 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCubic phases of garnet-type Li7La3Zr2O12: the role of hydration†
G. Larraz, A. Orera and M. L. Sanjuán*
Instituto de Ciencia de Materiales de Aragón, Universidad de Zaragoza-CSIC, Facultad de Ciencias, Universidad de Zaragoza, Pza. San Francisco s/n, 50009 Zaragoza, Spain. E-mail: sanjuan@unizar.es; Tel: +34 876553354
First published on 23rd July 2013
Abstract
We address the controversial issue of the structural stability of Li7La3Zr2O12 garnets, focusing on the mechanisms that result in the transformation from tetragonal to cubic symmetry. We show that undoped tetragonal Li7La3Zr2O12 not exposed to humidity at any moment undergoes a reversible phase transition to cubic symmetry at Tc ≃ 645 °C that we ascribe to lithium dynamic effects. On the other hand, a close correlation has been found between the appearance of a cubic phase between 100 and 200 °C in X-ray diffractograms and the presence of water, either in the atmosphere in which experiments are performed or already in the starting material. The natures of the high and low-temperature cubic garnets are totally different: the one found above the phase transition does not involve any change in the stoichiometry, whereas the cubic phase formed at low temperature is a hydrated, lithium defective phase, due to the combined effect of water insertion into the garnet structure and the H+/Li+ exchange mechanism. Differences in the actual compositions of the samples depending on their thermal history are corroborated by TG-MS experiments. Chemical reactions and phases formed along the thermal evolution are elucidated with the help of Raman spectroscopy.
1 Introduction
Lithium conductors with garnet-type structure are promising candidates to be used as electrolytes in all solid-state batteries.1 They fulfill at least two of the basic requirements of such components: good chemical stability in the presence of lithium anodes and a wide electrochemical window. As regards ionic conductivity, progressive increase is being achieved, much improving the values from the early compounds Li5La3Nb2O12 and Li5La3Ta2O12, which had σbulk ∼ 10−6 S cm−1 at room temperature (RT),2 to the Li rich compounds Li5+xAxLa3−xM2O12 (A = Sr, Ba; M = Nb, Ta) which reached σbulk ≳ 10−5 S cm−1, also at RT.3–5The discovery of very high conductivity (σ = 2.4 × 10−4 S cm−1 at RT) in the Zr analogue, with the formula Li7La3Zr2O12 (LLZO),6 stimulated intense research in these high lithium content materials. This soon put into evidence that LLZO has two polymorphs: a cubic one, with a structure similar to that of the early compounds (Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d space group), and a tetragonal form, with the I41/acd space group.7 Only the former presents appropriate σ values, the conductivity of the tetragonal polymorph being almost two orders of magnitude lower.
d space group), and a tetragonal form, with the I41/acd space group.7 Only the former presents appropriate σ values, the conductivity of the tetragonal polymorph being almost two orders of magnitude lower.
Producing the cubic form by standard ceramic methods has been elusive till now. Several groups reported that following the recipe described by Murugan et al. in ref. 6 yielded the decomposition of the garnet phase, whereas the tetragonal form seems to be the thermodynamically stable one and is quite easily obtained by solid state reaction at T ≃ 950–980 °C.
Efforts towards improving the conductivity of LLZO garnets have focused on two directions. First, in obtaining the cubic form by low temperature synthesis methods.8–12 Second, to obtain highly conducting Zr-based cubic garnets through doping with different metals, either at the Zr site, such as Nb,13 Ta,14 and Y,15 at the Li sites, mainly with Al (ref. 16–18) and Ga,19 or even at the La site.20 The case of Al doping is the most studied, since it was found that accidental contamination from Al2O3 crucibles prevented decomposition at the high temperatures required to synthesize cubic LLZO and resulted in the stabilization of the cubic form.21
Despite the interest in highly conducting materials, very little has been done in studying the thermal or aging behavior of Li7La3Zr2O12 garnets. A phase transition from tetragonal to cubic forms has been reported with the transition temperature between 100 and 200 °C.21–23 On the other hand, a tetragonal → cubic transition has been found in the Sn analogue but at a much higher temperature, above 750 °C.24
The possibility of stabilizing a cubic form just by moderate heating at temperatures below 200 °C is appealing, since that phase might have a higher conductivity than the tetragonal form. However, the origin of this transition, its reversibility and the nature of the resulting cubic form, have not been investigated in detail. In fact, to date, it has not yet been ascertained whether it is a true phase transition, as Adams et al. claim,22 driven by Li disordering between tetrahedral and octahedral sites, or just a partially or totally irreversible transformation arising from external factors, such as stoichiometry variations due to water and/or CO2 uptake, as proposed in ref. 23, in which case it may dramatically affect the possible use of LLZO as an electrolyte. It is a general concern regarding materials containing lithium as a constituent that, when exposed to air, they tend to lose some Li content through the combined processes of hydration and carbonation.25 Proton/lithium exchange has also been reported in Li conducting garnets, either by aging under ambient conditions, or by treatment in humid or acid medium.23,26–30 In this respect, it is interesting to note that Li7La3Sn2O12, when stored in air for some weeks, spontaneously exchanges Li with protons from atmospheric water to yield the protonated compound Li7-xHxLa3Sn2O12.29 A change from tetragonal to cubic symmetry accompanies proton insertion in this case.
In this work, we address the question of the compositional and structural stability of Li7La3Zr2O12 garnets, focusing on the mechanisms responsible for the tetragonal to cubic transformation occurring between 100 and 200 °C. A close correlation has been found between the formation of a cubic phase in that temperature range and the presence of water, either in the atmosphere in which the experiment is performed or already in the starting material. A sample not exposed to humidity at any moment keeps the tetragonal symmetry up to about 645 °C, when it undergoes a phase transition to cubic symmetry. This phase transition is reversible and shows hysteresis: the transition temperature on cooling is about 625 °C. Phase content and thermal evolution are characterized by means of room and high temperature X-ray-diffraction (XRD), thermal analysis and Raman spectroscopic techniques.
2 Experimental
A tetragonal Li7La3Zr2O12 garnet has been prepared by solid state reaction at 980 °C using dried Li2CO3 (99% purity), La2O3 (99.99% purity, pre-dried at 900 °C for 12 h) and ZrO2 (99% purity) as reagents. 15% excess of Li2CO3 was added to the stoichiometric ratio to compensate for lithium loss during the synthesis. That amount was found to be the optimal one not yielding detectable carbonate impurities either by XRD or by Raman spectroscopy. For comparison purposes, the high temperature cubic phase was synthesized at 1020 °C by doping with 0.25 mol of Al and a lithium excess of only 5%. Alumina crucibles were used in all cases. The chemical composition of the synthesized materials was determined by ICP-OES. RT-XRD experiments were performed on a Rigaku D/max 2500 diffractometer equipped with Cu Kα radiation and working at 40 kV and 100 mA. High-temperature X-ray diffraction studies were performed using an X'Pert PRO MPD diffractometer with an Anton Paar HTK1200 heating stage working at 45 kV and 40 mA. In both series of experiments, data were collected in a step mode (Δ2θ = 0.03°) and a counting time of 1 s per step. XRPD patterns were analyzed by the Rietveld method using the FullProf software.31 Raman dispersion measurements were performed using a DILOR XY spectrometer with a CCD detector and a spectral resolution of 1.4 cm−1. The 496.5 nm line of an Ar+ ion laser was used as the excitation source, power at the sample surface being ≤25 mW. Scattered light was collected through an X50 microscope objective lens. A Linkam TS1500V stage was used for in situ thermal treatments. Thermogravimetric analysis experiments were carried out in a Q-500 thermobalance provided with a mass spectrometer (TG-MS), with a heating rate of 10 °C min−1 in the temperature range from 25 to 750 °C under flowing air. Differential scanning calorimetry (DSC) was performed using a TA Instruments SDT Q600 TA/DSC.3 Results and discussion
3.1 XRD measurements
Several synthesis attempts have been made in order to get a tetragonal garnet as pure as possible, because some impurities, mainly La2O3, Li6Zr2O7, La2Zr2O7 or Li2ZrO3 could be detected depending on temperatures, times, heating and cooling rates. The presence of these secondary phases has been monitored by Raman spectroscopy, since it gives a better resolution than XRD. The XRD pattern of the fresh-synthesized tetragonal Li7La3Zr2O12 garnet is shown in Fig. 1. The XRD pattern was indexed in the I41/acd space group by using the FullProf package,31 and the lattice constants were refined to a = 13.1030 (7) Å and c = 12.6542 (7) Å, which are in agreement with the published data.7 Its chemical composition was determined by ICP-OES. Within the limits of that technique, it was in agreement with the stoichiometric formula.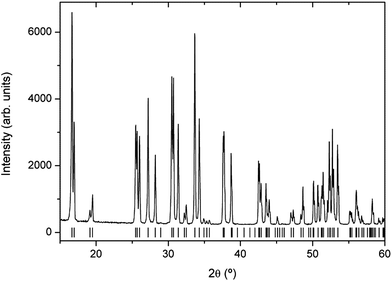 | ||
| Fig. 1 Powder X-ray diffractogram of a freshly synthesized tetragonal Li7La3Zr2O12 garnet. Vertical bars denote the Bragg reflection positions corresponding to the tetragonal phase with the I41/acd space group and lattice parameters a = 13.1030 (7) Å and c = 12.6542 (7) Å. | ||
Some authors have reported a phase transition from tetragonal to cubic symmetry at low temperature (100–200 °C), although this transition was not systematically observed.21–23 Since all these experiments were made in air atmosphere, the hydration effect from atmospheric water should not be overlooked and could play a key role in the evolution, as has been detected in other compounds.23,30 In order to verify the reproducibility of this observation, XRD patterns of Li7La3Zr2O12 were recorded as a function of temperature in air at temperatures ranging from RT to 600 °C. As Fig. 2 shows, the RT diffractogram indicates a tetragonal structure, but at 150 °C a cubic phase has developed, still coexisting with a small amount of the tetragonal phase. Above 150 °C, a purely cubic phase is found. Fig. 2 also shows that above 450 °C, pyrochlore La2Zr2O7 is formed, which indicates that the garnet is unstable in air even at such low temperatures. Though these results seem to confirm the observation of a low temperature phase transition made by other authors, the possibility of the result being conditioned by the atmosphere in which the experiment was performed cannot be ruled out. With the aim of observing the true thermal evolution of tetragonal Li7La3Zr2O12 without the influence of hydration and reaction with air, in situ XRD patterns of the just-synthesized tetragonal garnet (sample 1) were recorded as a function of temperature in dynamic vacuum. Extreme care was taken to avoid exposure of the sample to air at any moment. The XRD patterns from RT to 700 °C presented in Fig. 3 show that the structure remains tetragonal up to at least 550 °C, whereas a cubic phase is observed at 650 °C, still coexisting with a small amount of tetragonal phase that has clearly disappeared at 700 °C. These data evidence a phase transition from tetragonal to cubic symmetry occurring around 650 °C. After cooling to RT, the sample recovers the starting tetragonal symmetry, showing that the transition is reversible.
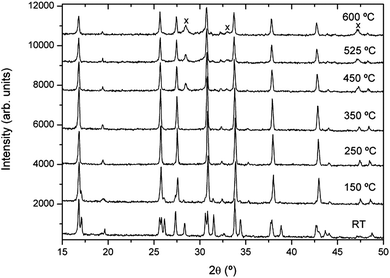 | ||
| Fig. 2 Thermal evolution in air of the X-ray diffractograms of sample 1. The crosses indicate the formation of the La2Zr2O7 pyrochlore phase upon heating. | ||
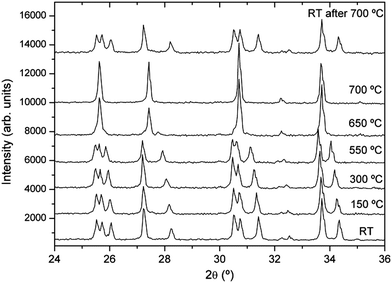 | ||
| Fig. 3 X-ray diffractograms of sample 1 (protected from air) as a function of temperature in dynamic vacuum. | ||
The lattice parameters obtained from Rietveld refinements of the diffractograms shown in Fig. 3 are given in Fig. 4 and listed in Table 1. The cell parameters increase with increasing temperature, as expected, but the tetragonality decreases (c/a → 1) as temperature is increased. This trend is enhanced above 550 °C. Table 1 also shows that, within error, the cell volume remains constant at the phase transition.
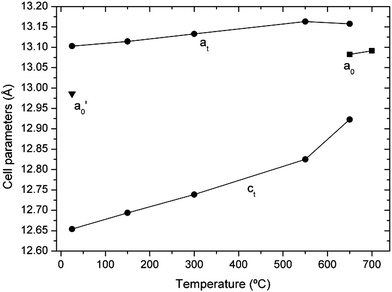 | ||
| Fig. 4 Lattice parameters of Li7La3Zr2O12 (sample 1, protected from air) as a function of temperature in dynamic vacuum. Circles: tetragonal phase. Squares: high-temperature cubic phase. The lattice parameter of a cubic garnet obtained by annealing at 300 °C (sample 3, triangle) is also included for comparison. | ||
| T (°C) | Tetragonal phase | Cubic phase | ||||
|---|---|---|---|---|---|---|
| at (Å) | ct (Å) | ct/at | Vt (Å3) | a0 (Å) | V0 (Å3) | |
| RT | 13.1030 (7) | 12.6542 (7) | 0.966 | 2172.6 (2) | ||
| 150 | 13.1145 (9) | 12.6938 (9) | 0.968 | 2183.2 (3) | ||
| 300 | 13.1329 (8) | 12.7386 (9) | 0.970 | 2197.1 (2) | ||
| 550 | 13.1633 (8) | 12.8252 (9) | 0.974 | 2222.3 (2) | ||
| 650 | 13.1576 (16) | 12.9230 (35) | 0.982 | 2237.3 (7) | 13.0826 (6) | 2239.1 (3) |
| 700 | 13.0913 (6) | 2243.6 (3) | ||||
In order to clarify if, apart from the atmosphere in which the experiment is carried out, the initial hydration state of the sample is also a determinant, several combinations of samples with different degrees of hydration and environmental conditions have been tested.
As an example, we show in Fig. 5 the XRD patterns of a sample that has not been stored in dry conditions (sample 2) but recorded from RT to 650 °C under vacuum to avoid the influence of the experiment atmosphere, with a heating rate of 10 °C min−1 and ∼20 min per diffractogram. Only the 24–36° range is shown to highlight the structural changes. Wider diffractograms, as well as FullProf fits, are given as the ESI.† The lattice parameters obtained from Rietveld refinements of the diffractograms shown in Fig. 5 are given in Fig. 6 and listed in Table 1 of the ESI;†Fig. 7 shows the cubic phase fraction obtained from Rietveld refinements. At RT, we can observe the tetragonal phase. At 150 °C, the diffractogram shows a cubic phase coexisting with the tetragonal one. At 250 °C, a purely cubic phase is found but, as Fig. 5 and 7 show, upon heating above 250 °C, the cubic phase fraction decreases until about 55% of the tetragonal phase is recovered at 450 °C. At 525 °C, the cubic phase fraction increases again and at 600 °C and above, a purely cubic diffractogram is obtained.
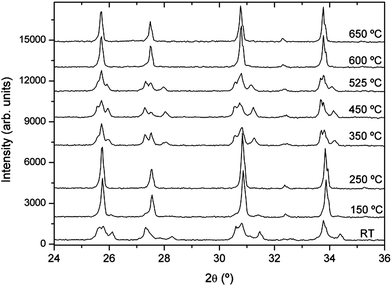 | ||
| Fig. 5 X-ray diffractograms of sample 2 (aged) as a function of temperature in dynamic vacuum. Wider range diffractograms and fitting parameters are given as ESI.† | ||
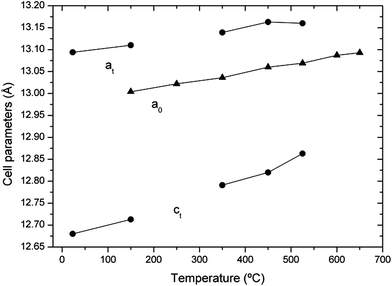 | ||
| Fig. 6 Lattice parameters of sample 2 as a function of temperature in dynamic vacuum. Circles: tetragonal phase. Triangles: cubic phase. | ||
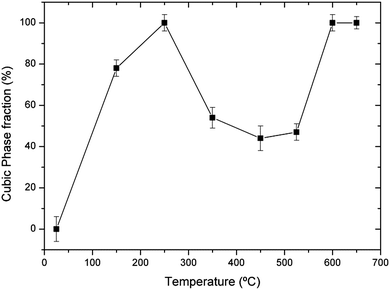 | ||
| Fig. 7 Cubic phase fraction of sample 2 as a function of temperature in dynamic vacuum. | ||
This experiment, together with the results for sample 1 (prevented from hydration) show that both the atmosphere in which the experiment is performed and the hydration state of the sample have an influence on the thermal evolution of Li7La3Zr2O12 garnets. However, these two factors are not completely independent, since a dynamic vacuum may modify the hydration state of the sample and alter its thermal evolution. We have also found that the results of the thermal evolution, either in air or in vacuum, depend on a number of experimental parameters such as the heating rate and the dwell times at each temperature.
3.2 TG-DSC-MS
The high sensitivity of Li-containing garnets to the presence of humidity is well known. In the previous section, we have shown that exposure of LLZO samples to moisture either before or during high-temperature XRD experiments is the determinant in the appearance of an early cubic phase much below the high temperature phase transition. In order to confirm the correlation between the initial state of the sample and its thermal evolution as well as to get more precise information about the phase transition detected at ∼650 °C in XRD experiments, TG and DSC measurements were performed.Fig. 8 shows the TG curve of the fresh sample (sample 1) that has been protected from any contact with air and keeps the tetragonal symmetry up to the tetragonal → cubic phase transition at 650 °C, as shown in Fig. 3. Its total weight loss is as low as 0.5%. On the other hand, the DSC curve of that sample (Fig. 8) provides relevant information concerning the high-temperature structural changes detected by XRD. The two broad endothermic peaks near 200 °C and 400 °C can be ascribed to the release of some H2O and CO2, as detected by the TG-MS analysis explained below. More interesting is the sharp reversible peak observed at 620–650 °C, that we relate to the reversible phase transition seen in the high temperature XRD experiments. Hysteresis is found between the heating and cooling cycles, the transition temperature being 645 °C on heating (T → C) and 620 °C on cooling (C → T).
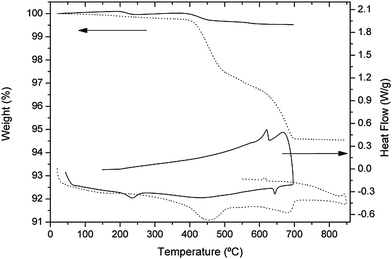 | ||
| Fig. 8 TG/DSC curves of the sample protected from air (sample 1, solid line) and sample obtained by long annealing at 300 °C (sample 3, dotted line). | ||
In order to explain the origin of the low temperature cubic phase observed in sample 2, as well as to understand its thermal evolution, TG+MS measurements have been performed. Fig. 9 shows the TG curves of sample 2 and the MS curves corresponding to H2O (m/z = 18) and CO2 (m/z = 44). The difference between the total weight loss of sample 2 (2.35%) and sample 1 (0.5%), reflects the more pronounced aging of the former. Three different steps can be distinguished in the TG curve of sample 2, at temperatures of about 250 °C (step 1), 400–450 °C (step 2) and from 550 to 700 °C (step 3). The accompanying MS curves show that the first weight loss is almost exclusively due to water, whereas in the second step, both H2O and CO2 are released and CO2 is the main contributor to the mass loss above 500 °C. The same steps are, in fact, observed in the TG curve of sample 1, but much weaker than in sample 2. Analogously, the species released along the thermal evolution of sample 1 are qualitatively the same as for sample 2 but of a much smaller magnitude. MS results are crucial in order to explain the mechanisms behind the three steps observed in TG experiments. The first mass loss is attributed to the removal of water molecules inserted in the garnet structure. Its intensity is highly variable from one sample to another; for sample 2, it amounts to ∼0.2 molecules of H2O per garnet formula, whereas for sample 1, it is almost negligible (<0.05 water molecules). The observation of a tetragonal to cubic structural change at around 150 °C by XRD even under vacuum and the recovery of the tetragonal phase above 250 °C, coincident with the water loss, suggest that these water molecules have entered the garnet structure and are not merely adsorbed at the surface. It also suggests that water is involved in the appearance of the cubic phase below 200 °C in sample 2.
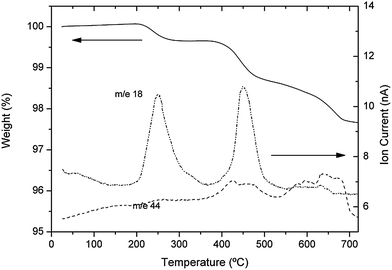 | ||
| Fig. 9 TG/MS curves of sample 2 (aged). | ||
The second and third steps are reminiscent of the thermal evolution shown by other Li-containing materials after prolonged aging in air,25,30,32 which is usually attributed to the removal of water and CO2, subsequent to a Li/proton exchange mechanism. In air, the process is thought to consist of the sequential steps of proton/lithium exchange, forming LiOH as the intermediate, followed by the carbonation of the lithium hydroxide to give Li7-xHxLa3Zr2O12 and x/2 Li2CO3 as the final products.25 Evidence that Li+/H+ exchange is indeed taking place in our samples is provided by the detection of lithium carbonate in Raman experiments presented in the next section. On heating, the reverse reaction would take place in two steps that can be monitored by TG experiments:
| Li7−xHxLa3Zr2O12 (+x/2 Li2CO3) → Li7−xLa3Zr2O12−x/2 (+x/2 Li2CO3) + x/2H2O | (1a) |
| Li7−xLa3Zr2O12−x/2 + x/2 Li2CO3 → Li7La3Zr2O12 + x/2 CO2. | (1b) |
Note that, at the end, the garnet stoichiometry is recovered.
At this point, a discussion about the order in which (1a) and (1b) processes occur is pertinent. In LLTO25 and the Ruddlesden–Popper compound Li2SrTa2O7 (ref. 32), both processes occur simultaneously; i.e. lithium is reinserted at the same time as the protons leave the sample in the form of water. As regards the lithium-conducting garnets, a mention is made in ref. 29 to lithium reinsertion in Li7La3Sn2O12 upon heating the exchanged sample above 800 °C and no information about lithium reinsertion from Li2CO3 is given for aged cubic garnets. Our own results for Li5La3Nb2O12 show that both processes occur almost simultaneously in this compound.33 We will show in the following that in Li7La3Zr2O12, the two processes are separated by at least 200 °C, so that (1a) occurs between 400 and 500 °C (in step 2) and (1b) between 600 and 700 °C (in step 3).
If the exchange mechanism were the only one taking place, a mass relation of (1a)/(1b) ≃ 0.4 would hold between steps 2 and 3. That ratio, however, is not verified experimentally. The exchange mechanism does not explain the release of CO2 in the second step evidenced by TG+MS experiments, since we have seen by Raman spectroscopy (see next section) that Li2CO3 disappears at much higher temperatures, well within step 3. Therefore, another mechanism involving a carbonate reaction is needed. The nature of such a carbonate has been elucidated again with the help of Raman spectroscopy (see next section), which shows that, besides Li2CO3, another CO2 source arises from a small amount of La2O2CO3 formed on heating just above the second step of TG. The origin of the oxycarbonate can be explained as follows:
Small amounts of La2O3 may remain from the synthesis as impurities undetectable at the XRD level. In air, lanthanum oxide reacts with water and CO2 to give quasiamorphous hydroxycarbonates such as La2(OH)2(CO3)2, which decomposes in two steps as34
| (450–500 °C): La2(OH)2(CO3)2 → La2O2CO3 + H2O + CO2 | (2a) |
| (650–700 °C): La2O2CO3 → La2O3 + CO2 | (2b) |
The relation between the masses released in both steps would be (2a)/(2b) = 1.41, which, when appropriately combined with the exchange process, may explain both the detection of CO2 in step 2 as well as the relative magnitude of steps 2 and 3. Note that the temperatures of processes (2a) and (2b) are coincident with those proposed for the exchange mechanisms (1a) and (1b).
A quantitative analysis of the different processes taking place in the garnet, hydration, H+ exchange and carbonation, can be achieved by combining the TG and MS data. The CO2 yield arising from the decomposition of La and Li carbonated phases can be worked out separately by assuming that eqn (1) and (2) hold. For the as-grown samples, as sample 1, where no Li2CO3 is detected by Raman spectroscopy, the garnet composition would be the stoichiometric one, Li7La3Zr2O12. Note that, in this case, the weak mass losses reflected in the TG+MS data (Fig. 8) agree almost exactly with the expectations from eqn (2) assuming a content of 0.05 moles of La-hydroxycarbonate per garnet formula. Applied to the slightly hydrated sample (sample 2), this procedure gives a lithium carbonate content of 0.07 moles which, together with the 0.2 water molecules released at 250 °C, results in a garnet composition of about Li6.86H0.14La3Zr2O12·(0.2H2O). For this sample, the calculated CO2 yield derived from La-related phases through processes (2a) and (2b) is ∼0.1 moles per garnet formula at each step. Following eqn (1a) and (1b) and allowing for the previous dehydration step, the thermal evolution of this garnet would be
| At 200–250 °C: Li6.86H0.14La3Zr2O12·(0.2H2O) → Li6.86H0.14La3Zr2O12 + 0.2H2O |
| At 400–450 °C: Li6.86H0.14La3Zr2O12 → Li6.86La3Zr2O11.93 + 0.07H2O |
| At 650–700 °C: Li6.86La3Zr2O11.93 + 0.07Li2CO3 → Li7La3Zr2O12 + 0.07CO2 |
If we assume that the TG experiment in air has been fast enough so that it can be compared to the XRD high temperature experiment in vacuum, where only the initial state of the sample plays a role in the appearance of the cubic phase, and putting side by side the phase transformations and the mass losses, both in magnitude as in temperature, we can deduce that the presence of water molecules in the sample is responsible for the low temperature transformation to cubic symmetry. Upon heating above 250 °C, water is released and the tetragonal symmetry is partially recovered. On the basis of the simultaneous occurrence of hydration and exchange mechanisms in the aged garnet, the recovery of tetragonal symmetry above 250 °C would come just from local areas where proton exchange has not occurred or has occurred very weakly. In contrast, areas where proton exchange has been important will remain cubic upon the first dehydration process. In fact, we shall show in the next section that exchange alone also results in a cubic phase. With these ideas in mind, we can understand the results of the high temperature experiments under air where the incorporation of additional H2O during the experiment and the presence of CO2 are probably favoring the proton/lithium exchange thus preventing the reversal to tetragonal symmetry.
The phase evolution of sample 2 above 300 °C can also be interpreted in terms of the coexistence of regions with varying lithium stoichiometry after the first dehydration step. According to the model that relates the T → C phase transition with lithium dynamics and stoichiometry,35 the lithium deficient regions would reach the cubic phase at temperatures lower than the lithium rich regions, thus resulting in a progressively increasing proportion of the cubic phase as temperature is increased, until the whole sample reaches the cubic state. This behavior is fully in agreement with the phase percentage evolution shown in Fig. 7.
We conclude that TG results can be explained through the combined effect of garnet hydration, lithium/proton exchange and carbonate decomposition. The relative contribution of each mechanism to the thermal evolution will depend on the initial stoichiometry (hydration degree, presence of impurities) and the thermal history of each sample. We note that the mass losses shown in Fig. 8 and 9 are in fact small, so that secondary phases such as lithium or lanthanum carbonates may be undetectable by XRD.
3.3 Raman spectroscopy
XRD and TG results show that the thermal evolution of tetragonal Li7La3Zr2O12 is determined by the sample condition and also by the atmosphere in which the experiments are performed.Different chemical reactions are proposed to explain the TG results, but the relation between such reactions and the structural evolution of the garnet is not evident. Reactions involving residual impurities such as lanthanum carbonates, for instance, may be required to explain some TG features and get an accurate quantification of the H+/Li+ exchange level, although not being related to the evolution of the garnet compound. Since no secondary phases have been detected by XRD, we have drawn upon the sensitivity of Raman spectroscopy to help in the identification of such phases as a means to understand the mechanisms of structural evolution in the garnet.
In order to identify the Raman spectra of both tetragonal and cubic phases, an Al-stabilized cubic Li7La3Zr2O12 garnet has been synthesized at 1020 °C by doping with 0.25 at. per formula and using Al2O3 crucibles, as previously reported by other authors.16 As shown in the inset of Fig. 10, the XRD pattern of the resulting product is that of a cubic garnet (space group Iad). The lattice constant was refined by the FullProf program to be a′ = 12.9651 (2) Å; ICP-OES gives an Al content of 0.225 at. per formula. Fig. 10 shows the Raman spectra of both tetragonal and cubic Li7La3Zr2O12 garnets, which agree with the spectra reported recently.36 Raman spectra have been used in this paper for phase identification purposes and no attempt has been made to attribute the origin of each band in detail. According to group theory, 81 and 51 modes are expected for the tetragonal and cubic Li7La3Zr2O12 garnets, respectively. Group theory also tells us that Zr cations are not Raman active since they occupy a site with inversion symmetry. Qualitatively, we may assign the bands between 100 and 150 cm−1 to the vibration of the heavy La cations and the band at about 640 cm−1 to the Zr–O bond stretching, which is expected to have the strongest force constant. By analogy with other systems, lithium vibrations are expected between 300 and 600 cm−1. These may, of course, be mixed with oxygen displacements. Oxygen bending modes are likely to be responsible for the bands appearing between 200 and 300 cm−1. The broad aspect of the spectrum of the cubic garnet is attributed both to static and (possibly) dynamic disorder.
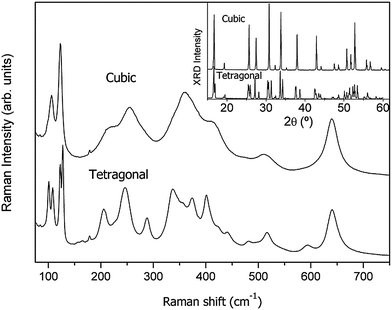 | ||
| Fig. 10 Raman spectra and X-ray diffractograms (inset) of as-synthesized tetragonal and cubic (Al-doped) Li7La3Zr2O12. | ||
In the TG-DSC experiments of LLZO garnets, several steps of weight loss were observed. To elucidate the processes or reactions involved in each step, portions of sample 2 were heated at selected temperatures and then cooled down to RT. Since thermal broadening effects obscure spectral details at high temperature, we present in Fig. 11 spectra measured at RT after cooling from 300, 550 and 700 °C (heating at 10 °C min−1 and cooling at 30 °C min−1; dwell times between 4 min (at 300 °C) and 15 minutes (at the other temperatures)), together with the spectrum of the starting material. After a short treatment at 300 °C, just after the first dehydration step (curve b), the spectrum is similar to that of the starting tetragonal sample (curve a) although some broadening and intensity increase is observed between 300 and 400 cm−1. By subtracting, using the appropriate factors, the tetragonal spectrum from curve b, we obtain curve c, which closely resembles the cubic reference spectrum of Fig. 10, thus highlighting the coexistence of the tetragonal and cubic phases of Li7La3Zr2O12 after heating at 300 °C. The subtracted spectrum also evidences lithium carbonate bands at 97, 156 and 195 cm−1,37 which are attributed to the exchange and carbonation mechanism explained in the previous section. A close look at the spectrum of the starting material shows that these bands were weakly present in curve a, implying that some exchange has already been produced by aging at RT and, in fact, a certain degree of proton exchange was required to explain the TG+MS experiments of this sample described in Section 3.2. The increase of the carbonate band upon cooling from 300 °C indicates that the process is enhanced by heating at that temperature.
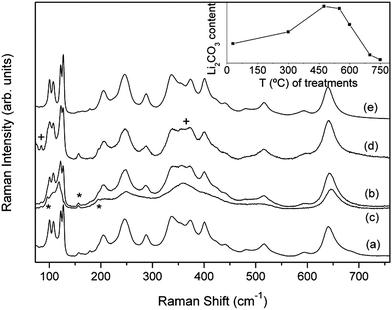 | ||
| Fig. 11 Room temperature Raman spectra of aged sample 2 after a short treatment at different temperatures in air. (a) Starting material; (b), (d), and (e) after cooling from 300, 550 and 700 °C, respectively. Curve “c” is obtained by subtracting the initial spectrum from the spectrum after 300 °C (curve b). (*) Li2CO3; (+) II-La2O2CO3. The inset shows the integrated intensity relation between the Li2CO3 band at 156 cm−1 and the area of the whole spectrum measured after cooling from each temperature. | ||
When the sample is cooled after 475 °C (not shown) or 550 °C (curve d), the amount of Li2CO3 increases significantly and an extra band appears (or is enhanced) at 83 cm−1. Intensity also increases around 363 cm−1, both frequencies being characteristic of II-La2O2CO3.38 The finding of lanthanum oxycarbonate just after the second step of weight loss together with the detection of CO2 as a byproduct in TG-MS analysis at the same temperatures suggest that La2O2CO3 may be formed in the second step of TG through the decomposition of hydrated lanthanum carbonates, as described in the previous section.
After cooling from 750 °C (curve e), the sample recovered a purely tetragonal spectrum and both lithium carbonate and lanthanum oxycarbonate disappeared, thus supporting that the weight loss observed in the TG curve of this sample above 600 °C is associated with the CO2 removal from La2O2CO3 and Li2CO3.
The amount of Li2CO3 formed at each temperature has been evaluated qualitatively by working out the integrated intensity of the carbonate band at 156 cm−1 relative to the area of the whole spectrum (see the inset of Fig. 11). This procedure gives interesting results: above the carbonate content of the starting material, segregation of Li2CO3 is remarkably enhanced on heating above 300 °C, which confirms the activated character of the exchange process. Moreover, it shows that carbonate formation is still enhanced on heating above the second TG step, where water release from exchanged protons occurs. Fig. 11 also shows that Li2CO3 disappears completely only after heating at 700–750 °C, confirming that the exchange process is reversible, as expected from eqn (1a) and (1b), but requires temperatures much higher in Li7La3Zr2O12 than in cubic garnets or lithium-conducting oxides.25,32,33
We now deal with the relationship between the lithium/proton exchange mechanism and the formation of a cubic phase. To verify to what extent both observations are related, we have submitted a fresh tetragonal sample to prolonged annealing at 300 °C in air (sample 3). The RT Raman spectrum of that sample after 30 hours at 300 °C shown in Fig. 12 is typical of a cubic phase of Li7La3Zr2O12 with a remarkable amount of Li2CO3 as the secondary phase. The cubic symmetry of the garnet is confirmed by its XRD pattern (see the inset of Fig. 12). FullProf fitting yields a′0 = 12.9859 (2) Å, which means that the lattice is slightly expanded (Vc = 2190 Å3) compared to the tetragonal one (Vt = 2173 Å3, see Table 1), a fact that is attributed to proton insertion. We note that a similar effect of lattice expansion was observed in Li+/H+ exchanged Li7La3Sn2O12.29 The enhancement of the bands corresponding to Li2CO3 in parallel with the development of the cubic phase confirms the correlation between the exchange of lithium ions by protons and the stabilization of a cubic garnet.
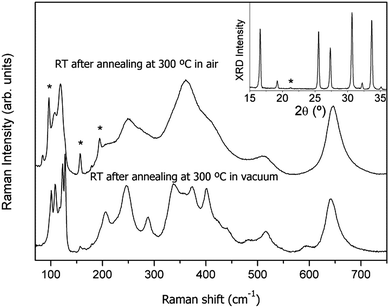 | ||
| Fig. 12 Room temperature Raman spectra of Li7La3Zr2O12 after annealing at 300 °C in air and in vacuum. Inset: X-ray diffractogram of Li7La3Zr2O12 after annealing at 300 °C in air. (*) Li2CO3. | ||
The TG and DSC curves of sample 3 are shown in Fig. 8. Two aspects are remarkable in these curves: the total mass loss of the exchanged sample amounts to 5.4%, to be compared with that of the non-hydrated sample 1 (0.5%) or the aged sample 2 (2.35%), a fact that is attributed to the increased amount of water and CO2 trapped by the sample through the exchange process. A quantitative analysis of H2O and CO2 losses, performed in a similar manner as for sample 2, gives a Li2CO3 content of 0.34 moles per garnet formula, from which a composition close to Li6.32H0.68La3Zr2O12 is derived for the cubic garnet exchanged at 300 °C. It is interesting to note that, for this sample, the number of CO2 moles related to the La-based mechanism (∼0.3) is several times higher than in the as-grown samples, which suggests that La ions from the garnet may also be involved, probably through a surface reaction with water and CO2.
The DSC curve of sample 3 shows no endothermic peak in the region of 645 °C, where the phase transition appears for stoichiometric samples, but an exothermic peak is clearly observed in the cooling run at Tc = 615 °C. The absence of the peak in the heating run is in agreement with the cubic symmetry of the exchanged sample, and the reappearance of the peak in the cooling run is attributed to lithium reinsertion occurring, as we have seen, between 650 and 700 °C in Li7La3Zr2O12.
Annealing experiments were also performed under dynamic vacuum conditions (3.2 × 10−5 mbar). As shown in Fig. 12, after 8 h at 300 °C under vacuum, the fresh sample retains the initial tetragonal symmetry and the amount of Li2CO3 is negligible, confirming that no exchange has taken place.
3.4 Phase transition in Li7La3Zr2O12
Li7La3Zr2O12, together with Sn and Hf analogues, are the only members of the family of lithium conducting garnets presenting stable tetragonal symmetry at RT.7,24,39 The initial report of a cubic modification synthesized above 1100 °C (ref. 6) was later unambiguously ascribed to the incorporation of Al at lithium sites.21 Later work focused on achieving cubic phases with high conductivity by doping the LLZO garnet with a number of cations, either at the Li, the Zr or even the La site.13–16,19,20 Attempts to obtain a cubic garnet by low temperature methods have also been successful.8According to data presented in this work, the undoped tetragonal Li7La3Zr2O12 garnet undergoes a reversible first-order phase transition to a cubic structure with Tc ≃ 645 °C on heating and Tc ≃ 625 °C on cooling. According to density-functional theory plus molecular dynamics calculations,35 the mechanism producing the phase transition would be the activation of lithium dynamics, resulting in a disordering of the lithium sublattice and, as a consequence, a change from tetragonal to cubic symmetry with no change in cell volume, as observed in this work. A direct relation between lithium stoichiometry and the Tc value is proposed in ref. 35. The critical temperature found in this work is in agreement with the range (800 K ≤ Tc ≤ 1000 K) proposed by Bernstein et al. for stoichiometric Li7La3Zr2O12. It is interesting to compare the critical temperature for the T–C phase transition in Li7La3Zr2O12 (645 °C) with that of the Sn analogue (between 750 and 800 °C).24 Since the phase transition is attributed to lithium disorder,35 the lower Tc of Li7La3Zr2O12 should be correlated with a lower temperature for the activation of lithium dynamics in the zirconate compared with Li7La3Sn2O12. In fact, we can also compare Tc values with the conductivities in both garnets, which are of the order of 10−6 and 10−8 S cm−1 for the tetragonal Zr and Sn garnets at RT, respectively.7,24
On the other hand, we have proved that the appearance of a cubic phase between 100 and 200 °C, previously reported by a number of authors,21–23 is due to garnet hydration either during the experiments or due to storage conditions and cannot be strictly considered a phase transition. The influence of the hydration degree of the starting material is proved by comparing the XRD thermal evolution in vacuum of a freshly synthesized sample and a sample shortly exposed to air (Fig. 4 and 5). The influence of the atmosphere is tested by comparing the XRD evolution of the same sample in air and in vacuum (Fig. 3 and 5).
A sample not exposed to humidity at any moment keeps the tetragonal symmetry up to the high temperature T → C phase transition. In the presence of water, a cubic phase is always formed below 200 °C. The relative amount of the cubic phase and the temperature at which it appears depend not only on the environmental humidity and the hydration degree of the sample, but also on experimental parameters such as the heating rates and the dwell times at each temperature.
Two hydration mechanisms are proposed to explain the formation of a cubic phase well below Tc: the first one involves the insertion of water molecules into the garnet structure and the second one is protonation through the H+/Li+ exchange mechanism, as explained in Section 3.2. Evidence of direct hydration is provided by the detection of a mass loss around 200 °C consisting only of water molecules, whereas evidence of the exchange process is provided by the detection of lithium carbonate in samples exposed to moisture, either by storage conditions at RT or upon heating in air. Each of these two mechanisms separately may yield cubic symmetry. Though they can occur simultaneously, they operate in different temperature ranges: direct hydration acts typically below 200 °C whereas the exchange is more efficiently activated at higher temperatures. Allowing for the simultaneous occurrence of both processes, we may write the hydrated garnet in generic form as Li7−δHδLa3Zr2O12·nH2O.
The evolution of the phase proportion in the partially hydrated sample after the first dehydration step is interpreted in terms of the coexistence of regions with varying stoichiometry, so that the lithium-poor regions would undergo the T–C phase transition at lower temperatures than the lithium-rich regions, thus resulting in a continuous increase of the C/T ratio, in agreement with the predictions of ref. 35.
A cubic proton-exchanged garnet has been formed by prolonged annealing of LLZO at 300 °C in air. About 10% of the lithium ions have been exchanged after annealing for 30 h. The thermal analysis of that sample, as well as the Raman spectra of a partially hydrated sample submitted to heating–cooling cycles, show that the exchange is reversible, but lithium reinsertion occurs at temperatures much higher in LLZO than in other garnets or lithium-conducting oxides.
4 Conclusions
A reversible tetragonal to cubic phase transition has been detected for the Li7La3Zr2O12 garnet, with Tc around 620–650 °C. This phase transition is ascribed to lithium disorder and does not involve any change in the stoichiometry of the material.On the other hand, the Li7La3Zr2O12 garnet has been shown to be very sensitive to ambient conditions, especially to exposure to moisture. Garnet hydration results in the appearance of a cubic phase at temperatures much below Tc, either by direct insertion of water molecules into the garnet structure or through a H+/Li+ exchange mechanism.
The nature of both cubic garnets is totally different: the one found above the phase transition does not involve any change in the stoichiometry, whereas the cubic phase formed at low temperature is a hydrated, lithium defective phase.
Acknowledgements
This work has been supported by the Spanish Ministerio de Economía and Feder funds through grant MAT2010-19837-C06-06. A. Orera acknowledges the financial support provided by CSIC and ESF through a JAE-Doc contract. G. Larraz acknowledges the financial support provided by Gobierno de Aragón through a PhD grant (B108/11). The authors wish to thank CAI DRX (U. Complutense, Madrid) and Servicio General de Apoyo a la Investigación-SAI (Universidad de Zaragoza) for technical support in XRD experiments, and the Servicio Interdepartamental de Investigación (Universidad Autónoma de Madrid) for technical support in TG-MS experiments. The authors also acknowledge J. Percival and P.R. Slater for fruitful discussions.References
- E. J. Cussen, J. Mater. Chem., 2010, 20, 5167–5173 RSC.
- V. Thangadurai, H. Kaack and W. J. F. Weppner, J. Am. Ceram. Soc., 2003, 86, 437–440 CrossRef CAS.
- V. Thangadurai and W. Weppner, J. Am. Ceram. Soc., 2005, 88, 411–418 CrossRef CAS.
- V. Thangadurai and W. Weppner, Adv. Funct. Mater., 2005, 15, 107 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Ionics, 2007, 13, 195–203 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS.
- J. Awaka, N. Kijima, H. Hayakawa and J. Akimoto, J. Solid State Chem., 2009, 182, 2046–2052 CrossRef CAS.
- N. Janani, S. Ramakumar, L. Dhivya, C. Deviannapoorani, K. Saranya and R. Murugan, Ionics, 2011, 17, 575–580 CrossRef CAS.
- H. Xie, J. A. Alonso, Y. Li, M. T. Fernández-Díaz and J. B. Goodenough, Chem. Mater., 2011, 23, 3587–3589 CrossRef CAS.
- H. Xie, Y. Li and J. B. Goodenough, Mater. Res. Bull., 2012, 47, 1229–1232 CrossRef CAS.
- Y. Shimonishi, A. Toda, T. Zhang, A. Hirano, N. Imanishi, O. Yamamoto and Y. Takeda, Solid State Ionics, 2011, 183, 48–53 CrossRef CAS.
- I. Kokal, M. Somer, P. H. L. Notten and H. T. Hintzen, Solid State Ionics, 2011, 185, 42–46 CrossRef CAS.
- S. Ohta, T. Kobayashi and T. Asaoka, J. Power Sources, 2011, 196, 3342–3345 CrossRef CAS.
- A. Logeat, T. Koehler, U. Eisele, B. Stiaszny, A. Harzer, M. Tovar, A. Senyshyn, H. Ehrenberg and B. Kozinsky, Solid State Ionics, 2012, 206, 33–38 CrossRef CAS.
- R. Murugan, S. Ramakumar and N. Janani, Electrochem. Commun., 2011, 13, 1373–1375 CrossRef CAS.
- M. Kotobuki, K. Kanamura, Y. Sato and T. Yoshida, J. Power Sources, 2011, 196, 7750–7754 CrossRef CAS.
- H. Buschmann, J. Doelle, S. Berendts, A. Kuhn, P. Bottke, M. Wilkening, P. Heitjans, A. Senyshyn, H. Ehrenberg, A. Lotnyk, V. Duppel, L. Kienle and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 19378–19392 RSC.
- E. Rangasamy, J. Wolfenstine and J. Sakamoto, Solid State Ionics, 2012, 206, 28–32 CrossRef CAS.
- M. A. Howard, O. Clemens, E. Kendrick, K. S. Knight, D. C. Apperley, P. A. Anderson and P. R. Slater, Dalton Trans., 2012, 41, 12048–12053 RSC.
- E. Rangasamy, J. Wolfenstine, J. Allen and J. Sakamoto, J. Power Sources, 2013, 230, 261–266 CrossRef CAS.
- C. A. Geiger, E. Alekseev, B. Lazic, M. Fisch, T. Armbruster, R. Langner, M. Fechtelkord, N. Kim, T. Pettke and W. Weppner, Inorg. Chem., 2011, 50, 1089–1097 CrossRef CAS.
- S. Adams and R. P. Rao, J. Mater. Chem., 2012, 22, 1426–1434 RSC.
- J. Percival, Synthesis and Characterisation of Novel Lithium Ion Containing Garnet-related Materials for Potential Lithium Ion Battery Applications, Ph.D. thesis, University of Surrey at Guilford, July 2009.
- J. Percival, E. Kendrick, R. I. Smith and P. R. Slater, Dalton Trans., 2009, 1–5 Search PubMed.
- A. Boulant, J. F. Bardeau, A. Jouanneaux, J. Emery, J. Y. Buzare and O. Bohnke, Dalton Trans., 2010, 39, 3968–3975 RSC.
- L. Truong and V. Thangadurai, Chem. Mater., 2011, 23, 3970–3977 CrossRef CAS.
- L. Truong and V. Thangadurai, Inorg. Chem., 2012, 51, 1222–1224 CrossRef CAS.
- M. Nyman, T. M. Alam, S. K. McIntyre, G. C. Bleier and D. Ingersoll, Chem. Mater., 2010, 22, 5401–5410 CrossRef CAS.
- C. Galven, J. L. Fourquet, M. P. Crosnier-Lopez and F. Le Berre, Chem. Mater., 2011, 23, 1892–1900 CrossRef CAS.
- C. Galven, J. Dittmer, E. Suard, F. Le Berre and M. P. Crosnier-Lopez, Chem. Mater., 2012, 24, 3335–3345 CrossRef CAS.
- J. Rodríguez-Carvajal, Physica B, 1993, 192, 55–69 CrossRef.
- C. Galven, J. L. Fourquet, E. Suard, M. P. Crosnier-Lopez and F. Le Berre, Dalton Trans., 2010, 39, 4191–4197 RSC.
- A. Oreraet al., unpublished.
- M. Akinc and D. Sordelet, Adv. Ceram. Mater., 1987, 2, 232–238 CAS.
- N. Bernstein, M. D. Johannes and K. Hoang, Phys. Rev. Lett., 2012, 109, 205702 CrossRef CAS.
- F. Tietz, T. Wegener, M. T. Gerhards, M. Giarola and G. Mariotto, Solid State Ionics, 2013, 230, 77–82 CrossRef CAS.
- N. Koura, S. Kohara, K. Takeuchi, S. Takahashi, L. A. Curtiss, M. Grimsditch and M. L. Saboungi, J. Mol. Struct., 1996, 382, 163–169 CrossRef CAS.
- A. Orera, G. Larraz and M. L. Sanjuán, J. Eur. Ceram. Soc., 2013, 33, 2103–2110 CrossRef CAS.
- J. Awaka, N. Kijima, K. Kataoka, H. Hayakawa, K. I. Ohshima and J. Akimoto, J. Solid State Chem., 2010, 183, 180–185 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ta11996c |
| This journal is © The Royal Society of Chemistry 2013 |