 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible hydrogen storage in Mg(BH4)2/carbon nanocomposites†
Yigang
Yan‡
*a,
Yuen S.
Au‡
b,
Daniel
Rentsch
c,
Arndt
Remhof
a,
Petra E.
de Jongh
*b and
Andreas
Züttel
a
aEMPA, Swiss Federal Laboratories for Materials Science and Technology, CH 8600 Dübendorf, Switzerland. E-mail: yigang.yan@empa.ch; Fax: +41 58 765 40 22; Tel: +41 58 765 40 82
bInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: p.e.dejongh@uu.nl
cEMPA, Swiss Federal Laboratories for Materials Science and Technology, Functional Polymers, 8600 Dübendorf, Switzerland
First published on 19th July 2013
Abstract
Mg(BH4)2 exhibits a high hydrogen content of 14.9 wt% and thermodynamic stability in the overall decomposition reaction that corresponds to hydrogen desorption at around room temperature. However, the potential applications in hydrogen storage are restricted by high kinetic barriers. In this study, we show the synthesis of Mg(BH4)2/carbon nanocomposites by ball milling of MgH2 nanoparticles supported on carbon aerogel in a B2H6/H2 atmosphere. The nanocomposite exhibits a lower kinetic barrier as compared to bulk Mg(BH4)2. The temperature for major hydrogen desorption is decreased to 160 °C. Furthermore, re-formation of Mg(BH4)2 after full dehydrogenation is achieved under mild conditions (200 °C and 80 to 150 bar H2) in the nanocomposite. This work demonstrates nanoengineering as an effective approach to realize the reversible hydrogen storage of Mg(BH4)2 under mild conditions.
Introduction
Hydrogen is regarded as a clean energy carrier for the future.1,2 Achieving safe and efficient hydrogen storage is one of the key technological challenges for the wide use of hydrogen as a fuel. Metal borohydrides, with high gravimetric and volumetric densities of hydrogen, have been extensively investigated for solid state hydrogen storage.3–5 Among them, magnesium borohydride, i.e. Mg(BH4)2, shows a hydrogen capacity of 14.9 wt% and an attractive enthalpy change of −39 kJ mol−1 H2 in dehydrogenation according to eqn (1).6–8| Mg(BH4)2 → MgB2 + 4H2 | (1) |
Although the dehydrogenation enthalpy change allows for hydrogen desorption at around room temperature, the major dehydrogenation reaction was observed above 250 °C.9–11 Rehydrogenation of MgB2 requires high hydrogen pressure and high temperature (e.g. 95 MPa H2, 400 °C).12 Numerous studies have revealed that the decomposition reaction (1) occurs in multiple steps involving the formation of Mg–B–H ternary intermediates such as Mg(B3H8)213 and MgB12H12.11,13–15 The partial decomposition of Mg(BH4)2 to Mg(B3H8)2 can be reversed at 250 °C and 120 bar with a hydrogen release amount of 2.5 wt%. The stable [B12H12]2− species is an unwanted intermediate that reduces the rehydrogenation capacity. The migration of atoms such as Mg and B in the formation process of [B12H12]2− from the reconstruction of [BH4]− and the breaking of B–H covalent bonds in [BH4]− are considered to result in slow kinetics of the dehydrogenation reaction of Mg(BH4)2.15
An effective approach to accelerate the reaction kinetics is to reduce the particle size and form nanomaterials.16–19 The faster kinetics is attributed to the shortened diffusion distances and lowered activation energy. By ball milling of MgB2 in a high pressure hydrogen atmosphere, nanostructured Mg(BH4)2 was formed with enhanced hydrogen desorption properties.20,21 Incorporation in a nanoporous scaffold can be used to avoid sintering in the de-/absorption cycles of nanostructured Mg(BH4)2.22,23 Infiltration of Mg(BH4)2 in activated carbon via wet impregnation was carried out, resulting in a reduced hydrogen release temperature.22 The drawbacks of solvent residue and unconfined Mg(BH4)2 were reported in this method. In recent research, the temperature of hydrogen desorption was reduced to 155 °C by combining nanoconfinement and a Ni catalyst.23 However, the regeneration of Mg(BH4)2 under mild conditions from the fully dehydrogenated state has not been reported so far.
Previous studies have demonstrated that solvent free metal borohydrides, such as LiBH4, Mg(BH4)2, Ca(BH4)2 and Y(BH4)3, can be synthesized via reaction between the corresponding metal hydrides and B2H6.24,25 In this study, we applied this method to synthesize nanostructured Mg(BH4)2. MgH2 nanoparticles supported on carbon served as precursors and were ball milled in a B2H6/H2 atmosphere to form Mg(BH4)2 according to eqn (2).
| MgH2 + B2H6 → Mg(BH4)2 | (2) |
To investigate the influence of different carbon additives, carbon aerogel (porous structure) and graphite (nonporous structure) were compared.
We observed the formation of nanostructured Mg(BH4)2, which exhibits a lower temperature for hydrogen release as compared to bulk Mg(BH4)2. Re-formation of the nanostructured Mg(BH4)2 also occurred under mild conditions.
Experimental
Carbon aerogel (CA) was synthesized through resorcinol–formaldehyde condensation.26 Resorcinol (Sigma Aldrich 99%, 8.65 g, 79 mmol), formaldehyde (Fisher Chemical, analytical reagent 37–41%, stabilized by 12% methanol, 12.89 g, 158 mmol) and sodium carbonate (Acros Organics, anhydrous pure, 0.017 g, 0.16 mmol) were dissolved in deionized water. After curing (1 day at room temperature, 1 day at 60 °C and 3 days at 90 °C), the obtained gel was cooled, powdered and subsequently washed with acetone. The gel was pyrolized in a tubular oven at 800 °C for 10 hours in an Ar flow and stored in an Ar purified glovebox (MBraun Labmaster) after cooling to room temperature. Non-porous graphite (KS-6), obtained from Timcal, was pre-treated at 600 °C overnight in an Ar flow. Unless otherwise stated, all sample handlings were carried out in the Argon purified glovebox (O2 < 0.1 ppm, H2O < 0.1 ppm) to prevent (hydro-)oxidation.MgH2 nanoparticles supported on carbon were prepared via melt infiltration.27 In a typical experiment, 0.9 g of CA or KS-6 was mixed and ground with 0.1 g of MgH2 in a mortar to obtain a mixture with 10 wt% MgH2. The mixture was placed in a graphite cup which was inserted in a stainless steel sample holder. The sample was then heated in a tubular oven at 10 °C min−1 to 658 °C and dwelled for 24 min under an Ar flow. Hydrogenation of the samples was performed at 80 bar H2 and 300 °C for 10 h in an autoclave (Parr). The obtained MgH2 supported on carbon aerogel (CA) and on non-porous graphite (KS-6) are labeled as MH-CA and MH-KS6, respectively. All samples investigated in this study are listed in Table 1.
| Carbon supports | Infiltrated with MgH2 | Ball milled under B2H6 |
|---|---|---|
| Carbon aerogel (CA) | MH-CA | MBH-CA |
| Non-porous graphite (KS-6) | MH-KS6 | MBH-KS6 |
LiZn2(BH4)5, synthesized by ball milling a mixture of ZnCl2 (Sigma-Aldrich, 99.9%) and LiBH4 (Katchem, 95%), was used as a B2H6 source releasing B2H6 and H2 above 100 °C according to eqn (3).
| LiZn2(BH4)5 → LiH + 2Zn + 5/2B2H6 + 2H2 | (3) |
500 mg of MH-CA or MH-KS6 was filled in the milling vial connected to the B2H6 source. The whole system was purged with hydrogen and evacuated. Subsequently, the diborane source was heated up to 150 °C to fill the system with a B2H6/H2 (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) atmosphere. After the completion of diborane source desorption, ball milling was carried out for 3 days and the progress of the subsequent reaction was monitored by the pressure change in the milling vial. The samples of MH-CA and MH-KS6 after B2H6 treatment were labeled as MBH-CA and MBH-KS6, respectively. Ideally, 10 wt% of MgH2 in MH-CA could result in 18 wt% of Mg(BH4)2 (according to eqn (2)) in MBH-CA. In order to compare the effect of different carbon addition methods, a physical mixture of Mg(BH4)2 with CA, labeled as MBH-CA-PM, was prepared by ball milling of the bulk Mg(BH4)2 reference (Sigma-Aldrich, 95%) and CA in the mass ratio of 1 to 5 under 10 bar H2 for 3 days. Also, pure CA was ball milled in B2H6/H2 and H2 atmosphere, respectively, to investigate the possible reactions between CA with B2H6 and H2.
:4) atmosphere. After the completion of diborane source desorption, ball milling was carried out for 3 days and the progress of the subsequent reaction was monitored by the pressure change in the milling vial. The samples of MH-CA and MH-KS6 after B2H6 treatment were labeled as MBH-CA and MBH-KS6, respectively. Ideally, 10 wt% of MgH2 in MH-CA could result in 18 wt% of Mg(BH4)2 (according to eqn (2)) in MBH-CA. In order to compare the effect of different carbon addition methods, a physical mixture of Mg(BH4)2 with CA, labeled as MBH-CA-PM, was prepared by ball milling of the bulk Mg(BH4)2 reference (Sigma-Aldrich, 95%) and CA in the mass ratio of 1 to 5 under 10 bar H2 for 3 days. Also, pure CA was ball milled in B2H6/H2 and H2 atmosphere, respectively, to investigate the possible reactions between CA with B2H6 and H2.
N2-physisorption (Micromeritics TriStar 3000) was performed at 77 K for porosity analysis. The total pore volume was obtained at P/P0 = 0.997. The samples were prepared in a capped quartz sample tube in the glovebox and were directly measured. The mesopore size distribution was analyzed with the Barrett–Joyner–Halenda (BJH) method from the adsorption branch of the isotherm with a carbon black thickness equation as reference.
X-ray diffraction (XRD) measurements were performed using a Bruker D8 diffractometer equipped with a Goebel mirror selecting Cu Kα radiation (λ = 1.5418 Å) and a linear detector system (Vantec). Samples for XRD measurements were filled and sealed in an inert atmosphere into glass capillaries (diameter, 0.7 mm; wall thickness, 0.01 mm).
For transmission electron microscopy (TEM) observation, a small amount of the sample was placed on a 200 mesh Cu grid coated with a carbon polymer film in the glovebox. The sample was exposed for 5 s to air during the insertion of the sample holder into the microscope. Images were recorded with an FEI Tecnai 20F (equipped with a Field Emission Gun) and operated at 200 kV in bright field mode. Elemental analysis was performed with an energy-dispersive X-ray spectrometer (EDX) and an electron energy loss spectrometer (EELS), which were connected to the microscope.
Solid state 11B magic angle spinning nuclear magnetic resonance (MAS NMR) experiments were performed on a Bruker Avance-400 NMR spectrometer using a 4 mm CP-MAS probe. The 11B NMR spectra were recorded at 128.38 MHz at 12 kHz sample rotation applying a Hahn echo pulse sequence to suppress the broad background resonance of boron nitride of the probe. Pulse lengths of 1.5 μs (π/12 pulse) and 3.0 μs were applied for the excitation and echo pulses, respectively. 11B NMR chemical shifts are reported in parts per million (ppm) externally referenced to a 1 M B(OH)3 aqueous solution at 19.6 ppm as an external standard sample. The reference sample K2B12H12 was purchased from Katchem.
Thermogravimetric (TG) measurements were carried out by using a magnetic suspension balance (Rubotherm), with a heating rate of 5 °C min−1 and an Ar flow of 200 mL min−1. Temperature programmed desorption combined with mass-spectrometry (TPD-MS) (Micromeritics AutoChem II 2920 and Pfeiffer Vacuum OmniStar™) measurements were performed to analyze the decomposition reaction of the samples. Approximately 50 mg of the sample was taken for each measurement. The experiments were carried out in an Ar flow of 25 mL min−1 with a temperature ramp of 5 °C min−1 up to 500 °C. The masses of B2H6 and H2 were recorded simultaneously over time.
The apparent activation energy (Ea) of hydrogen desorption reaction was determined by using the Kissinger method involving the application of eqn (4).28
 | (4) |
Results
Characterization
CA exhibits a porous structure with a surface area (SBET) of 605 m2 g−1, a total pore volume of 0.52 cm3 g−1 and an average pore size of 10 nm. After infiltration with Mg, the pore volume of CA is slightly decreased, as shown in Fig. 1. The pore volume is reduced to zero in MBH-CA, indicating that the porous structure of CA was destroyed during the ball milling process. | ||
| Fig. 1 Pore size distribution of CA, MH-CA and MBH-CA. | ||
The XRD patterns of MH-CA and MH-KS6 before and after ball milling in B2H6 are shown in Fig. 2. Diffraction peaks of MgH2 and Mg are observed in samples MH-CA and MH-KS6, indicating the presence of crystalline MgH2 and Mg on the surfaces of CA and KS6. After ball milling in B2H6/H2, the reflections of Mg and MgH2 disappear in MBH-CA, while the peak intensities of graphite, MgH2 and Mg are reduced in MBH-KS6. No observation of new reflections implies that the newly formed compounds by ball milling in a B2H6/H2 atmosphere are amorphous or nanocrystalline. Thereby, NMR spectroscopy is a suitable choice for further phase identification.
 | ||
| Fig. 2 XRD patterns of carbon supported MgH2 samples: MH-CA and MH-KS6, and samples after ball milling in B2H6/H2: MBH-CA and MBH-KS6. | ||
The 11B MAS NMR spectrum of MBH-CA in Fig. 3(a) shows two major resonances at −41 and −16 ppm. The resonance at −41 ppm shows the same chemical shift as the reference sample Mg(BH4)2, indicating the formation of Mg(BH4)2 in MBH-CA. The signal at −16 ppm belongs to the [B12H12]2− species, which may be formed by a reaction between the newly formed Mg(BH4)2 compound and B2H6. The presence of boron in MBH-CA is also confirmed by EELS as shown in Fig. S1 (ESI†). By integrating the individual peak area in Fig. 3(a), the mass ratio of Mg(BH4)2 to MgB12H12 is estimated to be 1 to 4.5. Considering that the initial amount of Mg is 10 wt% in MH-CA, the amount of Mg(BH4)2 and MgB12H12 in MBH-CA is estimated to be 6 and 27 wt%, respectively, with the remaining 67 wt% being carbon. 11B MAS NMR measurements on MBH-KS6 were not successful, due to the presence of unpaired electrons in graphite leading to a loss of the NMR signal.29,30
 | ||
| Fig. 3 11B MAS NMR spectra of (a) as-synthesized MBH-CA, and MBH-CA heated at (b) 90 °C, (c) 250 °C and (d) 360 °C, respectively, compared to the reference samples K2B12H12 and Mg(BH4)2. | ||
Hydrogen desorption properties
The hydrogen desorption behaviors of MBH-CA, MBH-KS6 and two reference samples were examined by TPD and MS. In Fig. 4(a), the desorption of the bulk Mg(BH4)2 reference starts from 250 °C. The three desorption peaks present within the temperature range of 270 to 400 °C apparently correspond to three decomposition steps.9 Only traces of B2H6 are detected by MS, as shown in Fig. 4(b). MBH-CA-PM shows a broad desorption peak within the same temperature region as bulk Mg(BH4)2.
The hydrogen desorption behaviors of MBH-CA and MBH-KS6 (Fig. 4(a)) are quite different from those of the reference samples. The desorption of MBH-CA starts around 100 °C, approximately 150 °C lower than that of bulk Mg(BH4)2, which is in good agreement with TG results in Fig. S2 (ESI†). The first desorption step of MBH-CA occurs at 160 °C and the second step at 335 °C. The simultaneous MS measurement in Fig. 4(b) reveals that the gas release in the TPD measurement is mainly due to the release of H2 and only a slight amount of B2H6 is detected. The first desorption peak of MBH-KS6 is also found at 160 °C (Fig. 4(a)). In addition, two other desorption peaks are observed at 285 and 336 °C, within a similar temperature range as the decomposition of bulk Mg(BH4)2. In summary, Mg(BH4)2 supported on different carbon matrixes shows different desorption behaviors.
Note that the release of B2H6 occurs at a lower temperature (below 100 °C) than that of H2 for both MBH-CA and MBH-KS6 (shown in Fig. 4(b)). The released B2H6 is possibly attributed to physisorption or trapping of B2H6 on carbon during the ball milling process. To verify this hypothesis, pure CA was ball milled under a B2H6/H2 and a pure H2 atmosphere, respectively, and examined by MS. In Fig. 5, MS data of MBH-CA and milled CA are compared. It is clearly observed that H2 is also desorbed from the ball milled CA in B2H6/H2 but without distinct peaks, and the H2 release profile of CA appears to follow the baseline of that of MBH-CA. Defects could be created in the carbon structure during the ball milling process, possibly leading to the formation of C–B–H chemical bonds. Ball milling of CA in the pure H2 atmosphere only results in traces of hydrogen release above 350 °C, possibly due to the formation of stable C–H bonds. Therefore, the hydrogen desorption peaks of MBH-CA at 160 and 335 °C (Fig. 5) can be ascribed to the decomposition of Mg(BH4)2. The ball milled CA under B2H6/H2 also shows a different B2H6 release behavior compared to MBH-CA, i.e. two B2H6 peaks at 120 and 250 °C, respectively, and the B2H6 release at 120 °C is very limited. This indicates that the B2H6 release of MBH-CA mainly originates from Mg(BH4)2.
 | ||
| Fig. 5 MS profiles of MBH-CA (blue, dashed line), and CA ball milled in a B2H6/H2 (gray line) and CA ball milled in a H2 atmosphere (black line) for 3 days. | ||
To clarify the evolution of the boron species in MBH-CA during the decomposition process, MBH-CA was decomposed at different temperatures and examined by 11B MAS NMR. As shown in Fig. 3, the resonance assigned to [BH4]− is present at 90 °C and disappears at 250 °C. The resonance centered at −16 ppm assigned to the [B12H12]2− species is present up to 360 °C, indicating the high stability of MgB12H12.
Reversibility
According to the results in Fig. 4 and 5, the hydrogen desorption from Mg(BH4)2 in MBH-CA shows apparently two steps and is completed below 360 °C. To examine the reversibility of Mg(BH4)2 supported on CA, the sample of MBH-CA was decomposed at 360 °C and rehydrogenated under 150 bar H2 at 200 or 270 °C and under 80 bar at 200 °C, respectively. The 11B MAS NMR spectra of the rehydrogenated samples are shown in Fig. 6. The re-formation of Mg(BH4)2 is observed at both 200 and 270 °C under 150 bar. By carrying out rehydrogenation at a lower H2 pressure of 80 bar at 200 °C, traces of Mg(BH4)2 were formed as well. In contrast, the rehydrogenation of bulk Mg(BH4)2 requires higher pressures (e.g. 900 to 950 bar of H2) and higher temperatures (e.g. 390 to 400 °C).12,29 | ||
| Fig. 6 11B MAS NMR spectra of MBH-CA rehydrogenated at 200 °C and 270 °C, respectively. | ||
Fig. 7 shows the hydrogen desorption of the sample rehydrogenated at 270 °C. In the 2nd cycle, less hydrogen is released and the hydrogen desorption temperature is increased to 200 °C, indicating the deterioration of the H2 desorption properties. In addition, desorption of B2H6 was also observed at 120 °C, similar to that of the as-synthesized sample (Fig. 4).
 | ||
| Fig. 7 MS profiles of MBH-CA rehydrogenated at 270 °C and 150 bar for 20 h. | ||
Discussion
Mg(BH4)2/carbon nanocomposites were successfully synthesized by ball milling of MgH2 nanoparticles supported on carbon aerogel in a B2H6/H2 atmosphere at room temperature, exhibiting a lower desorption temperature and reversibility under milder conditions as compared to bulk Mg(BH4)2.The reduced hydrogen desorption temperature of MBH-CA is attributed to the formation of nanostructured Mg(BH4)2. Reducing the particle size of hydrides to nanoscale improves the H2 sorption kinetics and decreases the reaction temperature.30–34 The improvement in kinetics by a “nanosize effect” arises from the lowered activation barrier and the shortened diffusion distance of atoms in the nanoscale structures. The activation energies (Ea) of the first hydrogen desorption step of MBH-CA and bulk Mg(BH4)2 are compared in Fig. 8. In the sample MBH-CA, the nanostructured Mg(BH4)2 shows a markedly lowered Ea of 102 ± 6 kJ mol−1, compared to 340 ± 11 kJ mol−1 for bulk Mg(BH4)2.
 | ||
| Fig. 8 Kissinger plot of the first hydrogen desorption step for MBH-CA and bulk Mg(BH4)2. | ||
Possibly due to the accelerated kinetics, the hydrogen desorption process of Mg(BH4)2 is altered from an apparently 3-step reaction for the bulk Mg(BH4)2 to a 2-step reaction for MBH-CA. A catalytic effect of the carbon matrix was not found for improving the kinetics in MBH-CA. When carbon aerogel was introduced into bulk Mg(BH4)2 in the mass ratio of 5 to 1 by ball milling, the hydrogen desorption temperature was only slightly reduced, as observed in the sample MBH-CA-PM (shown in Fig. 4).
Since the porous structure of carbon aerogel in MBH-CA was destroyed during the ball milling process, a possible effect due to nanoconfinement cannot be determined. The newly formed Mg(BH4)2 is very likely dispersed homogeneously on the non-porous carbon matrix with this synthesis method (see Fig. S1a†). In this sense, ball milling of MgH2 supported on nonporous graphite in B2H6/H2 could also lead to the formation of nanostructured Mg(BH4)2, which is responsible for the hydrogen desorption reaction occurring at 160 °C in Fig. 4. The hydrogen desorption reactions at 285 and 336 °C that occur within a temperature range similar to the desorption of bulk Mg(BH4)2 (Fig. 4(b)) imply the existence of bulk Mg(BH4)2 in MBH-KS6. This could be related to the observation of some crystalline MgH2 in the XRD pattern of MBH-KS6 (Fig. 2).
Fig. 9 summarizes schematically the images of bulk Mg(BH4)2, MBH-CA-PM, MBH-KS6 and MBH-CA, and their temperature ranges for hydrogen desorption are compared. Apparently, the smaller the particle size, the lower the desorption temperature; this tendency implies a relationship between particle size and hydrogen sorption performance of Mg(BH4)2, similar to the observations in other hydrides such as LiBH431,35 and MgH2.36 The argument of a nanosize effect may also apply to the improved rehydrogenation performance of MBH-CA. The rehydrogenation reaction of bulk Mg(BH4)2 from MgB2 according to eqn (1) needs a high temperature (e.g. 400 °C) due to the large kinetic barrier.12 According to the thermodynamic properties of Mg(BH4)2,6-8 a high hydrogen pressure over 660 bar is required for rehydrogenation at 400 °C, as displayed in Fig. 10. Following the van't Hoff plot of eqn (1), partial re-formation of Mg(BH4)2 was achieved under milder conditions (200 to 270 °C under 150 bar H2) for the nanostructured sample. By applying a lower H2 pressure of 80 bar at 200 °C, less Mg(BH4)2 was formed (shown in Fig. 6) due to the lower driving force.
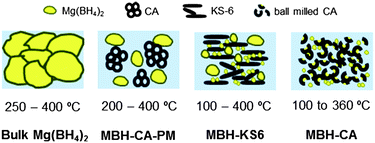 | ||
| Fig. 9 Schematic images of bulk Mg(BH4)2, MBH-CA-PM, MBH-KS6 and MBH-CA, with corresponding temperature ranges for hydrogen desorption. | ||
 | ||
| Fig. 10 van't Hoff plot of reaction Mg(BH4)2 ↔ MgB2 + 4H2 and comparison of rehydrogenation conditions (e.g. temperature and hydrogen pressure) of bulk Mg(BH4)2 (circle)12 and nanostructured Mg(BH4)2 (stars). The reduction in pressure and temperature for rehydrogenation reaction is attributed to a nanosize effect. | ||
About 30% of hydrogen can be re-absorbed in the second cycle at 270 °C and 150 bar, as shown in Fig. S3.† The decay in H2 desorption properties could be attributed to sintering of nanoparticles during the desorption and reabsorption processes, since the pore structure of the carbon matrix in MBH-CA was destroyed by ball milling. However, it is difficult to observe the change in morphology of nanoparticles before (Fig. S1†) and after recycling (Fig. S4†) by TEM, due to the low contrast between carbon, boron and Mg. Despite that, the B2H6 desorption in the 2nd cycle (Fig. 7) remains at the same temperature compared to the as-synthesized MBH-CA (Fig. 4(b)), indicating the formation of Mg(BH4)2 nanoparticles after rehydrogenation.
Conclusions
We present the synthesis of Mg(BH4)2/carbon composites by ball milling of MgH2 supported on the carbon matrix in a B2H6/H2 atmosphere. When carbon aerogel (average pore size of 10 nm) was taken as the carbon support, a Mg(BH4)2/carbon nanocomposite (MBH-CA) was obtained. 11B MAS NMR measurements confirmed the formation of Mg(BH4)2. MBH-CA exhibits a markedly reduced activation energy of 102 ± 6 kJ mol−1 compared to 340 ± 11 kJ mol−1 for bulk Mg(BH4)2, and a lowered temperature (160 °C) for the major hydrogen desorption. When nonporous graphite was used as the carbon support, the formed Mg(BH4)2/carbon composite (MBH-KS6) also shows a hydrogen desorption peak at 160 °C, indicating the presence of nanostructured Mg(BH4)2. In addition, the desorption reactions occurring at 285 and 336 °C within a similar temperature range as the decomposition of bulk Mg(BH4)2 imply the presence of bulk Mg(BH4)2 in MBH-KS6 as well.Re-formation of Mg(BH4)2 from dehydrogenated MBH-CA was achieved under mild conditions (200 °C and 80 to 150 bar H2). The deterioration in hydrogen desorption performance during the 2nd sorption cycle is possibly due to sintering of nanoparticles. Therefore, maintenance of the nanostructures is considered to be important for the reversible hydrogen sorption performance of Mg(BH4)2 under mild conditions.
Acknowledgements
The authors would like to acknowledge the financial support granted by Switzerland through the Swiss Contribution to the enlarged European Union, a grant from the Dutch organization for scientific research (NWO Vidi (016.072.316)), and by the Korean Research Council. Part of this work was supported by COST Action MP1103 “Nanostructured materials for solid-state hydrogen storage”. We thank J.D. Meeldijk for acquiring the TEM images and J. Geus for his advice on the EELS measurements. We are grateful to Timcal for providing us with the carbon support materials.Notes and references
- L. Schlapbach, Nature, 2009, 460, 809–811 CrossRef CAS.
- A. Zuttel, A. Remhof, A. Borgschulte and O. Friedrichs, Philos. Trans. R. Soc., A, 2010, 368, 3329–3342 CrossRef CAS.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- H. W. Li, Y. G. Yan, S. Orimo, A. Zuttel and C. M. Jensen, Energies, 2011, 4, 185–214 CrossRef CAS.
- M. J. van Setten, G. A. de Wijs, M. Fichtner and G. Brocks, Chem. Mater., 2008, 20, 4952–4956 CrossRef CAS.
- V. Ozolins, E. H. Majzoub and C. Wolverton, J. Am. Chem. Soc., 2009, 131, 230–237 CrossRef CAS.
- Y. Yan, H. W. Li, Y. Nakamori, N. Ohba, K. Miwa, S. Towata and S. Orimo, Mater. Trans., 2008, 49, 2751–2752 CrossRef CAS.
- H. W. Li, K. Kikuchi, Y. Nakamori, N. Ohba, K. Miwa, S. Towata and S. Orimo, Acta Mater., 2008, 56, 1342–1347 CrossRef CAS.
- N. Hanada, K. Chlopek, C. Frommen, W. Lohstroh and M. Fichtner, J. Mater. Chem., 2008, 18, 2611–2614 RSC.
- G. L. Soloveichik, Y. Gao, J. Rijssenbeek, M. Andrus, S. Kniajanski, R. C. Bowman, S. J. Hwan and J. C. Zhao, Int. J. Hydrogen Energy, 2009, 34, 916–928 CrossRef CAS.
- G. Severa, E. Ronnebro and C. M. Jensen, Chem. Commun., 2010, 46, 421–423 RSC.
- M. Chong, A. Karkamkar, T. Autrey, S. Orimo, S. Jalisatgi and C. M. Jensen, Chem. Commun., 2011, 47, 1330–1332 RSC.
- S. J. Hwang, R. C. Bowman, J. W. Reiter, J. Rijssenbeek, G. L. Soloveichik, J. C. Zhao, H. Kabbour and C. C. Ahn, J. Phys. Chem. C, 2008, 112, 3164–3169 CAS.
- H. W. Li, K. Miwa, N. Ohba, T. Fujita, T. Sato, Y. Yan, S. Towata, M. W. Chen and S. Orimo, Nanotechnology, 2009, 20, 204013 CrossRef.
- X. B. Chen, C. Li, M. Gratzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC.
- M. Fichtner, Adv. Eng. Mater., 2005, 7, 443–455 CrossRef CAS.
- J. J. Vajo, Curr. Opin. Solid State Mater. Sci., 2011, 15, 52–61 CrossRef CAS.
- P. Adelhelm and P. E. de Jongh, J. Mater. Chem., 2011, 21, 2417–2427 RSC.
- S. Gupta, I. Z. Hlova, T. Kobayashi, R. V. Denys, F. Chen, I. Y. Zavaliy, M. Pruski and V. K. Pecharsky, Chem. Commun., 2013, 49, 828–830 RSC.
- C. Pistidda, S. Garroni, F. Dolci, E. G. Bardaji, A. Khandelwal, P. Nolis, M. Dornheim, R. Gosalawit, T. Jensen, Y. Cerenius, S. Surinach, M. D. Baro, W. Lohstroh and M. Fichtner, J. Alloys Compd., 2010, 508, 212–215 CrossRef CAS.
- M. Fichtner, Z. Zhao-Karger, J. J. Hu, A. Roth and P. Weidler, Nanotechnology, 2009, 20, 204029 CrossRef.
- M. A. Wahab, Y. Jia, D. J. Yang, H. J. Zhao and X. D. Yao, J. Mater. Chem. A, 2013, 1, 3471–3478 Search PubMed.
- A. Remhof, A. Borgschulte, O. Friedrichs, P. Mauron, Y. Yan and A. Zuttel, Scr. Mater., 2012, 66, 280–283 CrossRef CAS.
- O. Friedrichs, A. Remhof, A. Borgschulte, F. Buchter, S. I. Orimo and A. Zuttel, Phys. Chem. Chem. Phys., 2010, 12, 10919–10922 RSC.
- R. W. Pekala and F. M. Kong, J. Phys., 1989, 50, C433–C440 Search PubMed.
- P. E. de Jongh, R. W. P. Wagemans, T. M. Eggenhuisen, B. S. Dauvillier, P. B. Radstake, J. D. Meeldijk, J. W. Geus and K. P. de Jong, Chem. Mater., 2007, 19, 6052–6057 CrossRef CAS.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- R. J. Newhouse, V. Stavila, S. J. Hwang, L. E. Klebanoff and J. Z. Zhang, J. Phys. Chem. C, 2010, 114, 5224–5232 CAS.
- A. Gutowska, L. Y. Li, Y. S. Shin, C. M. M. Wang, X. H. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS.
- A. F. Gross, J. J. Vajo, S. L. Van Atta and G. L. Olson, J. Phys. Chem. C, 2008, 112, 5651–5657 CAS.
- Y. F. Liu, K. Zhong, K. Luo, M. X. Gao, H. G. Pan and Q. D. Wang, J. Am. Chem. Soc., 2009, 131, 1862–1870 CrossRef CAS.
- P. Ngene, M. van Zwienen and P. E. de Jongh, Chem. Commun., 2010, 46, 8201–8203 RSC.
- T. Mueller and G. Ceder, ACS Nano, 2010, 4, 5647–5656 CrossRef CAS.
- X. F. Liu, D. Peaslee, C. Z. Jost, T. F. Baumann and E. H. Majzoub, Chem. Mater., 2011, 23, 1331–1336 CrossRef CAS.
- M. Paskevicius, D. A. Sheppard and C. E. Buckley, J. Am. Chem. Soc., 2010, 132, 5077–5083 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: TEM observation and EDX and EELS analysis of MBH-CA and MBH-KS6, thermogravimetry (TG) curves of MBH-CA, MBH-KS6 and bulk Mg(BH4)2, and cycling property of MBH-CA. See DOI: 10.1039/c3ta12222k |
| ‡ Both authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2013 |