 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation into the effect of Si doping on the performance of SrFeO3−δ SOFC electrode materials†
Jose M. Porras-Vazquez*a,
Thomas Pikea,
Cathryn A. Hancocka,
Jose F. Marcob,
Frank J. Berrya and
Peter R. Slater*a
aSchool of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: p.r.slater@bham.ac.uk; j.m.porras@bham.ac.uk; Fax: +44 (0)1214144403; Tel: +44 (0)1214148672
bInstituto de Química Física “Rocasolano”, CSIC, Serrano 119, 28006, Madrid, Spain
First published on 1st August 2013
Abstract
In this paper we report the successful incorporation of silicon into SrFeO3−δ perovskite materials for potential applications as electrode materials for solid oxide fuel cells. It is observed that Si doping leads to a change from a tetragonal cell (with partial ordering of oxygen vacancies) to a cubic one (with the oxygen vacancies disordered). Annealing experiments in 5% H2/95% N2 (up to 800 °C) also showed the stabilization of the cubic form for the Si-doped samples under reducing conditions, suggesting that they may be suitable for both cathode and anode applications. In contrast to the cubic cell of the reduced Si doped system, reduction of undoped SrFeO3−δ leads to the formation of a brownmillerite structure with ordered oxide ion vacancies. SrFe0.90Si0.10O3−δ and SrFe0.85Si0.15O3−δ were analysed by neutron powder diffraction, and the data confirmed the cubic cell, with no long range oxygen vacancy ordering. Mössbauer spectroscopy data were also recorded for SrFe0.90Si0.10O3−δ, and indicated the presence of only Fe3+ and Fe5+ (i.e. disproportionation of Fe4+ to Fe3+ and Fe5+) for such doped samples. Conductivity measurements showed an improvement in the conductivity on Si doping. Composite electrodes with 50% Ce0.9Gd0.1O1.95 were therefore examined on dense Ce0.9Gd0.1O1.95 pellets in two different atmospheres: air and 5% H2/95% N2. In both atmospheres an improvement in the area specific resistance (ASR) values is observed for the Si-doped samples. Thus the results show that silicon can be incorporated into SrFeO3−δ-based materials and can have a beneficial effect on the performance, making them potentially suitable for use as cathode and anode materials in symmetrical SOFCs.
1 Introduction
Perovskite transition metal containing oxides have attracted considerable interest due to potential applications as cathode materials in the field of Solid Oxide Fuel Cells (SOFCs). Traditionally doping strategies for such materials have focused on substitution with cations of similar size, e.g. Sr for La.1–5 In addition, perovskite-type anodes, such as Sr1−xTi1−2xNb2xO3, La0.75Sr0.25Cr0.5Mn0.5O3−δ, La4Sr8Ti11Mn0.5Ga0.5O37.5−δ and Sr2MgMoO6, have also been investigated in recent years as alternatives to Ni-cermets to allow the direct utilization of commercial hydrocarbon fuels (with their associated impurities, e.g. S).6–11Perovskite SrFeO3−δ is an interesting material which exhibits high mixed oxide ionic and electronic conductivity and therefore can be potentially used in electrochemical devices such as oxygen permeation membranes, oxygen sensors, and SOFCs.12–14 Iron cations in this system are in a mixed valence state ranging from +4 to +3, corresponding to a wide range of oxygen nonstoichiometry, while the structure changes from tetragonal to orthorhombic brownmillerite type, as the iron oxidation state reduces to 3+ and hence the composition changes to SrFeO2.5, with associated long range ordering of oxide ion vacancies.15–17 The formation of ordered oxygen vacancies is not favourable for practical applications because it drastically reduces oxide ion conduction, while the oxygen deficiency also results in a decrease in both mobility and concentration of hole carriers.18,19 Therefore, in this work we propose the modifications of SrFeO3-based materials through the incorporation of silicate to stabilize the high symmetry cubic form, even at low oxygen partial pressures.
The approach employed stems from prior observations on the successful incorporation of oxyanions into perovskite-type cuprate superconductors and related phases.20–28 This work demonstrated that the perovskite structure can incorporate significant levels of oxyanions (carbonate, borate, nitrate, sulfate, and phosphate). In such samples, C, B, N, P, and S of the oxyanion group were shown to reside on the perovskite B cation site, with the oxide ions of this group filling 3 (C, B, N) – 4 (P, S) of the available 6 oxide ion positions around this site.
Recently we have illustrated this oxyanion doping strategy in perovskite-type materials with potential for use in solid oxide fuel cells.29–31 For instance, borate, phosphate and sulphate were successfully incorporated into different cathode materials such as SrCoO3−δ, La1−xSrxCo0.8Fe0.2O3−δ, Ba1−xSrxCo0.8Fe0.2O3−δ, CaMnO3 and La1−xSrxMnO3-type materials, leading to stabilization of high symmetry forms, enhancements in the electronic conductivity, as well as the electrode performance, with respect to the parent compounds.
The introduction of silicate groups is of particular interest, since silica is widely considered a detrimental contaminant of SOFC materials, particularly electrolyte materials, as it has been reported to segregate at the grain boundaries where it forms insulating siliceous phases, lowering the conductivity, such that overall performance is degraded.32–38 For example, it has been reported that several hundreds of ppm of SiO2 can increase the electrolyte grain boundary resistance by over one order of magnitude.39–41
Our preliminary studies on Si incorporation were performed in cobalt-based perovskite electrode materials, showing the successful incorporation of Si into La0.6Sr0.4Co0.8Fe0.2O3−δ and Sr1−xYxCoO3−δ-based materials, with significant results in terms of improvements in the conductivity and an enhancement in the stability towards CO2.42 The enhancement in performance is in contrast to previous assumptions of the detrimental effect of Si and can be attributed to the incorporation of the Si into the perovskite structure, whereas in prior studies of the effect of Si on fuel cell materials researchers have typically examined its addition as a secondary phase. Very recently, SryCa1−yMn1−xSixO3−δ cathode materials have been prepared by solid state reaction, and direct evidence for the incorporation of Si into the structure is provided for the first time by 29Si NMR.43,44 In each case, Si doping is shown to enhance the conductivity, which can be attributed to electron doping (driven by the introduction of oxide ion vacancies due to the preference for Si to adopt tetrahedral coordination), as well as, for some systems, a change from a hexagonal (containing face sharing of octahedra) to a cubic perovskite (containing corner sharing of octahedra).
Therefore, in this work we have extended these studies to silicon doping into SrFeO3−δ systems, with a view to stabilize the cubic form of these systems at low oxygen partial pressures, avoiding its transition to the brownmillerite form, and thus, potentially allowing the use of these samples as anode and cathode materials in symmetrical fuel cells. In addition Si and Fe containing perovskites are of interest in Earth Science, where (Mg, Fe)SiO3, (Ca, Fe)SiO3 and Ca(Si, Fe)O3−x phases have attracted substantial interest due to their accepted presence in the Earth's interior.45–48 Such phases have been traditionally thought to require very high pressure synthesis conditions, and so the work here, showing for the first time synthesis of an Fe and Si containing perovskite at ambient pressure, is of significant relevance to a perovskite chemistry field in general.
2 Experimental
SrCO3 (Aldrich, 99.9%), Fe2O3 (Fluka, 99%) and SiO2 (Aldrich, 99.6%) were used to prepare SrFe1−xSixO3−δ (x ≤ 0.15). The powders were intimately ground and heated initially to 1100 °C for 12 h. They were then ball-milled (350 rpm for 1 hour, Fritsch Pulverisette 7 Planetary Mill) and reheated to 1150 °C for a further 12 h. Finally, they were then ball-milled (350 rpm for 1 hour) and reheated to 1200 °C for a further 12 h. All the heating processes were carried out in air.Powder X-ray diffraction (XRD) (Bruker D8 diffractometer with Cu Kα1 radiation in transmission mode, Debye–Scherrer) was used to demonstrate phase purity, as well as for cell parameter determination. For the latter, the GSAS suite of programs was used.49 In order to obtain further structural details, neutron diffraction data were collected on an HRPT diffractometer50 [SINQ neutron source at Paul Scherrer Institut, Villigen, Switzerland]. Rietveld refinement was performed using the GSAS suite of programs.49 Oxygen contents were estimated from thermogravimetric analysis (Netzsch STA 449 F1 Jupiter Thermal Analyser). Samples were heated at 10 °C min−1 to 1200 °C in N2 and held for 30 minutes to reduce the Fe oxidation state to 3+, with the original oxygen content and average Fe oxidation state then being determined from the mass loss observed.
To determine the potential use of these materials as SOFC anodes the samples were annealed for 24 hours at 800 °C in 5% H2–95% Ar to study the effect of silicon doping on the stability under reducing conditions.
In order to gather further information on the Fe oxidation state, 57Fe Mössbauer spectra were recorded in constant acceleration mode using a ca. 25 mCi57Co/Rh source and a helium closed-cycle cryo-refrigerator. All the spectra were computer fitted and the chemical isomer shift data are quoted relative to metallic iron at 298 K.
Pellets for conductivity measurements were prepared as follows: the powders were first ball-milled (350 rpm for 1 hour), before pressing (200 MPa) as pellets and sintering at 1200 °C for 12 h. Four Pt electrodes were attached with Pt paste, and then the sample was fired to 800 °C in air for 1 hour to ensure bonding to the sample. The samples were then furnace cooled to 350 °C in air and held at this temperature for 12 hours to ensure full oxygenation. Conductivities were then measured using the four probe dc method in two different atmospheres: air (for cathode applications) and 5% H2/95% N2 (for anode applications).
To elucidate the potential of these materials as SOFC electrodes, symmetrical electrodes were coated on both sides of dense Ce0.9Gd0.1O1.95 (CGO10, Aldrich) pellets (sintered at 1500 °C for 12 h) using a suspension prepared with a mixture of electrolyte and electrodes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 wt%) and Decoflux™ (WB41, Zschimmer and Schwarz) as a binder material. The symmetrical cells were fired at 900 °C for 1 h in air. Afterwards, a Pt-based ink was applied onto the electrodes to obtain a current collector layer and finally fired at 800 °C for 1 hour. Area-specific resistance (ASR) values were then obtained under both symmetrical air (cathodic conditions) and 5% H2/95% N2 (anodic conditions) atmospheres in a two electrode configuration. To avoid the reduction of the CGO electrolyte, the measurements were carried out only at 600 °C under reducing conditions. Impedance spectra of the electrochemical cells were collected using a HP4912A frequency analyser, at open circuit voltage (OCV), in the 5 Hz to 13 MHz frequency range with an ac signal amplitude of 100 mV. The spectra were fitted to equivalent circuits using the ZView software51 which allows an estimation of the resistance and capacitance associated with different cell contributions.
:1 wt%) and Decoflux™ (WB41, Zschimmer and Schwarz) as a binder material. The symmetrical cells were fired at 900 °C for 1 h in air. Afterwards, a Pt-based ink was applied onto the electrodes to obtain a current collector layer and finally fired at 800 °C for 1 hour. Area-specific resistance (ASR) values were then obtained under both symmetrical air (cathodic conditions) and 5% H2/95% N2 (anodic conditions) atmospheres in a two electrode configuration. To avoid the reduction of the CGO electrolyte, the measurements were carried out only at 600 °C under reducing conditions. Impedance spectra of the electrochemical cells were collected using a HP4912A frequency analyser, at open circuit voltage (OCV), in the 5 Hz to 13 MHz frequency range with an ac signal amplitude of 100 mV. The spectra were fitted to equivalent circuits using the ZView software51 which allows an estimation of the resistance and capacitance associated with different cell contributions.
3 Results and discussion
Solid solution range
For the SrFe1−xSixO3−δ series, single phase samples could be achieved up to higher levels of silicon, x ≤ 0.15. Exceeding this Si content led to the segregation of secondary phases, such as Sr2SiO4 (PDF 038-0271). The undoped sample showed a tetragonal symmetry due to partial ordering of the oxygen vacancies. This is clearly seen in the inset of Fig. 1, where we can observe the splitting of the peaks in the 45–60 2θ (°) region. Through Si doping there was no long range oxygen ordering and the samples showed a cubic cell, see Fig. 1. | ||
| Fig. 1 X-ray diffraction patterns of (a) SrFeO3−δ, (b) SrFe0.95Si0.05O3−δ, (c) SrFe0.90Si0.10O3−δ and (d) SrFe0.85Si0.15O3−δ. The inset shows the expanded region from 45 to 60° 2θ for SrFeO3−δ. | ||
Cell parameters for these systems were determined from the X-ray diffraction data using the Rietveld method (see Table 1), showing an increase in the cell volume as the Si content increases. The Rietveld plots for SrFeO3−δ and SrFe0.9Si0.1O3−δ are shown in Fig. S1.† In addition, hkl values for the main diffraction peaks are shown in Tables S1 and S2† for SrFeO3−δ and SrFe0.9Si0.1O3−δ. Similar results were reported in a previous study where (Ca, Sr)MnO3-based compounds were successfully doped with silicon.44 The change in cell parameters for these oxyanion doped perovskite materials is a balance between the effect of the smaller size of Si4+ ions, which would be expected to lead to a reduction in cell volume, and the associated reduction of Fe4+ to give a higher concentration of Fe3+, which would be expected to lead to an increase in cell volume. The formation of 3+ species through Si doping is predicted by the following defect equation:
 | (1) |
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m)
m)
| SrFe1−xSixO3−δ | ||||
|---|---|---|---|---|
| Si (x) | 0 | 0.05 | 0.10 | 0.15 |
| a (Å) | 10.9235(1) | 3.8636(1) | 3.8723(1) | 3.8755(1) |
| c (Å) | 7.6965(1) | — | — | — |
| V/Z (Å3) | 57.40(1) | 57.67(1) | 58.06(1) | 58.34(1) |
| Rwp (%) | 3.43 | 3.08 | 2.82 | 2.66 |
As can be seen from the above equation, a key driving force for the reduction of Fe4+ to Fe3+ is the introduction of oxide ion vacancies due to the lower coordination (tetrahedral rather than octahedral) preference of the Si dopant (i.e. for x = 0, the B cation site is completely occupied by Fe, while for x > 0 some Si is on this site, which is tetrahedrally coordinated, and thus leads to a reduction in the total oxygen content). Thus, while we are nominally performing an isovalent (Si4+ in place of Fe4+) substitution, the generation of oxide ion vacancies results in partial reduction, i.e. electron doping.
The average oxidation states of Fe in these systems reported in Table 3 showed a decrease in the average iron oxidation state and increase in the oxygen vacancies as the Si content increases, in good agreement with the defect equation given above illustrating the predicted effect of Si-doping in these systems.
Keeping in mind the use of these materials in symmetrical fuel cells, their stability was also examined under reducing conditions (24 h at 800 °C in 5% H2–95% N2), to examine their possible application as SOFC anode materials. The undoped sample, SrFeO3−δ, showed a transition from a tetragonal to an orthorhombic symmetry (see Fig. 2). This behaviour is due to the loss of oxygen, leading to the formation of a phase (Sr2Fe2O5) with the brownmillerite structure and ordered oxygen vacancies. On Si-doping we see a stabilization of the cubic symmetry under reducing conditions for silicon contents higher than or equal to x = 0.10. Therefore, through silicon doping we have achieved the stabilisation of the high symmetry form under reducing conditions, which is likely to help maintain its electrical properties and avoid anisotropic changes of dimensions that may provoke a mechanical failure in the working cell.
 | ||
| Fig. 2 X-ray diffraction patterns of (a) SrFeO3−δ and (b) SrFe0.90Si0.10O3−δ, as prepared; and (c) SrFeO3−δ and (d) SrFe0.90Si0.10O3−δ, annealed at 800 °C for 24 h in 5% H2–95% N2. | ||
Neutron diffraction structural study
The structures of the SrFe1−xSixO3−δ series, x = 0.1 and 0.15, samples were examined using neutron diffraction data. The data indicated a cubic cell (Pmm), with no evidence for the presence of extra peaks indicative of oxide vacancy ordering (see Fig. 3 and 4). The R factors were very low, Rwp = 3.73% and RF = 1.17% for SrFe0.9Si0.1O3−δ and Rwp = 3.47% and RF = 1.31% for SrFe0.85Si0.15O3−δ, indicating a good fit to the data. The refined structural data are shown in Table 2. The usual parameters: histogram scale factors, background coefficients, unit cell parameters, zero error and peak shape coefficients were varied. For the refinement, the atomic displacement parameters for Fe and Si were constrained to be equal. The Fe and Si occupancies were refined, with the constraint that their sum equalled 1.0, and the final values were in good agreement with those expected from the starting composition. Atomic displacement parameters were refined as isotropic. As can be seen from Table 2 the oxygen atomic displacement parameters are higher than the other atoms which can be related to the local distortions caused by the presence of the silicate groups. The data confirm the importance of silicon doping in these strontium ferrites showing the stabilization of the cubic form, with no long range oxide vacancy ordering. As future work, we are planning to carry out total scattering experiments to study the local structure of these samples and the effect of the introduction of a silicate group.
 | ||
| Fig. 3 Observed, calculated and difference neutron diffraction profiles of SrFe0.90Si0.10O3−δ. The structure was refined in a cubic space group (Pmm). | ||
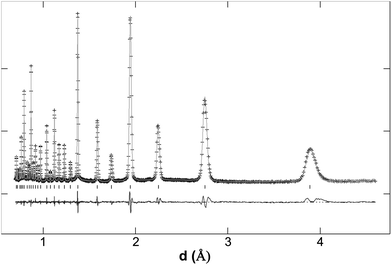 | ||
| Fig. 4 Observed, calculated and difference neutron diffraction profiles of SrFe0.85Si0.15O3−δ. The structure was refined in a cubic space group (Pmm). | ||
m)
| x = 0.10 | x = 0.15 | |||
|---|---|---|---|---|
| Atom (x, y, z) | 100 UISO | Occupancy | 100 UISO | Occupancy |
| Sr (0.5, 0.5, 0.5) | 1.07(1) | 1(−) | 1.22(1) | 1(−) |
| Fe (0, 0, 0) | 0.77(1) | 0.93(1) | 0.94(1) | 0.88(1) |
| Si (0, 0, 0) | 0.77(1) | 0.07(1) | 0.94(1) | 0.12(1) |
| O (0.5, 0, 0) | 1.65(1) | 0.90(1) | 1.75(1) | 0.87(1) |
| SrFe1−xSixO3−δ | ||||
|---|---|---|---|---|
| Si (x) | 0 | 0.05 | 0.10 | 0.15 |
| Oxygen deficiency (δ) | 0.10 | 0.20 | 0.23 | 0.25 |
| Oxidation state | 3.80 | 3.60 | 3.48 | 3.42 |
| Conductivity at 700 °C (S cm−1) | 26.3 | 21.2 | 35.3 | 17.8 |
| Conductivity at 800 °C (S cm−1) | 17.2 | 14.6 | 24.1 | 13.4 |
| ASR at 700 °C (Ω cm2) | 1.65 | 1.19 | 0.90 | 1.88 |
| ASR at 800 °C (Ω cm2) | 0.25 | 0.14 | 0.08 | 0.28 |
57Fe Mössbauer spectroscopy
57Fe Mössbauer data were collected in order to gain further information on the oxidation state and environment of iron. The spectra recorded from SrFe0.9Si0.1O3−δ at 298 K showed three quadrupole split absorptions (Fig. 5). The doublet with a chemical isomer shift of δ ≈ −0.05 mm s−1 can be associated with Fe5+ in perovskite-related structures.52–54 The component with chemical isomer shift δ ≈ 0.37 is typical of Fe3+ in octahedral coordination55 whilst the doublet with chemical isomer shift δ ≈ 0.18 mm s−1 is indicative of Fe3+ in a lower coordination55 (see Table 5). The results demonstrate that substitution of Fe4+ in SrFeO3−δ by Si4+ induces disproportionation of the remaining Fe4+ into Fe3+ and Fe5+. This has previously been observed by substituting the larger Sn4+ ions in place of Fe4+ ions in SrFeO3−δ, which causes lattice strain that can be mitigated by incorporating smaller Fe5+ ions.56,57 In this case the situation is most likely reversed with the smaller Si4+ ions causing significant local strain resulting in the Si4+ being surrounded by the large Fe3+ ions to relieve the strain with the adjacent cells incorporating the smaller Fe5+ ions.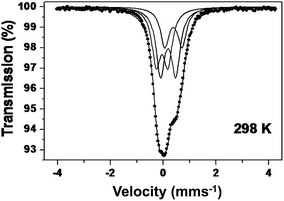 | ||
| Fig. 5 57Fe Mössbauer spectrum recorded from SrFe0.90Si0.10O3−δ at 298 K. | ||
The unfitted 57Fe Mössbauer spectra from SrFe0.9Si0.1O3−δ between 300 and 15 K (Fig. S2†) show that the material becomes magnetically ordered between 68 and 42 K. The spectra recorded from the magnetically ordered materials were complex and their resistance to fitting is consistent with the complexity of the interactions between Fe3+ and Fe5+ at low temperatures.
The spectra at 298 and 16 K from the H2 reduced parent material (Fig. 6) were best fitted to three sextets with chemical isomer shifts characteristic of Fe3+ (see Table 5). The results show that the reducing conditions cause the conversion of Fe5+ to Fe3+ resulting in a composition of SrFe0.9Si0.1O2.55. As can be seen, the spectra at 16 K for the reduced material are less complex compared to its prereduced counterpart since only Fe3+ superexchange interactions were observed, compared to the complex interactions in the prereduced material between the iron in different oxidation states. Previous studies have shown that the reduction of undoped SrFeO3−δ results in brownmillerite-related Sr2Fe2O5 where the oxide ion vacancies are ordered.58 The 57Fe Mössbauer spectra recorded here from reduced SrFe0.9Si0.1O3−δ were significantly different from those recorded previously from Sr2Fe2O5 which showed two sextets of equal area characteristic of equal amounts of octahedrally and tetrahedrally coordinated Fe3+.59–61 Attempts to fit our data to two sextets were not satisfactory. This can be explained by the extra oxygen in SrFe0.9Si0.1O2.55 (Sr2Fe1.8Si0.2O5.1) leading to disorder on the oxygen sublattice.
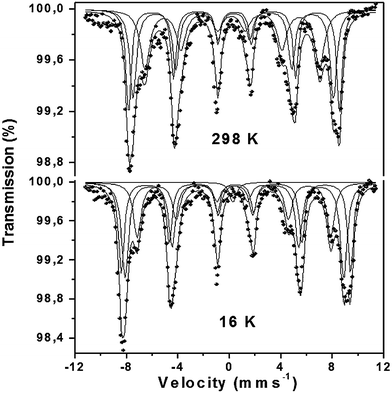 | ||
| Fig. 6 57Fe Mössbauer spectra recorded from reduced SrFe0.90Si0.10O3−δ at 298 and 16 K. | ||
Conductivity measurements
The conductivities of the samples were measured in two different atmospheres: air (for cathode applications) and 5% H2/95% N2 (for anode applications).Air atmosphere
An increase in conductivity was observed for the 10% doped (x = 0.1) sample, with further increases in Si content lowering the conductivity (see Fig. 7). The initial increase in conductivity for x = 0.1 may result from the observed changes in the Fe oxidation state as well as possibly an improvement in the conduction pathways from the change from a tetragonal to a more symmetrical cubic system. When the doping level increases further, the conductivity then decreases due to the silicate disrupting the Fe–O network. All samples showed a decrease in the conductivity above ∼400 °C, due to oxygen loss at these higher temperatures reducing the Fe4+ content.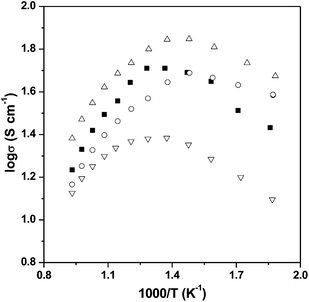 | ||
| Fig. 7 Plot of logσ vs. 1000/T for SrFeO3−δ (■), SrFe0.95Si0.05O3−δ (○), SrFe0.90Si0.10O3−δ (△) and SrFe0.85Si0.15O3−δ (▽) in air. | ||
5% H2/95% N2 atmosphere
Under these conditions we can see an important drop in the conductivity values, with respect to those obtained in air, due to the reduction in the iron oxidation state. The best conductivity values in the SrFe1−xSixO3−δ series were obtained for x ≥ 0.10 (see Table 4). This behaviour is in good agreement with the fact that, for this series, the x ≤ 0.05 samples showed a transition to a brownmillerite phase, detrimental to the electrical properties.| SrFe1−xSixO3−δ | ||||
|---|---|---|---|---|
| Si (x) | 0 | 0.05 | 0.10 | 0.15 |
| Conductivity at 700 °C (S cm−1) | 0.9 | 0.5 | 8.3 | 3.3 |
| Conductivity at 600 °C (S cm−1) | 0.6 | 0.3 | 5.4 | 3.0 |
| ASR at 600 °C (Ω cm2) | 1.86 | 1.51 | 0.17 | 0.24 |
| Material | Temperature of measurement (K) | Assignment | δ ± 0.02 (mm s−1) | Δ or e2qQ/2 ± 0.05 (mm s−1) | H ± 5 (T) | Area ± 5 (%) |
|---|---|---|---|---|---|---|
| As-prepared SrFe0.9Si0.1O3−δ | 300 | Fe5+ | −0.05 | 0.44 | 34 | |
| Fe3+ | 0.38 | 0.65 | 24 | |||
| Fe3+ | 0.18 | 0.58 | 42 | |||
| 300 | Fe3+ | 0.38 | −0.04 | 51 | 32 | |
| Fe3+ | 0.32 | −0.04 | 48 | 35 | ||
| Fe3+ | 0.19 | 0.06 | 42 | 33 | ||
| 16 | Fe3+ | 0.49 | 0.02 | 55 | 33 | |
| Fe3+ | 0.43 | −0.08 | 53 | 41 | ||
| Fe3+ | 0.29 | 0.13 | 47 | 26 |
Area-specific resistance study
Following the conductivity results, cathode testing was performed for the SrFe1−xSixO3−δ series. These experiments used a composite of the perovskite and CGO10 (1:1 wt%) on dense CGO10 pellets. The composite was deposited at 900 °C, and at this temperature there was no evidence of any segregation of secondary phases in perovskite–CGO10 mixtures.
Air atmosphere
The dependences of ASR in air on temperature are shown in Fig. 8 and Table 3. For instance, for SrFe0.95Si0.05O3−δ and SrFe0.90Si0.10O3−δ, the values obtained at 800 °C were 0.14 and 0.08 Ω cm2, respectively. These results entail a significant improvement with respect to the undoped sample: 0.25 Ω cm2. For the highest silicon content, SrFe0.85Si0.15O3−δ, there is no improvement, which is likely due to the disruptive effect of silicon on a conducting Fe–O network. We can see that there is a non-linear behaviour of the ASR data, with a bigger drop in values at higher temperatures. This behaviour is likely due to the fact that these systems show loss of oxygen at high temperature, causing an increase in oxide vacancies and hence a better oxide ion mobility and ASR values. | ||
| Fig. 8 Plot of log(area-specific resistance (ASR)) vs. 1000/T for SrFeO3−δ (■), SrFe0.95Si0.05O3−δ (○), SrFe0.90Si0.10O3−δ (△) and SrFe0.85Si0.15O3−δ (▽) in air. | ||
5% H2–95% H2 atmosphere
The dependences of ASR in 5% H2–95% H2 at 600 °C are shown in Table 4. For SrFe0.95Si0.05O3−δ, SrFe0.90Si10O3−δ and SrFe0.85Si0.15O3−δ the values obtained were 1.51, 0.17 and 0.24 Ω cm2, respectively. These results entail a significant improvement with respect to the undoped sample: 1.86 Ω cm2. This behaviour is likely due to the effect of silicon stabilising the cubic lattice and hence inhibiting long range oxide ion vacancy ordering, as previously discussed. In comparison, for current state-of-the-art Ni–CGO composites deposited by spray pyrolysis on CGO pellets, values of 0.34 Ω cm2 at 600 °C and 0.09 Ω cm2 at 550 °C have been reported in the literature,62,63 very close to the values observed for SrFe0.90Si10O3−δ and SrFe0.85Si0.15O3−δ in this work, where further improvements are likely to be possible through additional morphological control. The results therefore show promising potential for these Si doped perovskites in anode applications, and the fact that these systems are oxides means that problems of C or S poisoning, experienced by Ni containing anodes, may be mitigated in these systems. Further studies are therefore planned to investigate these SrFe1−xSxO3−δ anode materials in more detail.4 Conclusions
SrFe1−xSixO3−δ perovskite materials have been prepared by solid state reaction. A change from tetragonal symmetry (with ordered oxygen vacancies) to cubic (with the oxygen vacancies disordered) is observed through Si doping. Annealing experiments in 5% H2/95% N2 showed the stabilization of the cubic form for the Si-doped samples under reducing conditions, making them potentially suitable for anode applications. An improvement in the conductivity is observed on Si doping at the 10% level, although higher levels of Si are shown to decrease the conductivity, which can be attributed to the blocking effect of silicon on the electronic conduction pathways. Composites with 50% Ce0.9Gd0.1O1.95 were examined on dense Ce0.9Gd0.1O1.95 pellets in two different atmospheres: air and 5% H2/95% N2. In both atmospheres an improvement in the area specific resistance (ASR) values is observed for the Si-doped samples. Thus these preliminary results show that silicon can be incorporated into perovskite ferrites and can have a beneficial effect on the performance, making them potentially suitable for use as cathode and anode materials in symmetrical SOFCs. This work also demonstrates for the first time that Fe and Si containing perovskites can be prepared under ambient pressure conditions, contrary to previous studies where it was believed that very high pressures were necessary.Acknowledgements
We would like to express thanks to EPSRC for funding (grants EP/I003932 and EP/G009929). The Bruker D8 diffractometer and Netzsch STA 449 F1 Jupiter Thermal Analyser used in this research were obtained through the Science City Advanced Materials project: Creating and Characterising Next generation Advanced Materials project, with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). We would like to thank SINQ for neutron diffraction time and Vladimir Pomjakushin for help with the neutron diffraction experiments. We also thank the Spanish Ministry of Science and Innovation for financial support through project MAT2009-14578-C03-01.References
- A. Orera and P. R. Slater, Chem. Mater., 2010, 22, 675 CrossRef CAS
.
- A. J. Jacobson, Chem. Mater., 2010, 22, 660 CrossRef CAS
- A. Lashtabeg and S. J. Skinner, J. Mater. Chem., 2006, 16, 3161 RSC
- A. Martínez-Amesti, A. Larranaga, L. M. Rodríguez-Martínez, A. T. Aguayo, J. L. Pizarra, M. L. Nóa, A. Laresgoiti and M. I. Arriortua, J. Power Sources, 2008, 185, 401 CrossRef
- D. Kuscer, D. Hanzel, J. Holc, M. Hrovat and D. Kolar, J. Am. Ceram. Soc., 2001, 84, 1148 CrossRef CAS
- J. T. S. Irvine, P. R. Slater and P. A. Wright, Ionics, 1996, 2, 213 CrossRef CAS
- P. R. Slater, D. P. Fagg and J. T. S. Irvine, J. Mater. Chem., 1997, 7, 2495 RSC
- S. Tao and J. T. S. Irvine, Nat. Mater., 2003, 2, 320 CrossRef CAS
- J. C. Ruiz-Morales, J. Canales-Vázquez, C. Savaniu, D. Marrero-López, W. Zhou and J. T. S. Irvine, Nature, 2006, 439, 568 CrossRef CAS
- Y. H. Huang, R. I. Dass, Z. L. Xing and J. B. Goodenough, Science, 2006, 312, 254 CrossRef CAS
- Y. H. Huang, R. I. Dass, J. C. Denyszyn and J. B. Goodenough, J. Electrochem. Soc., 2006, 153, A1266 CrossRef CAS
- J. Yoo, A. Verma, S. Wang and A. J. Jacobson, J. Electrochem. Soc., 2005, 152, A497 CrossRef CAS
- A. Evdou, V. Zaspalis and L. Nalbandian, Fuel, 2010, 89, 1265 CrossRef CAS
- Y. Niu, W. Zhou, J. Sunarso, L. Ge, Z. Zhu and Z. Shao, J. Mater. Chem., 2010, 20, 9619 RSC
- J. P. Hodges, S. Short, J. D. Jorgensen, X. Xiong, B. Dabrowski, S. M. Mini and C. W. Kimball, J. Solid State Chem., 2000, 151, 190 CrossRef CAS
- V. Vashuk, L. Kokhanovskii and I. Yushkevich, Inorg. Mater., 2000, 36, 79 CrossRef CAS
- M. V. Patrakeev, I. A. Leonidov, V. L. Kozhevnikov and V. V. Kharton, Solid State Sci., 2004, 6, 907 CrossRef CAS
- P. Adler, A. Lebon, V. Damljanovic, C. Ulrich, C. Bernhard, A. V. Boris, A. Maljuk, C. T. Lin and B. Keimer, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 094451 CrossRef
- M. Schmidt and S. J. Campbell, J. Solid State Chem., 2001, 156, 292 CrossRef CAS
- C. Greaves and P. R. Slater, J. Mater. Chem., 1991, 1, 17 RSC
- C. Greaves and P. R. Slater, Physica C, 1991, 175, 172 CrossRef CAS
- P. R. Slater, C. Greaves, M. Slaski and C. M. Muirhead, Physica C, 1993, 208, 193 CrossRef CAS
- Y. Miyazaki, H. Yamane, N. Ohnishi, T. Kajitani, K. Hiraga, Y. Mori, S. Funahashi and T. Hirai, Physica C, 1992, 198, 7 CrossRef CAS
- A. Maignan, M. Hervieu, C. Michel and B. Raveau, Physica C, 1993, 208, 116 CrossRef CAS
- K. Kinoshita and T. Yamada, Nature, 1992, 357, 313 CrossRef CAS
- B. Raveau, M. Hervieu, D. Pelloquin, C. Michel and R. Retoux, Z. Anorg. Allg. Chem., 2005, 631, 1831 CrossRef CAS
- D. Pelloquin, M. Hervieu, C. Michel, N. Nguyen and B. Raveau, J. Solid State Chem., 1997, 134, 395 CrossRef CAS
- V. Caignaert, B. Domenges and B. Raveau, J. Solid State Chem., 1995, 120, 279 CrossRef CAS
- C. A. Hancock, R. C. T. Slade, J. R. Varcoe and P. R. Slater, J. Solid State Chem., 2011, 184, 2972 CrossRef CAS
- J. M. Porras-Vazquez and P. R. Slater, J. Power Sources, 2012, 209, 180 CrossRef CAS
- J. M. Porras-Vazquez, T. F. Kemp, J. V. Hanna and P. R. Slater, J. Mater. Chem., 2012, 22, 8287 RSC
- D. Ivanova, E. M. C. L. G. P. Lima, A. Kovalevsky, F. M. L. Figueiredo, V. V. Kharton and F. M. B. Marques, Ionics, 2008, 14, 349 CrossRef CAS
- X. Guo and R. Waser, Prog. Mater. Sci., 2006, 51, 151 CrossRef CAS
- M. J. Verkerk, A. J. A. Winnubst and A. J. Burggraaf, J. Mater. Sci., 1982, 17, 3113 CrossRef CAS
- S. P. S. Badwal and J. Drennan, J. Mater. Sci., 1987, 22, 3231 CrossRef CAS
- M. L. Mecartney, J. Am. Ceram. Soc., 1987, 70, 54 CrossRef CAS
- S. P. S. Badwal and J. Drennan, J. Mater. Sci., 1989, 24, 88 CrossRef CAS
- D. Ivanova, A. Kovalevsky, V. V. Kharton and F. M. B. Marques, Bol. Soc. Esp. Ceram. Vidrio, 2008, 47, 201 CrossRef CAS
- B. C. H. Steele, Solid State Ionics, 2000, 129, 95 CrossRef CAS
- T. S. Zhang, J. Ma, S. H. Chan, P. Hing and J. A. Kilner, Solid State Sci., 2004, 6, 565 CrossRef CAS
- S. P. S. Badwal, Solid State Ionics, 1995, 76, 67 CrossRef CAS
- J. M. Porras-Vazquez and P. R. Slater, Fuel Cells, 2012, 12, 1056 CrossRef CAS
- C. A. Hancock and P. R. Slater, Dalton Trans., 2011, 40, 5599 RSC
- J. M. Porras-Vazquez, E. R. Losilla, P. J. Keenan, C. A. Hancock, T. F. Kemp, J. V. Hanna and P. R. Slater, Dalton Trans., 2013, 42, 5421 RSC
- U. W. Blass, F. Langenhorst, T. Boffa-Ballaran, F. Seifert, D. J. Frost and C. A. McCammon, Phys. Chem. Miner., 2004, 31, 52 CrossRef
- M. Murakami, K. Hirose, K. Kawamura, N. Sata and Y. Ohishi, Science, 2004, 304, 855 CrossRef CAS
- A. R. Oganov and S. Ono, Nature, 2004, 430, 445 CrossRef CAS
- A. R. Oganov, R. Martonak, A. Laio, P. Raiteri and M. Parrinello, Nature, 2005, 438, 1142 CrossRef CAS
- A. C. Larson and R. B. V. Dreele, General Structure Analysis System (GSAS) program. Rep. No. LA-UR-86748, Los Alamos National Laboratory, Los Alamos, CA, 1994 Search PubMed
- P. Fischer, G. Frey, M. Koch, M. Konnecke, V. Pomjakushin, J. Schefer, R. Thut, N. Schlumpf, R. Burge, U. Greuter, S. Bondt and E. Berruyer, Physica B, 2000, 146, 276 Search PubMed
- D. Johnson, ZView: A Software Program for IES Analysis, Version 2.8, Scribner Associates, Inc. Southern Pines, NC, 2008 Search PubMed
- P. Adler, J. Solid State Chem., 1997, 130, 129 CrossRef CAS
- S. E. Dann, M. T. Weller, D. B. Currie, M. F. Thomas and A. D. Al-Rawas, J. Mater. Chem., 1993, 3, 1231 RSC
- G. S. Case, A. L. Hector, W. Levason, R. L. Needs, M. F. Thomas and M. T. Weller, J. Mater. Chem., 1999, 9, 2821 RSC
- F. Menil, J. Phys. Chem., 1985, 46, 763 CAS
- G. Demazeau, P. Fabritchnyi, L. Founes, S. Darracq, I. A. Presniakov, K. V. Pokholok, V. P. Gorkov and J. Etourneau, J. Mater. Chem., 1995, 5, 553 RSC
- F. J. Berry, A. F. Bowfield, F. C. Coomer, D. Jackson, E. A. Moore, P. R. Slater, M. F. Thomas, A. J. Wright and X. Ren, J. Phys.: Condens. Matter, 2009, 21, 256001 CrossRef
- P. K. Gallagher, J. P. Sanders, P. M. Woodward and I. N. Lokuhewa, J. Therm. Anal. Calorim., 2005, 80, 217 CrossRef CAS
- P. K. Gallagher, J. B. MacChesney and D. N. E. Buchanan, J. Chem. Phys., 1964, 41, 2429 CrossRef CAS
- T. C. Gibb, Dalton Trans., 1985, 1455 RSC
- A. Nemundry, M. Weiss, I. Gainutdinov, V. Boldyrev and R. Schollhorn, Chem. Mater., 1998, 10, 2403 CrossRef
- U. P. Muecke, K. Akiba, Anna Infortuna, Tomas Salkus, N. V. Stus and L. J. Gauckler, Solid State Ionics, 2008, 178, 1762 CrossRef CAS
- J.-C. Chen, C.-L. Chang, C.-S. Hsu and B.-H. Hwang, Mater. Res. Bull., 2007, 42, 1674 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ta12113e |
| This journal is © The Royal Society of Chemistry 2013 |