 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The room temperature formation of gold nanoparticles from the reaction of cyclohexanone and auric acid; a transition from dendritic particles to compact shapes and nanoplates†
Madeeha A.
Uppal
a,
Andreas
Kafizas
b,
Michael B.
Ewing
a and
Ivan P.
Parkin
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: i.p.parkin@ucl.ac.uk; Fax: +44 (0)20 7679 7463
bDepartment of Chemistry, Imperial College London, South Kensington Campus, London SW7 2AZ, UK
First published on 9th May 2013
Abstract
A new straightforward method for the synthesis of gold nanoparticles from addition of cyclohexanone to aqueous solutions of auric acid at room temperature is presented. By understanding this process we have discovered a new organic chemistry transformation reaction for converting cyclic ketones to α-chloro ketones and a mechanism for the nanoparticle formation. Contrary to conventional gold nanoparticle syntheses, the reaction “self-initiates” at room temperature and forms an increasingly red solution over ≈60 minutes. By studying the gold colloid's formation using transmission electron microscopy it was observed that large dendritic (63 ± 21 nm diameter) structures made of clustered particles (6 ± 1 nm) were initially formed. These dendritic particles then compacted into an array of denser shapes that slowly increase in size until the reaction is complete. The most prominent shapes observed were spheres (43 ± 7 nm); other shapes included dodecahedra (39 ± 10 nm) triangular (≈50 nm in height) and hexagonal (≈70 nm wide) nanoplates. The solution was stable to precipitation for over 3 months. During this period the nanoplate structures substantially increased in size (triangular ≈ 250 nm, hexagonal ≈ 320 nm) whereas other structures showed no further growth. X-ray diffraction studies demonstrated that the gold nanoparticles were crystalline. The formation of the 2-chlorocyclohexanone by-product was observed in solution phase 1H & 13C NMR, gas phase chromatography and IR spectroscopy. A mechanism is presented to account for this by-product and the reduction of auric acid to gold.
Introduction
Gold nanoparticle colloids have received a great deal of interest because of their intense and size/shape tuneable surface plasmon resonance (SPR) properties.1–3 Gold nanoparticles are used in a wide sector of applications as optical enhancers in composite semiconductor films,4–7 bactericides,8,9 biological markers,10 DNA and single molecule detectors11,12 and in catalysts.13 The most common method for preparing gold colloids is based on the thermal reduction of aqueous auric acid with tri-citrate, devised by Turkevich et al.;14,15 where solutions are typically heated to 100 °C for the reaction to initiate. Gold nanoparticles have been synthesized by heating auric acid solutions with a variety of fruit extracts.16 These fruit extracts contained several ketone functional groups and it may be that these functional groups mediate the formation of multi-shaped nanoparticles. Such multi-shaped gold nanoparticles have been used to investigate the biological toxicity of gold nanoparticles in zebrafish embryos17 and mammalian cells18 but are yet to be applied in a functional material. Silver nanoparticles of different shapes and sizes have also been found to induce multi-colour photochromism when photo-chemically loaded onto TiO2 thin-films.19The use of ketones in precious metal nanoparticle synthesis has been reported – although all routes involve significant heating and no mechanism for their formation has been proposed. Truncated gold nanocubes could be produced using an electrochemical approach and an acetone injection stage.20,21 Dendritic nanoparticle structures comprised of Pd have been synthesized by reaction of the Pd acetate salt mixed with ethylene glycol and KOH in a methyl isobutyl ketone solvent initiated by microwave heating.22 This formed stable ≈50 nm wide dendritic structures. The formation of large 2D triangular and hexagonal nanoplates has also been previously observed.23–25 By mixing gold seeds with a mixture of CTAB, PEG and the ketone polymer PVP a slow room temperature reaction over 8 hours produced platelet-type particles that were typically ≈5 nm thick and ≈200 to 500 nm wide. It was also shown that microwave heating (85 °C) of aqueous solutions of auric acid with long-chained PVPs yielded similar structures.26 Mixing aqueous auric acid solutions with a lemongrass plant extract (Cymbopogon flexuosus) at room temperature produced nanoplate mixtures with lengths on the micrometer scale, where the directional growth was related largely to the ketone groups present in the extract.27
To our knowledge, the use of cyclohexanone in initiating a self-propagating gold nanoparticle formation reaction has not previously been reported. Nevertheless, the use cyclohexanone–water–isopropanol mixtures in synthesizing a range of copper based nanoparticles28 and cyclohexanone as solvent and capping agent for forming gold nanoparticles through laser ablation of a submerged gold target.29 It was also shown that aryl ketones can photocatalytically reduce solutions containing Au3+ to Au0.30 The growth mechanisms for traditional gold nanoparticle formation methods have come to be somewhat understood through in situ monitoring methods such as UV-visible spectroscopy, dynamic light scattering, electrode potential measurements or through the extraction and transition electron microscopy analysis of aliquots at set times during the reaction.31,32
We show that cyclohexanone reacts with auric acid and causes a “self-initiated” room temperature nanoparticle formation reaction to commence. During this reaction the particles undergo several shape changes. Initially, large dendritic structures composed of smaller aggregates form that later compact primarily into spherical particles and a fraction form alternative 3D structures such as triangular prisms and dodecahedrons and large flat 2D hexagonal and triangular nanoplates. We also provide an insight into the underpinning mechanism showing that the cyclohexanone acts as the reducing agent; reducing Au3+ in auric acid to Au1+ (that later disproportionates to Au0) and being converted to 2-chlorocyclohexanone in this process. This new method of multi-shaped nanoparticle synthesis is reproducible and amongst the simplest reported33,34 in that it involves no heating stage, occurs at room temperature and just requires one stage of reagent mixing. Furthermore, the nanoparticles formed are stable against precipitation for more than 3 months stored at room temperature.
Experimental section
All reagents were purchased from Sigma-Aldrich UK. All glassware was washed with aqua regia and rinsed with copious amounts of de-ionized water before use. All reactions were performed in beakers.The nanoparticle solution was made by mixing auric acid (HAuCl4·3H2O – 0.85 mg, 0.28 mM) in distilled water (9.5 ml) with cyclohexanone ((CH2)5CO – 0.50 ml, 0.47 g, 0.48 M). This solution did not require thermal activation (or any alternative energy source) and “self-initiated” the uniform formation of gold nanoparticles throughout the solution when conducted under room light or in a dark cupboard. The reaction was accompanied by a darkening of the solution. A further experiment was conducted where the cyclohexanone was injected gently on top of the aqueous auric acid solution to form a separate organic layer on top of the aqueous. In this case, the nanoparticle formation reaction occurred only at the emulsion interface as judged by the change in colour. The effect of temperature on the reaction was investigated by mixing auric acid (2.6 mg, 0.28 mM) in distilled water (28.5 ml) with cyclohexanone (1.5 ml, 1.4 g, 0.48 M) at ≈20, 30, 40, 50 and 60 °C. After first heating the aqueous auric acid solution to the desired temperature, the cyclohexanone reducing agent was rapidly injected, causing a thorough mixing of the solutions. The rate of consumption of auric acid in the reaction was determined by titrating solution aliquots during the nanoparticle formation reaction.35 This was achieved by scaling the reaction up by a factor of 5 and taking 2 ml aliquots out every 5 minutes for analysis. The nanoparticle formation reaction was quenched as soon as the aliquots were extracted by addition of excess KI (0.02 g). This caused all auric acid present within the aliquot to be consumed to AuI, forming an I2 side-product. The level of I2 formed, and thus the amount of auric acid originally present, was measured by titration against K2S2O3 (1 mM) using potato starch indicator (0.02 g) where the disappearance of blue-black colour indicated completion.
The size and shape of nanoparticles formed at room temperature by rapid mixing of the solution were investigated using transmission electron microscopy (TEM) using a Jeol 4000EX microscope. Solution aliquots were taken out from the nanoparticle reaction after 5, 20 and 65 minutes of reaction and placed onto lacey carbon-coated copper grids and allowed to air dry before being investigated by TEM.
Nanoparticles solutions formed at room temperature by rapid mixing were freeze dried to remove the aqueous solvent and any excess cyclohexanone to form a nanoparticle powder. The powder was analyzed by X-ray diffraction (XRD) using a micro-focus Bruker GADDS powder X-ray diffractometer with a monochromated CuKα (1.5406 Å) source and CCD area detector. After mixing the powder within a pressed KBr disc, the infrared (IR) spectrum of the solid was taken using a PerkinElmer RX-I instrument.
Nanoparticle solutions were centrifuged on a Beckman Coulter Avanti Centrifuge J-26 XP at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (≈48000g) causing the gold particles to rest at the bottom of the solution. The liquid layer above was removed and passed through a gas chromatograph (Clarus 500, PerkinElmer) equipped with flame ionization detector and 30 m capillary column (Ellite-1, cross-bond 100% dimethyl polysiloxane) and compared with standards to elucidate the reaction by-products. The gases released from the nanoparticle reaction were analyzed by IR spectroscopy and compared with the pure gas phase spectra of cyclohexanone and 2-chlorocyclohexanone in a sealed gas cell under vacuum. Three samples were examined by both 1H and 13C nuclear magnetic resonance (NMR) spectroscopy using a Bruker AV600 (600 MHz) instrument: (i) a gold nanoparticle solution (diluted by a factor of 100) formed from the reaction of auric acid and cyclohexanone in D2O at room temperature, (ii) cyclohexanone in D2O and (iii) 2-chlorocyclohexanone in D2O.
000 rpm (≈48000g) causing the gold particles to rest at the bottom of the solution. The liquid layer above was removed and passed through a gas chromatograph (Clarus 500, PerkinElmer) equipped with flame ionization detector and 30 m capillary column (Ellite-1, cross-bond 100% dimethyl polysiloxane) and compared with standards to elucidate the reaction by-products. The gases released from the nanoparticle reaction were analyzed by IR spectroscopy and compared with the pure gas phase spectra of cyclohexanone and 2-chlorocyclohexanone in a sealed gas cell under vacuum. Three samples were examined by both 1H and 13C nuclear magnetic resonance (NMR) spectroscopy using a Bruker AV600 (600 MHz) instrument: (i) a gold nanoparticle solution (diluted by a factor of 100) formed from the reaction of auric acid and cyclohexanone in D2O at room temperature, (ii) cyclohexanone in D2O and (iii) 2-chlorocyclohexanone in D2O.
Results and discussion
The self-initiated formation of gold nanoparticles was induced by reacting a thousand fold excess of cyclohexanone with aqueous solutions of auric acid at room temperature. Several photographs were taken at set time intervals during the self-initiated nanoparticle formation reaction, as displayed in Fig. 1. Two reactions were studied; (i) the rapid mixing of cyclohexanone with auric acid and (ii) the layering of cyclohexanone on top of the aqueous auric acid layer. In the case of thorough pre-mixing of the cyclohexanone reducing agent with the auric acid solution (Fig. 1(a)) no colour change was observed until after ≈10 minutes when the solution turned slightly pink. This coloration became more intense as the reaction proceeded. After ≈40 minutes of reaction the solution appeared purple and was more strongly coloured. After ≈60 minutes of reaction, the solution did not further deepen in colour, signifying the end of the gold nanoparticle formation reaction. When the solution was viewed off-angle, a two-tone effect was revealed where the solution showed a more orange tone when viewed at high angles and a more purple tone when looked at directly. | ||
| Fig. 1 A series of images of the same solution taken at time intervals of 0, 10, 20, 30, 40, 50 and 60 minutes reaction time (from left to right) in the self-initiated gold nanoparticle formation reaction of auric acid solution with cyclohexanone that was (a) shaken into the solution or (b) layered gently on top of the aqueous layer. | ||
In the case of gentle layering of cyclohexanone at the surface of the aqueous auric acid solution, an emulsion layer was formed in which the colour change and hence nanoparticles formation occurred solely at the cyclohexanone–water interface. Fig. 1(b) shows that just after addition of the reducing agent (0 min), two separate immiscible layers can be observed. Approximately 20 minutes into the reaction a pink layer began to form within the aqueous layer at the interface. As the reaction progressed, the pink layer deepened in colour and penetrated deeper into the aqueous layer as the nanoparticles diffused. After 60 minutes of reaction, almost half of the aqueous layer had turned pink. On comparing the two reactions it is clear that nanoparticle formation is more rapid in the solution that was thoroughly pre-mixed. However, one can also infer the importance of the partial miscibility of the cyclohexanone on the mechanism of nanoparticle formation.
The nanoparticle formation reaction, where the cyclohexanone initiator was well-mixed, was also followed using UV-visible spectroscopy over the 190–1100 nm range, as shown in Fig. 2. The spectra showed the formation of a band at 540–575 nm corresponding to the surface plasmon resonance (SPR) of Au nanoparticles. The UV-visible spectrum was taken every ≈70 seconds until the SPR band stopped increasing. Approximately 10 minutes after the cyclohexanone was added a SPR band started to appear. This continued to increase at a relative steady rate until after ≈65 minutes after cyclohexanone addition, when the SPR band suddenly stopped increasing in magnitude. The overall baseline also increased its absorbance during the process – this stopped its upward shift at the same time as the SPR band showed no further increase in magnitude.
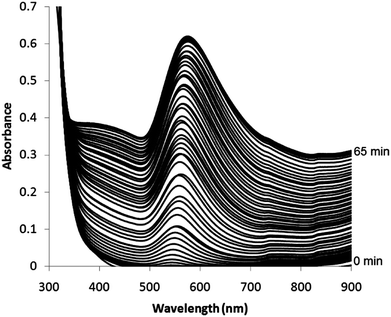 | ||
| Fig. 2 UV-visible spectrum from 300 to 900 nm monitoring the growth of gold nanoparticles in situ during their self-initiated formation reaction from an aqueous auric acid solution and cyclohexanone. Each spectrum was taken approximately every 70 seconds and the plasmon band appeared ≈10 minutes after the addition of cyclohexanone. The spectra are shown as recorded and no shift was applied to the baseline – this naturally shifted upwards with the growth of the SPR band. | ||
The centre (nm) and height (absorbance) of each progressively growing SPR band was extracted and plotted against reaction time (minutes), as shown in Fig. 3. As was seen in UV-visible absorbance spectra, an SPR band (543 nm, 0.01 absorbance units) first appeared 10 minutes after the addition of cyclohexanone. The SPR band then grew in height at a relatively steady rate of roughly 0.012 absorbance units per minute. The centre of the SPR also shifted to longer wavelengths. These two properties continued to increase until exactly 64 minutes after the addition of cyclohexanone, where they both reached a plateau. From 64 to 75 minutes reaction time both the SPR centre and height did not change significantly. Over the reaction period the SPR height increased from 0 to 0.61 absorbance units. In addition, the SPR centre shifted from 543 to 576 nm. The solutions were highly stable to precipitation and altered little after being stored for 3 months; although the (SPR centre decreased to 563 nm and the absorbance dropped to 0.59).
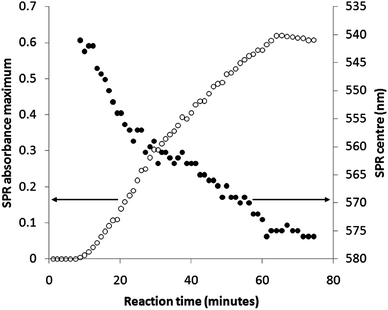 | ||
| Fig. 3 A plot of the centre (nm) and height (absorbance) of the progressively growing surface plasmon resonance band extracted from UV-visible spectra monitoring the growth of gold nanoparticles in situ during their self-initiated formation reaction from auric acid solution and cyclohexanone. | ||
It is well known that an increase in SPR centre wavelength signifies an increase in average nanoparticle size.15 Using the relationship derived by Haiss et al.,36 the increase in SPR centre from 543 to 576 nm signifies an average increase in nanoparticle diameter of ≈34 nm, assuming the particles are predominantly spherical.
The rate of consumption of auric acid in the reaction was determined by titrating solution aliquots during the nanoparticle formation reaction.35 The level of auric acid was found to decrease linearly over the time period (Fig. 4); decreasing from 0.26 mM at the start of the reaction (0 minutes) at a rate of 6.2 μM min−1 to there being no presence of auric acid after 45 minutes of reaction. This was in contrast with nanoparticles formed by the reaction of auric acid and citric acid (the Turkevich method), where during the reaction a sharp change in auric acid concentration is observed at a critical point.31 From our spectroscopic studies it was observed that surface plasmon band formation, indicating the formation of gold nanoparticles, terminated after ≈65 minutes of reaction, whereas our titrations showed that the auric acid was depleted after 45 minutes. This showed that there was a 20-minute lag between the complete reduction of gold ions and their subsequent nucleation to form nanoparticles with detectable surface plasmon resonance band.
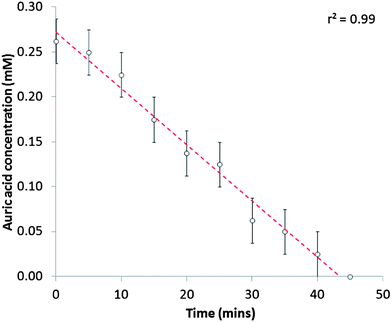 | ||
| Fig. 4 Auric acid (mM) depletion in the self-initiated gold nanoparticle formation reaction of auric acid (0.28 mM) with cyclohexanone (0.48 M). This was measured by quenching the reaction in sample aliquots with iodide salt and measuring the level of I2 side-product by titration with thiosulfate using starch indicator. The decrease in auric acid concentration was strongly linear (r2 = 0.99). | ||
The gold nanoparticle formation reaction was repeated with solution aliquots being extracted 5, 20 and 65 minutes after the addition of cyclohexanone and placed on lacey carbon-coated copper grids for transmission electron microscopy (TEM) analysis. Representative TEM images are shown in Fig. 5. Fig. 5(a) shows that after 5 minutes of reaction dendritic structures had formed, which ranged in size from 20 to 93 nm in diameter. The average diameter and standard deviation of these dendritic structures was 63 ± 21 nm. On closer inspection it can be seen that these dendritic structures are composed of clusters of smaller particles. These particles were predominantly spherical and ranged in size from 4.0 to 10 nm. The average diameter and standard deviation of these particles of which the dendritic structures were composed was 6 ± 1 nm. Fig. 5(b) shows a less magnified image of the same sample. Some well-formed gold nanoparticles had formed alongside the dendritic structures. Mainly spherical shapes (11 ± 7 nm) were observed, however dodecahedral (21 ± 2 nm) and rod-like (33 ± 17 nm in length) gold nanoparticle shapes were also present. Intriguingly, large nanoplates (flat structures) were also observed that were mainly triangular in shape (29 ± 12 nm in height). Fig. 5(c) shows that after 20 minutes of reaction these large dendritic structures had disappeared and only more compact looking and more regularly shaped gold nanoparticles were present. Spherical nanoparticles were still the most abundant shape, but had more than doubled in average size from 11 ± 7 nm after 5 minutes of reaction to 25 ± 7 nm after 20 minutes of reaction. Dodecahedral (28 ± 5 nm), rod-like (49 ± 23 nm in length) and triangular nanoplate (29 ± 5 nm in height) structures were also still present. However, some extra shapes, not previously observed after 5 minutes of reaction were now observed, such as decahedra (31 ± 4 nm), trigonal bipyramids (35 ± 6 nm) and triangular prisms (30 ± 5 nm). Fig. 5(d) shows the final state of the nanoparticles after they had been allowed to react to completion (65 minutes total reaction time). No new shapes were observed and all of the various kinds of nanostructure observed after 20 minutes of reaction were still present, except for triangular prisms. The diameter/length of each type of shape had increased on average by ≈20 nm. Spheres were still the most predominant shape, increasing in size from 25 ± 6.4 nm after 20 minutes of reaction to 43 ± 7 nm after 65 minutes of reaction. Colloidal solutions were stable to precipitation upon prolonged storage (at least 3 months). A TEM image size analysis of such a solution after 3 months of storage demonstrated how little the predominantly spherical nanoparticles changed in size (50 ± 10 nm). However, the primarily flat nanoplate structures showed an almost 5-fold increased in size (Fig. 5(e) and (f)). These nanoplates were similar in size and shape to those formed by Tsuji et al. from the microwave heating (85 °C) of aqueous solutions of auric acid with long-chained PVPs.26
 | ||
| Fig. 5 Transmission electron microscopy images of solution aliquots taken from the self-initiated gold nanoparticle formation reaction of auric acid solution and cyclohexanone after (a and b) 5 minutes, (c) 20 minutes and (d) 65 minutes of reaction. After 3 months storage larger (e) hexagonal and (f) triangular nanoplate structures were also present. | ||
The distribution of nanoparticle sizes and shapes are summarized in the histograms shown in Fig. 6. The histogram plots for the nanostructures observed after 20 and 65 minutes of reaction and 3 months storage were skewed distributions. The histogram after 5 minutes reaction was very much skewed to the left due to the large number of minute spherical particles that were present within the initial large dendritic structures. The overall average diameter of all nanostructures increased during the reaction period from ≈15 nm after 5 minutes, to ≈25 nm after 20 minutes, to ≈40 nm after 65 minutes of reaction. After 3 months in storage the average nanoparticle size did not increase greatly (≈40 nm) and signified that the nanoparticle formation reaction was primarily over after 65 minutes of reaction. Although a multitude of 3D shapes and flat nanoplate structures were observed, spherically shaped nanoparticles were present in majority (70% presence on average) throughout the reaction. For the duration of the reaction it was observed that the SPR band centre shifted from 543 to 576 nm (Fig. 3), denoting an average increase in size of ≈34 nm.36 From TEM studies, a similar size change was observed for the spherical particles that were present in majority, increasing in average size from 11 ± 7 nm after 5 minutes of reaction to 43 ± 7 nm after 65 minutes of reaction; a 32 nm average size increase, hence correlating well with the SPR shift.
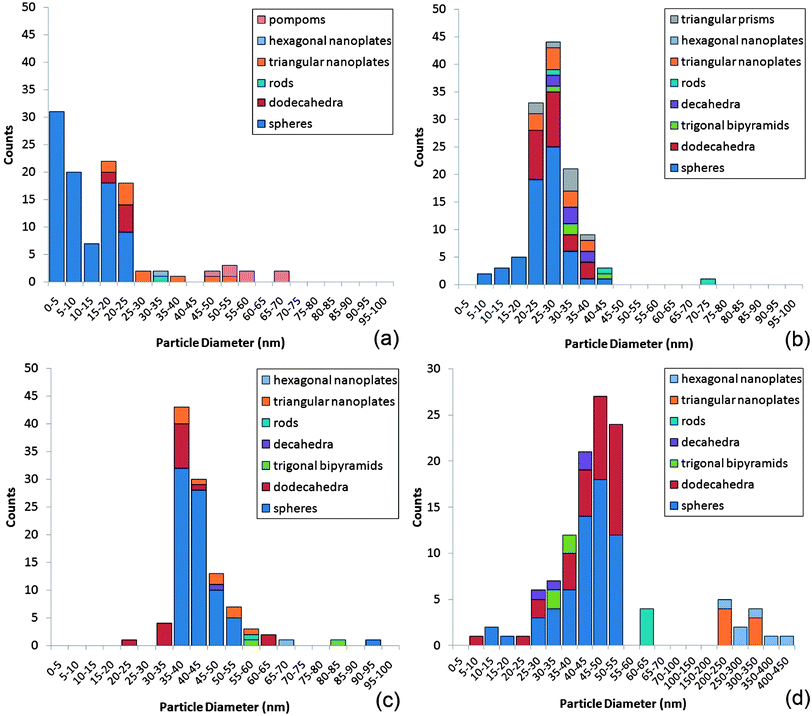 | ||
| Fig. 6 Histogram plots of nanoparticle size (nm) and count for the various nanostructures observed after (a) 5 minutes, (b) 20 minutes and (c) 65 minutes of reaction from the self-initiated gold nanoparticle formation reaction of auric acid solution and cyclohexanone and (d) after 3 months storage. | ||
A gold nanoparticle solution that had been allowed to react to completion (>65 minutes) was freeze dried to form a powder for XRD analysis. The pattern, shown in Fig. 7, was phase equivalent to that of bulk gold showing primitive face-centred cubic (FCC) symmetry (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, a = 4.0781 Å).37 Through fitting the pattern to a Le Bail refined model, a slightly larger unit cell was observed, where a = 4.1031 Å. This equated to an average unit cell expansion of 1.5% compared with bulk gold. In measuring the relative heights of each diffraction peak, slight preferred growth was observed in the (200) plane. Through applying the Scherrer equation to each diffraction peak, an average crystallite size of 14 nm with a standard deviation of 1.4 nm was observed. This indicates that the particles observed by TEM with average size ca. 40 nm are in fact on average composed of multiple crystal grains.
m, a = 4.0781 Å).37 Through fitting the pattern to a Le Bail refined model, a slightly larger unit cell was observed, where a = 4.1031 Å. This equated to an average unit cell expansion of 1.5% compared with bulk gold. In measuring the relative heights of each diffraction peak, slight preferred growth was observed in the (200) plane. Through applying the Scherrer equation to each diffraction peak, an average crystallite size of 14 nm with a standard deviation of 1.4 nm was observed. This indicates that the particles observed by TEM with average size ca. 40 nm are in fact on average composed of multiple crystal grains.
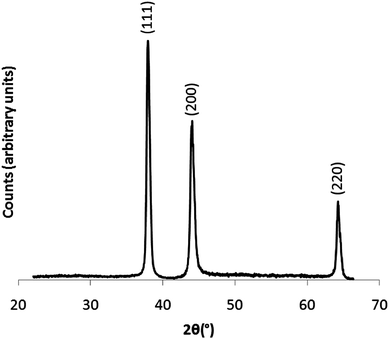 | ||
| Fig. 7 X-ray diffraction pattern of a colloidal powder suspension. The powder was acquired through freeze-drying a gold nanoparticle solution, formed from the self-initiated reaction of auric acid solution and cyclohexanone that was allowed to react to completion (>65 minutes). | ||
The freeze dried powder was also investigated using IR spectroscopy within a pressed KBr disc. Cyclohexanone solution was investigated as a standard for comparison. The presence of no organic compound other than cyclohexanone was observed in both spectra. This indicated that cyclohexanone not only acts as a reducing agent in the self-initiated formation of gold nanoparticles from auric acid, but also seems to play a surfactant-type role for stabilizing these particles. In addition, the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O dimer bond stretch did not within error shift in the gold nanoparticle powder (1714 cm−1) compared with that of pure cyclohexanone (1715 cm−1). In Sarkosh et al.'s IR study of gold nanoparticles formed by ablating a gold target submerged in cyclohexanone solution an ≈24 cm−1 decrease in this CO stretch was observed and was attributed to the capping of cyclohexanone molecules to the gold nanoparticles through their CO bond at oxygen. In comparison, this indicates that either cyclohexanone did not preferentially bind through this functional group to gold in the colloid reported in this paper or that a number of cyclohexanone layers were present, stacking randomly around each nanoparticle.
O dimer bond stretch did not within error shift in the gold nanoparticle powder (1714 cm−1) compared with that of pure cyclohexanone (1715 cm−1). In Sarkosh et al.'s IR study of gold nanoparticles formed by ablating a gold target submerged in cyclohexanone solution an ≈24 cm−1 decrease in this CO stretch was observed and was attributed to the capping of cyclohexanone molecules to the gold nanoparticles through their CO bond at oxygen. In comparison, this indicates that either cyclohexanone did not preferentially bind through this functional group to gold in the colloid reported in this paper or that a number of cyclohexanone layers were present, stacking randomly around each nanoparticle.
Mechanistic studies
Gold nanoparticles were formed by simply mixing cyclohexanone with an aqueous solution of auric acid. The reaction took place at room temperature, unlike typical Au nanoparticle syntheses, without the need for any initiation i.e. thermal activation, sonication, photo-activation, etc. Three components were present in the solution; water, auric acid and cyclohexanone.The reaction was repeated in D2O so that any by-products could be examined using 1H and 13C NMR. In the 13C NMR spectrum, several peaks not relating to the presence of the excess reducing agent, cyclohexanone [13C NMR (600 MHz, D2O) δ (ppm) 25.1, 27.2, 41.8, 128.2, 209.2 {ESI 1†}], were observed. These peaks [13C NMR (600 MHz, D2O) δ 22.7, 23.9, 33.8, 36.8, 39.2, 41.2, 65.2, 67.0, 95.2, 210.2, 216.3 {ESI 2†}] well-matched a pure spectrum of 2-chlorocyclohexanone [13C NMR (600 MHz, D2O) δ (ppm) 22.6, 24.7, 27.7, 30.9, 33.8, 37.4, 39.1, 41.2, 65.9, 67.0, 94.9, 210.1, 216.0 {ESI 3†}].
The two missing peaks at δ (ppm) 27.7 and 30.9 of 2-chlorocyclohexanone were masked by the large presence of cyclohexanone in this region. Additional peaks were observed but not identified. A similar case was observed in 1H NMR, where both cyclohexanone [1H NMR (600 MHz, D2O) δ (ppm) 1.18 (2H, dddd), 1.36 (4H, dddd), 1.98 (4H, dd)] and 2-chlorocyclohexanone [1H NMR (600 MHz, D2O) δ (ppm) 1.38 (1H, dtt), 1.51 (1H, dtt), 1.63 (1H, ddddd), 1.73 (1H, ddddd), 1.82 (1H, dddd), 1.97 (1H, dddd), 2.57 (1H, ddd), 2.63 (1H, ddd), 4.05 (1H, dd)] environments were observed. The majority by-product was thus identified as 2-chlorocyclohexanone, which was shown to be identical to literature spectra and a dilute solution of the reagent. This was supported by gas chromatography measurements. Gas chromatography of the nanoparticle solution (containing the by-product) and clean measurements of pure 2-chlorocyclohexanone was performed. Two matching peaks at the same retention time of 17.9 minutes were observed. Further confirmation of 2-chlorocyclohexanone being formed was found by IR spectroscopy. Measurements of the gas phase taken during the nanoparticle formation reaction in a gas-tight cell showed the progressive release of 2-chlorocyclohexanone (Fig. 7). A maximum concentration of 2-chlorocyclohexanone was reached after approximately 30 minutes, in line with titrations that indicated the complete reduction of auric acid in a similar time-frame (Fig. 8). Although cyclohexanone possesses a higher vapor pressure (4.5 mmHg, 25 °C) than 2-chlorocyclohexanone (0.28 mmHg, 25 °C), the far poorer solubility of 2-chlorocyclohexanone in water (0.02 g l−1) compared with cyclohexanone (87 g l−1) was the over-riding factor that led to its existence in the gas phase. Assuming the 1:1 formation of 2-chlorocyclohexanone from auric acid, 0.32 mg would have formed during the reaction; 0.16 mg over the solubility limit in 10 ml of water and encouraging vaporization. On the other hand, when mixed thoroughly, cyclohexanone (0.47 g) was fully soluble, being 0.4 g under the solubility limit.
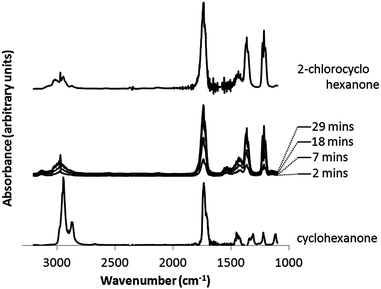 | ||
| Fig. 8 IR spectra of the gas phase above the solution taken during the reaction of auric acid (0.28 mM) with cyclohexanone (0.48 M). Gas phase measurements of 2-chlorocyclohexanone and cyclohexanone are shown for reference. | ||
The tautomerism of secondary ketones such as cyclohexanone is well known, forming an enol functional group with greater capacity for oxidation.38 The presence of this tautomer was indicated in this study as aqueous solutions of cyclohexanone discoloured bromine water (indicating an alkene group was present) and turned an acidified solution of K2Cr2O7 from orange to green upon slight heating (primary/secondary hydroxyl group present). In fact, a slight difference in the UV-visible absorption spectrum of aqueous cyclohexanone compared with pure was observed, when the peak at 913 nm in pure cyclohexanone disappeared in the aqueous mixture.
With the by-product and keto–enol chemistry in mind, a mechanism by which Au3+ could be reduced to Au0 is presented (Scheme 1). The mechanism was based in part on the similar chemistry observed between the pentavalent bismuth salt of Ph3Bi(V)Cl2 with cyclohexanone that leads to chlorine addition at the α position on cyclohexanone in a 2e− process forming 2-phenylcyclohexanone.39 Furthermore, Kochi demonstrated the reaction of CuCl2 with acetone in forming α-chloro ketones, again a 2e− transfer process.40 To our knowledge, the comparable reaction of auric acid (HAuCl4) and cyclohexanone has not been reported.
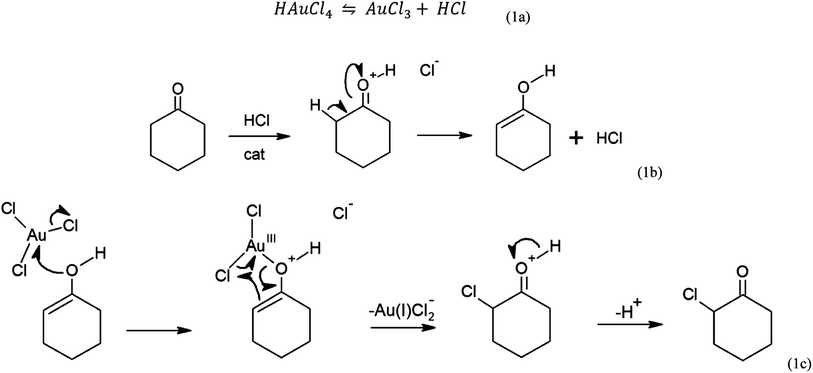 | ||
| Scheme 1 The mechanism for self-initiated gold nanoparticle growth from auric acid and cyclohexanone. | ||
Auric acid can release HCl freely in solution, leaving AuCl3 (Scheme 1a). The acid can tautomerise cyclohexanone to the enol catalytically (Scheme 1b). Trigonal planar AuCl3 is then nucleophilically attacked by the lone pair on the enol leading to the SN2 release of Cl− (Scheme 1c). The locality of the Cl–Au–O bond to the carbon α position induces a cyclic nucleophilic attack starting from the alkene bond. This leads to the reduction of gold from Au3+ (AuCl3) to Au1+ (AuCl2−) and the formation of 2-chlorocyclohexanone.
As the reduction of a metal chloride by a ketone is a 2e− transfer process,39,40 the direct reduction of AuCl2− to Au0 by cyclohexanone was not possible. Therefore, the disproportionation of AuCl2− to Au0 was the only viable route to Au0:
| 3AuCl2− → 2Au0 + AuCl4− + 2Cl− | (1) |
According to Gammons et al., the disproportionation of AuCl2− to Au0 takes place at room temperature.41 However, the rate of disproportionation increases rapidly with AuCl2− concentration and temperature. Moreover, the reaction is catalyzed in the presence of Au foil. The fact that Au0 catalyzes the disproportionation of AuCl2− to Au0 shows that particle growth on Au0 seed sites will be favoured; thus leading to the nucleation and growth of nanoparticles through the continued absorption and disproportionation of AuCl2−. The steps can be summarized as follows:
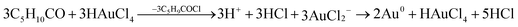 | (2a) |
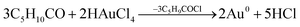 | (2b) |
Our TEM studies showed that large dendritic structures initially formed 5 minutes into the nanoparticle formation reaction. These structures then collapsed into smaller and denser particles with a variety of shapes. A similar growth mechanism has been observed in the Turkevich reaction (auric acid and tri-sodium citrate) between 70 and 100 °C.31 Initially, large particles formed that were ≈100 nm in diameter that then collapsed over the course of the reaction into predominantly spherical shaped nanoparticles that were ≈20 nm in size. The similar nanoparticle growth mechanism observed in this study demonstrates the similar behaviour of the surfactants. Nevertheless, the observed phenomenon of particle shrinkage is akin to an inverse-Ostwald ripening process.42 This phenomenon was explained by Rodríguez-González et al. in terms of the specific adsorption of steric/electrosteric layers.32 Assessing the Turkevich synthesis, Rodríguez-González et al. found that chloride ions primarily surround the large gold structures that initially form. However, when the AuCl4− is near consumed, the redox potential reduces allowing citrate ions to replace the surrounding chloride. This induces peptization as the gold surface is made more electrically neutral. We propose a similar phenomenon for the reaction of auric acid and cyclohexanone. We also suggest an extended Ostwald ripening43 process occurs over the 3 month storage period, causing the gold nanoplates to dramatically increase in size. We believe that the phenomenon is observed in the 2D shapes formed as opposed to the 3D shapes formed as growth in just a single plane facilitates significant increases in particle size.
Conclusions
A new and simple method for growing multi-shaped colloidal gold is presented. By adding cyclohexanone to aqueous solutions of auric acid, a self-initiating and self-propagating room temperature reaction occurs; resulting in the formation of gold nanoparticles. Such room temperature self-initiating nanoparticle synthesis reactions are different to traditional methods, such as the Turkevich method, where solutions require boiling temperatures to react. The formation proceeded in majority via large dendritic structures that collapsed to form more dense particles with a variety of shapes though most prominently spheres. The particles were highly crystalline and were stabilized by the cyclohexanone reducing agent, dually acting as a surfactant. Through identifying the 2-chlorocyclohexanone by-product formed in this nanoparticle synthesis a mechanism by which Au3+ was reduced to Au0 was proposed. Furthermore, a new method of converting cyclic ketones to α-chloro ketones was discovered. This new synthetic route to colloidal gold is straightforward and fast, forming nanoparticles with a range of shapes. The resulting nanoparticle solution is stable to precipitation for more than 3 months at room temperature. Surprisingly the flat nanoparticle sheets seem to grow over the 3 month period whereas the other nanoparticle shapes remain invariant after ca. 1 h of reaction.Acknowledgements
IPP wishes to thank the EPSRC for funding. Dr Daniel Ortega is thanked for his help with TEM studies and Dr Abil Aliev is thanked for his help with NMR studies. Prof. William Motherwell and Dr Matthew Powner are thanked for help with the mechanism.References
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS
.
- G. Walters and I. P. Parkin, J. Mater. Chem., 2009, 19, 574 RSC
- K.-T. Yong, M. T. Swihart, H. Ding and P. N. Prasad, Plasmonics, 2009, 4, 79 CrossRef CAS
- M. C. Ferrara, L. Mirenghi, A. Mevoli and L. Tapfer, Nanotechnology, 2008, 19, 365706 CrossRef CAS
- R. G. Palgrave and I. P. Parkin, Chem. Mater., 2007, 19, 4639 CrossRef CAS
- R. G. Palgrave and I. P. Parkin, J. Am. Chem. Soc., 2006, 128, 1587 CrossRef CAS
- R. Binions, C. Piccirillo, R. G. Palgrave and I. P. Parkin, Chem. Vap. Deposition, 2008, 14, 33 CrossRef CAS
- J. Gil-Tomas, S. Tubby, I. P. Parkin, N. Narband, L. Dekker, S. P. Nair, M. Wilson and C. Street, J. Mater. Chem., 2007, 17, 3739 RSC
- S. Perni, C. Piccirillo, J. Pratten, P. Prokopovich, W. Chrzanowski, I. P. Parkin and M. Wilson, Biomaterials, 2009, 30, 89 CrossRef CAS
- S. I. Stoeva, J.-S. Lee, J. E. Smith, S. T. Rosen and C. A. Mirkin, J. Am. Chem. Soc., 2006, 128, 8378 CrossRef CAS
- X.-M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912 RSC
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220 CrossRef CAS
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700 CrossRef CAS
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC
- Y. Tai, N. T. T. Tran, Y.-C. Tsai, J.-Y. Fang and L.-W. Chang, IET Nanobiotechnol., 2011, 5, 52 CrossRef CAS
- O. Bar-Ilan, R. M. Albrecht, V. E. Fako and D. Y. Furgeson, Small, 2009, 5, 1897 CrossRef CAS
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662 CrossRef CAS
- Y. Ohko, T. Tatsuma, T. Fujii, K. Naoi, C. Niwa, Y. Kubota and A. Fujishima, Nat. Mater., 2003, 2, 29 CrossRef CAS
- C. Huang, P. Chiu, Y. Wang, W. R. Chen and T. H. Meen, J. Electrochem. Soc., 2006, 153, D129 CrossRef CAS
- C. Huang, Y. Wang, P. Chiu, M. Shih and T. H. Meen, Mater. Lett., 2006, 60, 1896 CrossRef CAS
- X. Tong, Y. Zhao, T. Huang, H. Liu and K. Y. Liew, Appl. Surf. Sci., 2009, 255, 9463 CrossRef CAS
- B. K. Jena and C. R. Raj, Chem. Mater., 2008, 20, 3546 CrossRef CAS
- A. Ali Umar and M. Oyama, Cryst. Growth Des., 2009, 9, 1146 CAS
- C. Wang, C. Kan, J. Zhu, X. Zeng, X. Wang, H. Li and D. Shi, J. Nanomater., 2010, 2010, 1 Search PubMed
- M. Tsuji, M. Hashimoto, Y. Nishizawa, M. Kubokawa and T. Tsuji, Chem.–Eur. J., 2005, 11, 440 CrossRef CAS
- S. S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad and M. Sastry, Nat. Mater., 2004, 3, 482 CrossRef CAS
- S. Biswas, S. K. Hait, S. C. Bhattacharya and S. P. Moulik, J. Dispersion Sci. Technol., 2005, 25, 801 CrossRef
- L. Sarkhosh, H. Aleali, R. Karimzadeh and N. Mansour, Phys. Status Solidi A, 2010, 207, 2303 CrossRef CAS
- M. L. Marin, K. L. McGilvray and J. C. Scaiano, J. Am. Chem. Soc., 2008, 130, 16572 CrossRef CAS
- M. K. Chow and C. F. Zukowski, J. Colloid Interface Sci., 1994, 165, 97 CrossRef CAS
- B. Rodríguez-González, P. Mulvaney and L. M. Liz-Marzán, Z. Phys. Chem., 2007, 221, 415 CrossRef
- D. Ray, V. K. Aswal and J. Kohlbrecher, Langmuir, 2011, 27, 4048 CrossRef CAS
- A. I. Yanson, P. Rodriguez, N. Garcia-Araez, R. V. Mom, F. D. Tichelaar and M. T. M. Koper, Angew. Chem., Int. Ed., 2011, 50, 6346 CrossRef CAS
- V. Herschlog, Ind. Eng. Chem., Anal. Ed., 1941, 13, 561 CrossRef
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215 CrossRef CAS
- E. A. Owen and E. L. Yates, J. Chem. Phys., 1935, 3, 605 CrossRef
- A. K. Cederstav and B. M. Novak, J. Am. Chem. Soc., 1994, 116, 4073 CrossRef CAS
- D. H. R. Barton, J.-C. Blazejewski, B. Charpiot, J.-P. Finet, W. B. Motherwell, M. T. B. Papoula and S. P. Stanforth, J. Chem. Soc., Perkin Trans. 1, 1985, 2667 RSC
- J. K. Kochi, J. Am. Chem. Soc., 1955, 77, 5274 CrossRef CAS
- C. H. Gammons, Y. Yu and A. E. Williams-Jones, Geochim. Cosmochim. Acta, 1997, 61, 1971 CrossRef CAS
- D. Lee, S. Park, J. Lee and N. Hwang, Acta Mater., 2007, 55, 5281 CrossRef CAS
- P. W. Voorhees, J. Stat. Phys., 1985, 38, 231 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Zoomed-in 13C NMR spectra identify by-products in the reaction. See DOI: 10.1039/c3ta11546a |
| This journal is © The Royal Society of Chemistry 2013 |