) solution in ethanol (10 mL) was spread onto a cut filter membrane or coffee filter paper (2.33 g), which was dried at 80 °C.To load a PIL onto the filter membrane (entry 19), a dried filter membrane (3 g) was immersed in the PIL solution in acetonitrile (10 wt%, viscosity =1.4 mPa s). After 5 minutes, it was taken out. The residue solution on the surface was wiped off and the rest was removed by vacuum filtration. The filter membrane was then dried at 80 °C until constant weight. The fraction of the PIL was calculated as the weight difference between the filter membrane and the PIL-loaded filter membrane.
Loading of a PIL onto cotton
3 g of dried cotton was immersed in a PIL solution in acetonitrile (10 wt%) for 5 min. The wet cotton was then pressed into a round disc membrane in a Buchner funnel. The residue solution was removed by vacuum filtration via squeezing the wet cotton tightly into a dense foam. The cotton was then dried at 80 °C until constant weight. The fraction of the PIL was calculated as the weight difference between the cotton and the PIL-loaded cotton.Carbonization process
For carbonization, an IL, a PIL, or their mixtures with biomaterials were loaded into an alumina crucible in an alumina tube furnace, and carbonized under nitrogen flow. The sample was heated up to 400 °C (5 °C min−1) and kept for 1 h at 400 °C. In the case of entry 18, the sample was heated to 500 °C (5 °C min−1) and kept for 1 h at 500 °C. Afterwards, the furnace was slowly cooled down to room temperature.Dye sorption and fire-retardant experiments
For the dye-removal experiment, carbonaceous foam derived from the PIL/cotton composite was cut into a designed shape (0.8 cm × 0.8 cm × 0.2 cm), and immersed in a methylene blue aqueous solution (10 mg L−1). After a few seconds, the foam was taken out, and squeezed to repel the clean solution out of the foam. For removing crystal violet from aqueous solution, 20 mg of the carbonaceous cotton foam or PIL/cotton composite foam was added to crystal violet solution (10 mg L−1), and the mixture was shaken for 5 min. For the fire-retardant experiment, cylindrical carbonaceous foams were obtained directly by carbonization of cotton or PIL-coped cotton within a quasi-tube.Characterization methods
Nitrogen sorption experiments were performed with a Quantachrome Autosorb-1 or Quadrasorb at liquid nitrogen temperature, and data analysis was performed with Quantachrome software. The surface area was calculated using the BET equation. The pore size distribution was calculated using the Barrett–Joyner–Halenda (BJH) method. The samples were degassed at 120 °C for 15 h before measurements.Elemental analysis was performed for carbon, hydrogen, sulphur and nitrogen using a Vario EL Elementar. TGA experiments were accomplished on a Netzsch TG209-F1 apparatus at a heating rate of 10 K min−1 under nitrogen. SEM images were obtained on a LEO 1550-Gemini instrument after sputtering with platinum.
Mechanical tensile testing was performed at room temperature using a Zwick Z005 (Zwick GmbH & Co KG, Germany) tensile testing machine equipped with a 10 N load cell. All samples had identical geometry, the dog bone shape, achieved using a cutting die of a standard shape (DIN 53504) for the pure tensile uniaxial strain experiment. The viscosity measurement was performed with a MCR 301 rheometer (Anton Paar) attached with a coaxial cylindrical double-gap geometry (inner cup radius of 11.910 mm, outer cup radius of 13.795 mm, inner bob radius of 12.330 mm, outer bob radius of 13.334 mm and an internal bob length of 42 mm) at 25 °C.
Results and discussion
ILs are substances composed of paired, mostly organic cations and anions which melt below 100 °C.24–26 PILs are ionic polymers obtained by polymerization of monomeric ILs.27–29 The chemical structures of the IL and PIL studied in detail here are illustrated in Scheme 1. The unique structural feature of both substances is that either one or two cyanomethyl-groups are attached directly to the imidazolium cation ring, while a large-sized anion, bis(trifluoromethylsulfonyl)imide, Tf2N, was chosen for charge neutralization. This unique chemical combination is known to readily form microporous carbons upon pyrolysis of pure IL or PIL under nitrogen at unexpectedly low temperatures even without any external template, as reported by Dai et al.30–34 In our study, when the IL (3-cyanomethyl-1-vinylimidazolium Tf2N) was heated to only 400 °C at a heating rate of 5 °C min−1 under a nitrogen atmosphere, it produced a carbonaceous framework (entry 1 in Table 1) with an oven yield of 39.1 wt%. The N2 sorption measurement (Fig. 1) revealed that the product is essentially microporous with a specific surface area (SBET) ∼ 570 m2 g−1, which is in good accordance with the earlier reported values.32 Here, the ultramicropores (<2 nm) were left by the thermal removal of the large Tf2N anion, i.e. a molecular templating mechanism, also explaining the weight loss. This is supported by the observation of a low sulfur content in these products.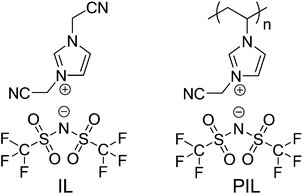 | ||
| Scheme 1 Chemical structures of the ionic liquid and poly(ionic liquid) applied as chemical activation agents. | ||
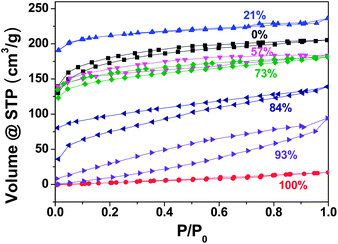 | ||
| Fig. 1 Nitrogen sorption isotherms of carbonaceous products (entries 1–3 and 5–8 in Table 1) obtained at 400 °C from a mixture of IL with different weight fractions of β-CD. | ||
β-Cyclodextrin, β-CD, here used as a cyclic oligosaccharide, on the other hand generates only a compact and nonporous solid when processed in the same manner. Unexpectedly, when β-CD was mixed in 21 wt% with this IL (molar ratio of β-CD/IL = 1:10), a microporous network formed with an even higher SBET ∼ 724 m2 g−1, while the oven yield remained practically untouched (entry 3 in Table 1). In a word, the mixing of β-CD and IL seems to promote pore formation. Only the nitrogen content drops from 39.1 to 22.9 wt% when β-CD was added. This is understandable, as β-CD is a nitrogen-free compound. The same result was obtained for α-CD as well (entry 4 in Table 1).
The β-CD-content was systematically increased in the carbonization mixture. At a β-CD weight fraction of 57 wt%, yet a microporous product with SBET ∼ 503 m2 g−1 (entry 5 in Table 1) was formed, which is very close to that of the pure IL. Upon replacing more IL with β-CD in the mixture, the SBET values of the formed products drop accordingly. Nevertheless, even at the highest β-CD weight content of 93 wt% in the mixture, the pyrolyzed product remained porous (SBET ∼ 116 m2 g−1, entry 8 in Table 1), although with a non-homogeneous pore distribution. Obviously, β-CD turns into (micro)porous networks with the assistance of an IL, and the IL acts as a “pore-inducing agent” at 400 °C. The oven yield remains almost constant throughout all experiments, ending up with 35.8 wt% at a high β-CD content of 93 wt% in the starting mixture.
To create a more complete picture of this unique behaviour, a wider reaction temperature range from 300 to 1000 °C was investigated at a constant β-CD content of 73 wt%. As shown in Fig. 2, the highest SBET value was observed at 500 °C. The solid achieved below 400 °C was nonporous, and above 800 °C it seems that the pores formed earlier at intermediate temperatures collapse. The effect of the dwelling time was also studied at 400 °C. The microporous framework, once built up, was proven to be stable upon further thermal annealing, and only a small deviation in SBET was seen (Fig. 2).
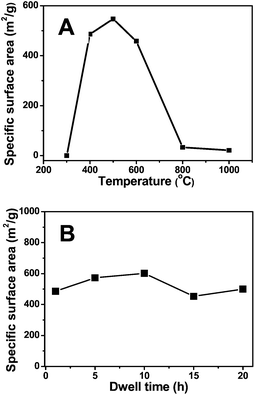 | ||
| Fig. 2 (A) Plot of the specific surface area vs. the carbonization temperature. (B) Plot of specific surface area vs. the dwelling time. The sample was prepared by mixing 73 wt% of β-CD with 27 wt% of IL. Heating program: 5 °C min−1 to the final temperature and dwelling for different periods at the final temperature. | ||
To judge the generality, glucose, cellulose and starch were also employed as a carbohydrate base at a component fraction of 57 wt% (entries 9–11 in Table 1). As expected, all products presented a high surface area of around 600 m2 g−1 with a satisfactory oven yield exceeding 35%. In contrast, when several synthetic polymers, such as poly(ethylene glycol) (PEG), polystyrene (PS) and poly(acrylic acid) (PAA), were treated in the same manner, the reaction ended up with poorly porous solids and low yields (entries 12–14 in Table 1).
As glucose and polysaccharides are abundant and industrially available for minimal prices as raw materials and “micro-structured” templates (such as paper), respectively, the accelerated carbonization/activation function of ILs can be readily expanded to produce porous carbonaceous materials from a multiplicity of pre-shaped templates. For practical reasons, we complemented the IL with its polymer form, poly(3-cyanomethyl-1-vinyl-imidazolium Tf2N).35–38 The advantage of PILs over ILs in this case lies in the superior processability of polymers, which allows us to coat also the inner surface of the hierarchical structures of biotemplates more conveniently, homogeneously and efficiently by simple impregnation; therefore a “dry” and touchable green body product is obtained for further processing. In contrast, it is difficult for ILs to form a thin film on a planar surface with thickness down to nanoscale, and dewetting is another problem due to their mobile/fluidic feature. The fact that a PIL also acts as an “activation agent” was quantified in a model reaction with a 57 wt% β-CD mixture at 400 °C. A microporous solid with SBET ∼ 530 m2 g−1 was received (entry 17 in Table 1). Similar results are obtained at 500 °C at 73 wt% β-CD (entry 18 in Table 1).
The conversion of a biotemplate with extended shape was first tested with a commercially available cellulose filter membrane and coffee filter paper (entries 15, 16 and 19), both of which are made mainly of natural cellulose fibers. In the doping step, an IL solution in ethanol was spread onto the biotemplate. This was carried out with caution to avoid inhomogeneity of IL deposition. After 400 °C treatment, the SBET reached 680 m2 g−1 for the filter membrane and 720 m2 g−1 for the coffee filter. In the case of a PIL, the procedure is more convenient. The filter membrane or paper was directly immersed in a PIL solution to allow the PIL macromolecules to soak into the interstitial voids of the cellulose paper. After removal of the excess solution via soft vacuum filtration, the PIL/cellulose composite paper was dried at 80 °C until constant weight to calculate the PIL content, which is 12 wt% (Fig. 3A). After thermal treatment at 400 °C, a flat carbonaceous membrane with SBET ∼ 400 m2 g−1 (Fig. 3B) was obtained (entry 19 in Table 1). The radial shrinkage of the membrane after pyrolysis was determined to be ca. 10–15%. To our surprise, the membrane preserved essentially the mechanical flexibility of the original paper. As illustrated in Fig. 3C, it can be bent to a large extent at will and cut into different forms, such as stripes shown in Fig. 3D and E. This feature is of high convenience to assemble it into filtration and separation devices, which require high mechanical stability and flexibility. It is noteworthy that without IL/PIL doping, the carbonaceous membrane showed only little porosity and a rather unsatisfying mechanical behavior. Fig. 3F shows the tensile testing experiment results. When produced from a PIL-doped filter membrane, the carbon membrane reaches not only a four times higher breaking strength but also a 50% larger rupture strain. This clearly illustrates the onset of a new condensation chemistry.
 | ||
| Fig. 3 Photographs of (A) PIL wetted filter paper, (B) after carbonization at 400 °C, (C) bending test of the carbonaceous membrane paper, and (D) a carbonaceous stripe cut from a carbonaceous membrane. (E) The bending test of the stripe. (F) Plots of the tensile testing experiments of carbon membranes derived from the pure cellulose filter membrane (dotted line) and from the PIL-doped filter membrane (solid line). | ||
Beside cellulose membranes or papers, natural cotton could also be taken to “carbonize” it under preservation of the outer shape and microtexture to end up in a condensed product with good mechanical performance (entry 20 in Table 1). The particular features of cotton are its very high absolute porosity and the assorted superior deformability, which can be pressed or engineered into various shapes (note that cotton is one of the oldest gas permeable sealant materials of chemistry). Typically, a handful of cotton in its original state (Fig. 4A) was soaked in a PIL solution for surface coating. The excess solution was mechanically pressed out or sucked away by vacuum filtration. After drying at room temperature, it was squeezed between two glass plates or into a cavity and in situ heat-transferred into a film (Fig. 4B) or into a differently shaped monolithic object. A carbonaceous monolith (Fig. 4C) was for instance generated by densely pressing the coated cotton precursor into a test tube. Carbonizing the PIL/cotton composite in its original form produces an irregularly shaped fluffy foam that can be conveniently cut into any well-defined geometry, such as a cuboid geometry shown in Fig. 4D. The carbonaceous cuboid foam is again sponge-like and deformable. In a mechanical test shown in Fig. 4E and F, it recovers itself readily after being compressed by 2000 times of its own weight.
 | ||
| Fig. 4 Photographs of (A) natural cotton, (B) carbonaceous thin film derived from a PIL/cotton composite film, (C) carbonaceous monolith derived from a PIL/cotton composite cylinder, and (D) cut carbonaceous cuboid from a carbonized PIL/cotton composite. (E and F) A shape retention test on the composite. | ||
With the fairly good preservation of the macroscopic shape, it is an interesting question what occurs to the interior microscopic texture of the cellulose throughout pyrolysis. To analyze this, the intact filter paper, PIL-coated-filter paper and the carbonaceous membrane copy were subjected to scanning electron microscopy (SEM) characterization. Fibers of 10–40 μm in diameter are clearly seen in these samples (Fig. 5A–C), essentially no changes of the microtexture are found even with careful observation. The fiber skeleton holds firmly its architecture when converted from cellulose to the carbonaceous fibers. Therefore, the described low temperature carbonization process retains the morphology well, both macro- and microscopically.
 | ||
| Fig. 5 SEM images of an intact cellulose filter membrane (A), a PIL-decorated cellulose filter membrane (B), and its corresponding carbonaceous membrane after PIL activation (C). | ||
To illustrate one of the many possible functions of these biotemplate-derived porous carbonaceous materials, we performed dye sorption experiments in aqueous solutions (Fig. 6). The porous carbonaceous sponge derived from the cotton/PIL pad quickly absorbed the aqueous solution of methylene blue dye and clean water was released by squeezing it.
 | ||
| Fig. 6 (A–C) Photographs illustrating the sorption of methylene blue dye from aqueous solution by a carbonaceous foam derived from the PIL/cotton composite. (D–F) Photographs of the initial aqueous solution of crystal violet (D), 5 min after treatment by pure cotton-based carbon foam (E), and 5 min after treatment by a cotton/PIL-based carbon foam (F). | ||
A video (Video 1) of the actual experiment is included in the ESI.† In an alternative example, an aqueous solution of methylene blue was decolorized after passing through a short column filled with the carbon sponge, which demonstrates the efficient removal of organic dyes from the aqueous phase. The PIL added to the cotton played an important role in accelerating the absorption kinetics. In a control experiment of absorbing crystal violet from aqueous solution, the porous carbon foam derived from cotton/PIL (Fig. 6F) presented a much faster kinetics in the dye sorption than the porous carbon foam derived from pure cotton (Fig. 6E).
Inertness against oxidation is another classical feature of N-heterocycles and N-doped carbons.39 Here, we tested our carbon foams (cylinders prepared at 400 °C from cotton with/without PIL) by wetting the foam with 500 wt% of ethanol and burning it in air (Fig. 7A). This test was repeated 4 times to monitor the shape and mass change after each cycle. As observed, the carbon foam derived from pure cotton collapsed after the 1st cycle already, while the PIL/cotton carbon remained intact. The mass vs. firing cycle plot (Fig. 7B) indicates that the 1st firing cycle burnt away 80.3 wt% of the cotton-based carbon product, while only 4.1 wt% of mass loss occurred for the cotton/PIL carbon foam. Within 3 cycles, only the cotton/PIL carbon foam preserved its shape and 85 wt% of its mass. Under even harsh conditions, for examples, when repeatedly exposed to the flame of a butane/propane (70:30) gas burner, the foam maintained well its shape and size, without quenching fire (Fig. 7C). In a real-time video (Video 2 in the ESI†), it is observed that the foam bottom could be turned to a red blaze in the central flame (∼1300 °C), but quenched immediately when pulled out (or even treated with blowing air). This illustrates that the material is even robust against co-firing, a really exceptional property for high surface area carbons.
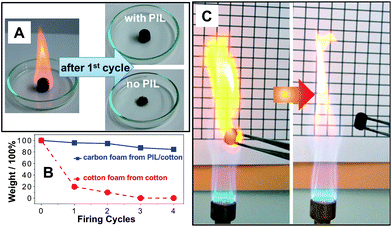 | ||
| Fig. 7 (A) Photographs of the 1st firing cycle test of the carbon foams (absorb 500 wt% of ethanol and burn in air). Left: a burning ethanol-wetted foam. Right: foams after the 1st firing cycle test. (B) Plot of the carbon foam mass vs. the firing cycle. (C) Photographs illustrating the fire-retardancy of a carbonaceous foam by repeatedly firing the sample using a butane/propane gas burner. | ||
The “low temperature carbonization/activation” function of ILs/PILs for biomolecules and biomass resembles a chemical activation process, but at much lower temperatures and lower chemical demand than standard carbonization. Certain intrinsic chemical reactions between the IL species and the sugar unit must occur at a molecular level at the described temperatures. Fig. 8 illustrates the thermogravimetric analysis (TGA) curves of IL, β-CD and a mixture of IL/β-CD (weight ratio ∼ 43/57, entry 5 in Table 1), which help understand the structural preservation of biotemplates during the carbonization. It is clearly found that the mixing lowers significantly the onset of mass loss, indicating that a new chemistry is involved. The primary weight loss starts at 240 °C, ca. 70 °C lower than in β-CD and even 90 °C than in the IL. This is exactly the critical and important temperature reduction as now carbonization sets in practically before the thermal degradation of cellulose starts, i.e. the biological structure can be preserved throughout the chemical conversions, while sintering is suppressed.
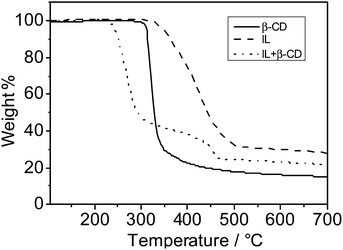 | ||
| Fig. 8 TGA curves of IL, β-CD and the mixture of IL/β-CD in a weight ratio of 43/57. The experiments were conducted under a nitrogen atmosphere at a heating rate of 10 °C min−1. | ||
Dehydration of sugar with sulfuric acid is a well-known chemical reaction, and several groups reported earlier that sugar units could be catalytically dehydrated at reduced temperatures in some ILs.31,40 Our TGA analysis matches well with these findings, implying that the early weight loss of the IL/β-CD mixture can be attributed to a catalytic dehydration step. Analysis of the fragmentation products indicated a rather complex decomposition scenario rather than simple water elimination. The nitrile groups of the IL moiety must play a key role in cross-linking the scaffold.30–33 A more detailed study on the ongoing chemical processes is in progress and will be reported elsewhere.
Conclusions
In conclusion, we demonstrated in this report a simple transformation of polysaccharide-based biomaterials into porous, shapable carbonaceous frameworks, simply by coating them with an IL/PIL, followed by thermal transformation under a nitrogen atmosphere at 400 °C. The IL and PIL act as efficient carbonization/activation agents to promote conversion and pore generation in the biomaterial even at comparably low temperatures. Their role can be understood as an “activation agent” with own structural contribution. It was shown that through this route, useful carbonaceous products of different geometries could be obtained from similarly shaped biotemplates acting as “green bodies” for this process. To our surprise, the carbonized hybrid still offers comparably good mechanical properties, such as bendability of membranes or recovery of foams, which attributes some flexibility to the as-made product. Here, it must be stated that elasticity and microporosity usually exclude each other, structure-wise, both in polymers as well as in carbons.41 The heteroatoms incorporated in the final products from the IL/PIL precursors, such as nitrogen here, add extra value to the carbon products, as shown here by the brought in extraordinary oxidation stability. We believe that the concept of PIL activation opens an alternative avenue to produce appealing functional carbonaceous materials in a rather sustainable way.Acknowledgements
The authors thank the Max Planck Society for the financial support. Y. Men would like to acknowledge the China Scholarship Council for a scholarship.Notes and references
- M.-M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- L. Liu, D. He, G.-S. Wang and S.-H. Yu, Langmuir, 2011, 27, 7199–7206 CrossRef CAS PubMed.
- X.-L. Wu, T. Wen, H.-L. Guo, S. Yang, X. Wang and A.-W. Xu, ACS Nano, 2013, 7, 3589–3597 CrossRef CAS PubMed.
- A. Walther, J. V. I. Timonen, I. Díez, A. Laukkanen and O. Ikkala, Adv. Mater., 2011, 23, 2924–2928 CrossRef CAS PubMed.
- J. Hilgert, N. Meine, R. Rinaldi and F. Schuth, Energy Environ. Sci., 2013, 6, 92–96 CAS.
- F. Schüth, Chem. Ing. Tech., 2011, 83, 1984–1993 CrossRef.
- K. Ariga, Q. Ji, M. J. McShane, Y. M. Lvov, A. Vinu and J. P. Hill, Chem. Mater., 2011, 24, 728–737 CrossRef.
- Z. Weng, Y. Su, D.-W. Wang, F. Li, J. Du and H.-M. Cheng, Adv. Energy Mater., 2011, 1, 917–922 CrossRef CAS.
- H.-W. Liang, Q.-F. Guan, L.-F. Chen, Z. Zhu, W.-J. Zhang and S.-H. Yu, Angew. Chem., Int. Ed., 2012, 51, 5101–5105 CrossRef CAS PubMed.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- R. Li, K. Parvez, F. Hinkel, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2013, 52, 5535–5538 CrossRef CAS PubMed.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- X. Sun, J. Liu and Y. Li, Chem.–Eur. J., 2006, 12, 2039–2047 CrossRef CAS PubMed.
- F. Khan, D. Walsh, A. J. Patil, A. W. Perriman and S. Mann, Soft Matter, 2009, 5, 3081–3085 RSC.
- E. M. Lambert, C. Viravaidya, M. Li and S. Mann, Angew. Chem., Int. Ed., 2010, 49, 4100–4103 CrossRef CAS PubMed.
- G. P. Mane, S. N. Talapaneni, C. Anand, S. Varghese, H. Iwai, Q. Ji, K. Ariga, T. Mori and A. Vinu, Adv. Funct. Mater., 2012, 22, 3596–3604 CrossRef CAS.
- Z.-Y. Wu, C. Li, H.-W. Liang, J.-F. Chen and S.-H. Yu, Angew. Chem., Int. Ed., 2013, 52, 2925–2929 CrossRef CAS PubMed.
- O. Ioannidou and A. Zabaniotou, Renewable Sustainable Energy Rev., 2007, 11, 1966–2005 CrossRef CAS PubMed.
- B.-H. Han, W. Zhou and A. Sayari, J. Am. Chem. Soc., 2003, 125, 3444–3445 CrossRef CAS PubMed.
- H. Wang, Q. Gao and J. Hu, J. Am. Chem. Soc., 2009, 131, 7016–7022 CrossRef CAS PubMed.
- F. Rodríguez-Reinoso, M. Molina-Sabio and M. T. González, Carbon, 1995, 33, 15–23 CrossRef.
- K. Mohanty, M. Jha, B. C. Meikap and M. N. Biswas, Chem. Eng. Sci., 2005, 60, 3049–3059 CrossRef CAS PubMed.
- D.-C. Guo, J. Mi, G.-P. Hao, W. Dong, G. Xiong, W.-C. Li and A.-H. Lu, Energy Environ. Sci., 2013, 6, 652–659 CAS.
- J. P. Paraknowitsch and A. Thomas, Macromol. Chem. Phys., 2012, 213, 1132–1145 CrossRef CAS.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS PubMed.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS PubMed.
- Y. Men, X.-H. Li, M. Antonietti and J. Yuan, Polym. Chem., 2012, 3, 871–873 RSC.
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS PubMed.
- J. S. Lee, H. Luo, G. A. Baker and S. Dai, Chem. Mater., 2009, 21, 4756–4758 CrossRef CAS.
- J. S. Lee, R. T. Mayes, H. Luo and S. Dai, Carbon, 2010, 48, 3364–3368 CrossRef CAS PubMed.
- J. S. Lee, X. Wang, H. Luo, G. A. Baker and S. Dai, J. Am. Chem. Soc., 2009, 131, 4596–4597 CrossRef CAS PubMed.
- J. S. Lee, X. Wang, H. Luo and S. Dai, Adv. Mater., 2010, 22, 1004–1007 CrossRef CAS PubMed.
- Q. Zhao, T.-P. Fellinger, M. Antonietti and J. Yuan, J. Mater. Chem. A, 2013, 1, 5113–5120 CAS.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS PubMed.
- Q. Zhao, T.-P. Fellinger, M. Antonietti and J. Yuan, Macromol. Rapid Commun., 2012, 33, 1149–1153 CrossRef CAS PubMed.
- Q. Zhao, M. Yin, A. P. Zhang, S. Prescher, M. Antonietti and J. Yuan, J. Am. Chem. Soc., 2013, 135, 5549–5552 CrossRef CAS PubMed.
- Q. Zhao, P. Zhang, M. Antonietti and J. Yuan, J. Am. Chem. Soc., 2012, 134, 11852–11855 CrossRef CAS PubMed.
- J. P. Paraknowitsch, A. Thomas and M. Antonietti, J. Mater. Chem., 2010, 20, 6746–6758 RSC.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738–755 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Videos of the dye sorption experiment and the fire-retardant experiment. See DOI: 10.1039/c3ta12302b |
| This journal is © The Royal Society of Chemistry 2013 |