 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diacetylene bridged triphenylamines as hole transport materials for solid state dye sensitized solar cells†
Miquel Planells‡a, Antonio Abate‡b, Derek J. Hollmanb, Samuel D. Stranksb, Vishal Bhartic, Jitender Gaurc, Dibyajyoti Mohantyc, Suresh Chandc, Henry J. Snaith*b and Neil Robertson*a
aEastChem – School of Chemistry, University of Edinburgh, Kings Buildings, Edinburgh EH9 3JJ, UK. E-mail: neil.robertson@ed.ac.uk
bDepartment of Physics, University of Oxford, Oxford OX1 3PU, UK. E-mail: h.snaith1@physics.ox.ac.uk
cCSIR-National Physical Laboratory, Dr. K.S. Krishnan Marg, New Delhi-110012, India
First published on 1st May 2013
Abstract
We have synthesized and characterized a series of triphenylamine-based hole-transport materials (HTMs), and studied their function in solid-state dye sensitized solar cells (ss-DSSCs). By increasing the electron-donating strength of functional groups (–H < –Me < –SMe < –OMe) we have systematically shifted the oxidation potential and ensuing photocurrent generation and open-circuit voltage of the solar cells. Correlating the electronic properties of the HTM to the device operation highlights a significant energy offset required between the Dye – HTM highest occupied molecular orbital (HOMO) energy levels. From this study, it is apparent that precise control and tuning of the oxidation potential is a necessity, and usually not achieved with most HTMs developed to date for ss-DSSCs. To significantly increase the efficiency of solid-state DSSCs understanding these properties, and implementing dye-HTM combinations to minimize the required HOMO offset is of central importance.
Introduction
Significant improvements in power conversion efficiency have been reported for solid-state solution-processed solar cells, which make this cost-effective technology competitive with established thin-film solar technologies.1–4 Snaith and co-workers have recently shown a record efficiencies of over 12%,5,6 using organometallic halide perovskites mesostructured at the interface with 2,2′-7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (Spiro-OMeTAD) as the hole-transport material (HTM).7,8 In addition, the highest reported efficiencies (approximately 7%) for solid-state dye sensitized solar cells (ss-DSSCs) have also been achieved employing Spiro-OMeTAD as the HTM.9 However, recent studies have demonstrated that the slow charge transport in Spiro-OMeTAD significantly limits the device performance near the maximum power point in the solar cell.10 In particular, we have shown that the losses due to series resistance in the HTM reduces the obtainable power in conventional ss-DSSCs by 10 to 20% when generating less than 10 mA cm−2 photocurrent.11 The recent significant enhancements in photocurrent generation with the perovskite absorbers,5,12,13 and further likely enhancements in ss-DSSCs will increase the IR losses,10 making enhanced HTMs of paramount importance for continued improvements.5,11 Therefore, together with the other main device components (light harvesters and electron transporting materials),8,14 the HTM requires novel solutions to further improve this technology. Many new materials have been proposed to improve hole mobility, yet the measured device efficiencies have always remained lower than Spiro-OMeTAD.15–17 A promising alternative will be to increase the pore-filling fraction of the mesostructured photo-electrode by melt infiltration,18,19 although this strongly limits the choice of the materials to those compatible with the dye thermal stability. The most significant progress has been through adding strong oxidants as p-dopants, thereby increasing the conductivity in Spiro-OMeTAD.9,11,20 However, controlled doping can require processing under inert atmosphere, which is an undesirable method for producing cost-effective ss-DSSC technologies, and still gives an upper limit on the conductivity.11Here we present the synthesis and characterization of triphenylamine derivatives with various electron-donating functional groups as HTM in ss-DSSCs (Fig. 1). These molecules are comprised of two triphenylamine moieties bridged with a diacetylene group (DATPA). The intramolecular electron transfer and electron valence properties of this type of system have been previously studied, but have not yet been utilized as a HTM.21,22 We measure the electrochemical and optical properties of DATPA derivatives and assess the impact of the electron donating substituent on ss-DSSC performances. In particular, we discuss the importance of the offset between the oxidation potential of the dye and the HTM on the device performances.
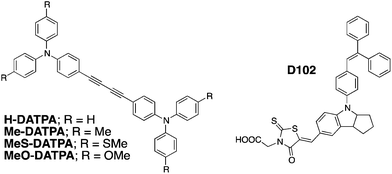 | ||
| Fig. 1 Chemical structure of HTMs (left) and D102 dye23,24 (right) used in this study. Each of the molecules is comprised of two triphenylamine moieties bridged with a diacetylene group (DATPA). | ||
Experimental section
Materials
All reagents were purchased from either Sigma-Aldrich or Alfa-Aesar and they were used as received without further purification unless otherwise stated. N-Bromosuccinimide (NBS) was recrystallized from water before use.Synthesis of N,N-diphenyl-4-((triethylsilyl)ethynyl)aniline (1)
Bis(triphenylphosphine)palladium dichloride (52 mg, 0.075 mmol) triphenylphosphine (39 mg, 0.15 mmol) and copper iodide (57 mg, 0.3 mmol) were placed in a Schlenk tube and dried under high vacuum for 30 minutes. Then, a previously degassed solution of 4-bromotriphenylamine (815 mg, 2.5 mmol), piperidine (2 mL, 20 mmol) and triethylsilylacetylene (920 μL, 5 mmol) in 15 mL of dry toluene was added via cannula. The mixture was degassed again by the pump/freeze technique and stirred at 90 °C overnight under N2. Then, a silica plug was run in CH2Cl2 and the solvent removed. The crude product was purified by column chromatography (SiO2, Hexanes/CH2Cl2 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford the product as yellow oil (933 mg, 97% yield). 1H NMR (500 MHz, CDCl3) δH: 7.33 (d, J = 8.6 Hz, 2H); 7.26 (m, 4H); 7.09 (d, J = 8.6 Hz, 4H); 7.05 (t, J = 7.4 Hz, 2H); 6.96 (d, J = 8.6 Hz, 2H); 1.05 (t, J = 7.9 Hz, 9H); 0.67 (q J = 7.9 Hz, 6H). 13C NMR (125 MHz, CDCl3) δC: 148.2; 147.4; 133.2; 129.6; 125.1; 123.7; 122.5; 116.5; 106.8; 90.6; 7.7; 4.7. MS EI (m/z): [M]+ calcd for C26H29NSi: 383.20638; found: 383.206001. Anal. calcd for C26H29NSi: C, 81.41; H, 7.62; N, 3.65; found: C, 81.31; H, 7.56; N, 3.69.
:1) to afford the product as yellow oil (933 mg, 97% yield). 1H NMR (500 MHz, CDCl3) δH: 7.33 (d, J = 8.6 Hz, 2H); 7.26 (m, 4H); 7.09 (d, J = 8.6 Hz, 4H); 7.05 (t, J = 7.4 Hz, 2H); 6.96 (d, J = 8.6 Hz, 2H); 1.05 (t, J = 7.9 Hz, 9H); 0.67 (q J = 7.9 Hz, 6H). 13C NMR (125 MHz, CDCl3) δC: 148.2; 147.4; 133.2; 129.6; 125.1; 123.7; 122.5; 116.5; 106.8; 90.6; 7.7; 4.7. MS EI (m/z): [M]+ calcd for C26H29NSi: 383.20638; found: 383.206001. Anal. calcd for C26H29NSi: C, 81.41; H, 7.62; N, 3.65; found: C, 81.31; H, 7.56; N, 3.69.Synthesis of 4-ethynyl-N,N-diphenylaniline (2)
1 (3 g, 8.8 mmol) was placed in a round bottom flask with 45 mL of CH2Cl2. Then, tetrabutylammonium fluoride 1 M in THF (12 mL, 12 mmol) was added and the mixture was stirred for 1.5 hours under N2 at r.t. After removing the solvent, the crude product was purified by column chromatography (SiO2, Hexanes/CHCl3 2:1) to afford the product as pale solid (2.1 g, 90% yield). 1H NMR (500 MHz, CDCl3) δH: 7.33 (d, J = 8.8 Hz, 2H); 7.26 (m, 4H); 7.10 (d, J = 8.3 Hz, 4H); 7.06 (t, J = 7.3 Hz, 2H); 6.97 (d, J = 8.7 Hz, 2H); 3.02 (s, 1H). 13C NMR (125 MHz, CDCl3) δC: 148.6; 147.3; 133.3; 129.6; 125.2; 123.8; 122.2; 114.9; 84.1; 76.3. MS EI (m/z): [M]+ calcd for C20H15N: 269.12100; found: 269.12054. Anal. calcd for C20H15N: C, 89.19; H, 5.61; N, 5.20; found: C, 89.21; H, 5.51; N, 5.17.Synthesis of 4,4′-(buta-1,3-diyne-1,4-diyl)bis(N,N-diphenylaniline) (H-DATPA)
2 (269 mg, 1 mmol) was placed in a round bottom flask with 50 mL of CH2Cl2. Tetramethylethylenediamine (TMEDA) (4.5 mL, 30 mmol) and copper(I) chloride (2.9 g, 30 mmol) were added and the mixture was stirred vigorously for 4 hours under air with a CaCl2 tube on top. Then, the solvent was removed and the crude product was purified by column chromatography (SiO2, Hexanes/CHCl3 2:1) to afford the product as yellow solid (251 mg, 95%). 1H NMR (500 MHz, CDCl3) δH: 7.34 (d, J = 8.8 Hz, 4H); 7.28 (m, 10H); 7.11 (dd, J = 8.3, 1.1 Hz, 8H); 7.08 (t, J = 7.4 Hz, 4H). 13C NMR (125 MHz, CDCl3) δC: 148.7; 147.1; 133.6; 129.7; 125.5; 124.1; 121.8; 114.4; 82.1; 73.7. MS EI (m/z): [M]+ calcd for C40H28N2: 536.22470; found: 536.224675. Anal. calcd for C40H28N2: C, 89.52; H, 5.26; N, 5.22; found: C, 89.47; H, 5.18; N, 5.25. m.p. = 246 °C.Synthesis of 4-bromo-N,N-di-p-tolylaniline (3)
4-Bromoaniline (4.9 g, 29 mmol), 4-iodotoluene (25 g, 115 mmol), potassium tert-butoxide (9.7 g, 87 mmol), copper iodide (221 mg, 1.16 mmol) and 2,2′-bipyridyl (174 mg, 1.16 mmol) were placed in a round bottom flask and dried under high vacuum for 30 minutes. Previously degassed dry toluene (120 mL) was added and the mixture was stirred at 120 °C overnight under N2. Then, a silica plug was run in CH2Cl2 and the solvent removed. The crude product was purified by column chromatography (SiO2, Hexanes up to Hexanes/CHCl3 4:1) to afford the product as pale yellow solid (3.8 g, 37% yield). 1H NMR (500 MHz, CDCl3) δH: 7.30 (d, J = 8.7 Hz, 2H); 7.09 (d, J = 8.4 Hz, 4H); 6.99 (d, J = 8.4 Hz, 4H); 6.91 (d, J = 8.8 Hz, 2H); 2.34 (s, 6H). 13C NMR (125 MHz, CDCl3) δC: 147.6; 145.1; 133.1; 132.1; 130.2; 124.9; 124.1; 113.8; 21.0. MS EI (m/z): [M]+ calcd for C20H18BrN: 351.06171; found: 351.061377. Anal. calcd for C20H18BrN: C, 68.19; H, 5.15; N, 3.98; found: C, 68.33; H, 5.20; N, 4.07.Synthesis of 4-methyl-N-(4-methylphenyl)-N-(4-((triethyllsilyl)ethynyl)phenyl)aniline (4)
Bis(triphenylphosphine)palladium dichloride (168 mg, 0.24 mmol) triphenylphosphine (125 mg, 0.48 mmol) and copper iodide (182 mg, 0.96 mmol) were placed in a round bottom flask and dried under high vacuum for 30 minutes. Then, a previously degassed solution of 3 (2.8 g, 8 mmol), piperidine (6.3 mL, 64 mmol) and triethylsilylacetylene (2 mL, 11 mmol) in 40 mL of dry toluene was added via cannula. The mixture was degassed again by the pump/freeze technique and stirred at 90 °C overnight under N2. Then, a silica plug was run in CH2Cl2 and the solvent removed. The crude product was purified by column chromatography (SiO2, Hexanes/CHCl3 3:1 up to 2:1) to afford the product as yellow oil (1.89 g, 57% yield). 1H NMR (500 MHz, CDCl3) δH: 7.28 (d, J = 8.8 Hz, 2H); 7.07 (d, J = 8.3 Hz, 4H); 6.98 (d, J = 8.3 Hz, 4H); 6.89 (d, J = 8.8 Hz, 2H); 2.31 (s, 6H); 1.03 (t, J = 7.9 Hz, 9H); 0.66 (q, J = 7.9 Hz, 6H). 13C NMR (125 MHz, CDCl3) δC: 148.6; 144.9; 133.4; 133.1; 130.2; 125.3; 121.3; 115.4; 107.1; 90.2; 21.1; 7.8; 4.8. MS EI (m/z): [M]+ calcd for C28H33NSi: 411.23768; found: 411.237326. Anal. calcd for C28H33NSi: C, 81.69; H, 8.08; N, 3.40; found: C, 81.58; H, 8.15; N, 3.51.Synthesis of 4-ethynyl-N,N-di-p-tolylaniline (5)
4 (950 mg, 2.3 mmol) was placed in a round bottom flask with 12 mL of CH2Cl2. Then, tetrabutylammonium fluoride 1 M in THF (3.2 mL, 3.2 mmol) was added and the mixture was stirred for 2 hours at r.t. under N2. After removing the solvent, the crude product was purified by column chromatography (SiO2, Hexanes/CH2Cl2 4:1) to afford the product as pale solid (634 mg, 93% yield). 1H NMR (500 MHz, CDCl3) δH: 7.29 (d, J = 8.7 Hz, 2H); 7.08 (d, J = 8.3 Hz, 4H); 7.00 (d, J = 8.3 Hz, 4H); 6.90 (d, J = 8.8 Hz, 2H); 3.00 (s, 1H); 2.32 (s, 6H). 13C NMR (125 MHz, CDCl3) δC: 148.9; 144.8; 133.6; 133.1; 130.2; 125.5; 120.9; 113.8; 84.3; 76.0; 21.1. MS EI (m/z): [M]+ calcd for C22H19N: 297.15120; found: 297.151344. Anal. calcd for C22H19N: C, 88.85; H, 6.44; N, 4.71; found: C, 88.84; H, 6.53; N, 4.83.Synthesis of 4,4′-(buta-1,3-diyne-1,4-diyl)bis(N,N-di-p-tolylaniline) (Me-DATPA)
5 (148 mg, 0.5 mmol) was placed in a round bottom flask with 25 mL of CH2Cl2. TMEDA (2.2 mL, 15 mmol) and copper(I) chloride (1.45 g, 15 mmol) were added and the mixture was stirred vigorously for 4 hours under air with a CaCl2 tube on top. Then, the solvent was removed and the crude was purified by column chromatography (SiO2, Hexanes/CHCl3 3:1) to afford the product as yellow solid (118 mg, 80% yield). 1H NMR (500 MHz, CDCl3) δH: 7.29 (d, J = 8.7 Hz, 4H); 7.09 (d, J = 8.3 Hz, 8H); 7.01 (d, J = 8.3 Hz, 8H); 6.87 (d, J = 8.8 Hz, 4H); 2.32 (s, 12H). 13C NMR (125 MHz, CDCl3) δC: 149.1; 144.6; 133.9; 133.5; 130.3; 125.7; 120.5; 113.3; 82.3; 73.5; 21.1. MS EI (m/z): [M]+ calcd for C44H36N2: 592.28730; found: 592.28732. Anal. calcd for C44H36N2: C, 89.15; H, 6.12; N, 4.73; found: C, 89.03; H, 6.06; N, 4.69. m.p. = 206 °C.Synthesis of 4-bromo-N,N-bis(4-methoxyphenyl)aniline (6)
4-Bromoaniline (2.8 g, 16.5 mmol), 4-iodoanisole (16.5 g, 66 mmol), potassium tert-butoxide (5.5 g, 49.5 mmol), copper iodide (125 mg, 0.7 mmol) and 2,2′-bipyridyl (100 mg, 0.7 mmol) were placed in a round bottom flask and dried under high vacuum for 30 minutes. Previously degassed dry toluene (66 mL) was added and the mixture was stirred at 120 °C overnight under N2. Then, a silica plug was run in CH2Cl2 and the solvent removed. The crude product was purified by column chromatography (SiO2, Hexanes/CHCl3 4:1 up to 2:1) to afford the product as yellow oil (1.87 g, 30% yield). 1H NMR (500 MHz, CDCl3) δH: 7.23 (d, J = 8.8 Hz, 2H); 7.02 (d, J = 9.1 Hz, 4H); 6.82 (d, J = 9.1 Hz, 4H); 6.79 (d, J = 8.8 Hz, 2H); 3.79 (s, 6H). 13C NMR (125 MHz, CDCl3) δC: 156.2; 148.1; 140.8; 131.9; 126.7; 122.2; 115.0; 112.5; 55.7. MS EI (m/z): [M]+ calcd for C20H18BrNO2: 383.05154; found: 383.051358. Anal. calcd for C20H18BrNO2: C, 62.51; H, 4.72; N, 3.65; found: C, 62.52; H, 4.74; N, 3.54.Synthesis of 4-methoxy-N-(4-methoxyphenyl)-N-(4-((triethyllsilyl)ethynyl)phenyl)aniline (7)
Bis(triphenylphosphine)palladium dichloride (65 mg, 0.09 mmol) triphenylphosphine (49 mg, 0.18 mmol) and copper iodide (71 mg, 0.37 mmol) were placed in a shlenck tube and dried under high vacuum for 30 minutes. Then, a previously degassed solution of 6 (1.2 g, 3.12 mmol), piperidine (2.5 mL, 25 mmol) and triethylsilylacetylene (780 μL, 4.37 mmol) in 15 mL of dry toluene was added via cannula. The mixture was degassed again by the pump/freeze technique and stirred at 90 °C overnight under N2. Then, a silica plug was run in CH2Cl2 and the solvent removed. The crude product was purified by column chromatography (SiO2, Hexanes/CHCl3 3:1 up to 2:1) to afford the product as yellow oil (1.36 g, 98% yield). 1H NMR (500 MHz, CDCl3) δH: 7.25 (d, J = 8.4 Hz, 2H); 7.04 (d, J = 8.9 Hz, 4H); 6.83 (d, J = 8.9 Hz, 4H); 6.79 (d, J = 8.8 Hz, 2H); 3.79 (s, 6H); 1.03 (t, J = 7.9 Hz, 9H); 0.65 (q, J = 7.9 Hz, 6H). 13C NMR (125 MHz, CDCl3) δC: 156.4; 149.0; 140.5; 133.1; 127.1; 119.4; 115.0; 114.4; 107.2; 89.9; 55.7; 7.7; 4.7. MS EI (m/z): [M]+ calcd for C28H33NO2Si: 443.22751; found: 443.227937. Anal. calcd for C28H33NO2Si: C, 75.80; H, 7.50; N, 3.16; found: C, 75.87; H, 7.40; N, 2.98.Synthesis of 4-ethynyl-N,N-bis(4-methoxyphenyl)aniline (8)
7 (1.35 g, 3 mmol) was placed in a round bottom flask with 30 mL of CH2Cl2. Then, tetrabutylammonium fluoride 1 M in THF (4.2 mL, 4.2 mmol) was added and the mixture was stirred for 1.5 hours at r.t. under N2. After removing the solvent, the crude product was purified by column chromatography (SiO2, Hexanes/CHCl3 3:1) to afford the product as yellow oil (930 mg, 93% yield). 1H NMR (500 MHz, CDCl3) δH: 7.26 (d, J = 8.8 Hz, 2H); 7.06 (d, J = 8.9 Hz, 4H); 6.84 (d, J = 9.0 Hz, 4H); 6.80 (d, J = 8.8 Hz, 2H); 3.80 (s, 6H); 2.98 (s, 1H). 13C NMR (125 MHz, CDCl3) δC: 156.6; 149.4; 140.3; 133.1; 127.4; 119.1; 115.0; 112.8; 84.5; 75.8; 55.7. MS EI (m/z): [M]+ calcd for C22H19NO2: 329.14103; found: 329.140097. Anal. calcd for C22H19NO2: C, 80.22; H, 5.81; N, 4.25; found: C, 80.10; H, 5.80; N, 4.35.Synthesis of 4,4′-(buta-1,3-diyne-1,4-diyl)bis(N,N-bis(4-methoxyphenyl)aniline) (MeO-DATPA)
8 (220 mg, 0.67 mmol) was placed in a round bottom flask with 33 mL of CH2Cl2. TMEDA (3 mL, 20 mmol) and copper(I) chloride were added and the mixture was stirred vigorously for 4 hours under air with a CaCl2 tube on top. Then, the solvent was removed and the crude product was purified by two column chromatography (SiO2, Hexanes/CHCl3 2:1 and then Hexanes/Et2O 1:1) to afford the product as yellow solid (66 mg, 30% yield). 1H NMR (500 MHz, CDCl3) δH: 7.27 (d, J = 8.5 Hz, 4H); 7.07 (d, J = 9.0 Hz, 8H); 6.84 (d, J = 9.0 Hz, 8H); 6.78 (d, J = 8.5 Hz, 4H); 3.80 (s, 12H). 13C NMR (125 MHz, CDCl3) δC: 56.7; 149.5; 140.0; 133.5; 127.5; 118.8; 115.1; 112.4; 82.3; 73.3; 55.7. MS EI (m/z): [M]+ calcd for C44H36N2O4: 656.26696; found: 656.266885. Anal. calcd for C44H36N2O4: C, 80.47; H, 5.52; N, 4.27; found: C, 80.54; H, 5.43; N, 4.38. m.p. = 186 °C.Synthesis of 4-(methylthio)-N-(4-(methylthio)phenyl)-N-phenylaniline (9)
4-bromo-N-(4-bromophenyl)-N-phenylaniline (4.7 g, 11.6 mmol) was added to a round bottom flask with 60 mL of anhydrous DMF and stirred under N2. Sodium thiomethoxide (3.25 g, 46.4 mmol) was added in small portions and the mixture heated up to 120 °C and stirred under N2 for 48 hours. Then, the crude product was poured into 200 mL of aqueous NaHCO3 satured solution and extracted with 200 mL of Et2O twice. The organic phase was washed with saturated NaHCO3 and brine, dried over Na2SO4 and the solvent removed. The crude product was purified by column chromatography (SiO2, Hexanes/CH2Cl2 10:1 up to 4:1) to afford the product as colorless oil (691 mg, 16% yield). Note that monoreacted product, 4-bromo-N-(4-(methylthio)phenyl)-N-phenylaniline, was also obtained with a high yield (2.35 g, 51% yield). 1H NMR (500 MHz, CDCl3) δH: 7.24 (t, J = 7.9 Hz, 2H); 7.18 (d, J = 8.6 Hz, 4H); 7.05 (m, 7H); 2.47 (s, 6H). 13C NMR (125 MHz, CDCl3) δC: 147.6; 145.6; 131.8; 129.4; 128.8; 124.8; 124.1; 123.0; 17.1. MS EI (m/z): [M]+ calcd for C20H19NS2: 337.09534; found: 337.095559. Anal. calcd for C20H19NS2: C, 71.17; H, 5.67; N, 4.15; found: C, 71.07; H, 5.62; N, 4.18.Synthesis of 4-bromo-N,N-bis(4-(methylthio)phenyl)aniline (10)
9 (691 mg, 2 mmol) was placed in a round bottom flask with 20 mL of anhydrous DMF and the mixture was cooled down to 0 °C. Then, NBS (365 mg, 2 mmol) was added in small amounts at 0 °C and the mixture stirred for 3 hours at r.t. The crude product was dissolved in 200 mL of Et2O and washed with 200 mL of water (×2) and brine. The organic layer was dried under Na2SO4 and the solvent removed. The crude product was purified by column chromatography (SiO2, Hexanes/CH2Cl2 3:1) to afford the product as colorless oil (515 mg, 60% yield). 1H NMR (500 MHz, CDCl3) δH: 7.31 (d, J = 0.5 Hz, 2H); 7.17 (d, J = 8.5 Hz, 4H); 6.99 (d, J = 8.5 Hz, 4H); 6.91 (d, J = 8.5 Hz, 2H); 2.47 (s, 6H). 13C NMR (125 MHz, CDCl3) δC: 146.8; 145.0; 132.6; 132.4; 128.7; 125.0 (×2); 115.1; 16.9. MS EI (m/z): [M]+ calcd for C20H18BrNS2: 415.00586; found: 415.005850.Synthesis of 4-(methylthio)-N-(4-(methylthio)phenyl)-N-(4-((triethylsilyl)ethynyl)phenyl)aniline (11)
Bis(triphenylphosphine)palladium dichloride (35 mg, 0.05 mmol) triphenylphosphine (26 mg, 0.1 mmol) and copper iodide (38 mg, 0.2 mmol) were placed in a Schlenk tube and dried under high vacuum for 30 minutes. Then, a previously degassed solution of 10 (515 mg, 1.25 mmol), piperidine (1 mL, 10 mmol) and triethylsilylacetylene (460 μL, 2.5 mmol) in 7.5 mL of dry toluene was added via cannula. The mixture was degassed again by the pump/freeze technique and stirred at 90 °C overnight under N2. Then, a silica plug was run in CH2Cl2 and the solvent removed. The crude product was purified by column chromatography (SiO2, Hexanes/CHCl3 4:1 up to 2:1) to afford the product as yellow oil (757 mg, 96% yield). 1H NMR (500 MHz, CDCl3) δH: 7.31 (d, J = 8.6 Hz, 2H); 7.17 (d, J = 8.6 Hz, 4H); 7.00 (d, J = 8.6 Hz, 4H); 6.93 (d, J = 8.6 Hz, 2H); 2.47 (s, 6H); 1.04 (t, J = 7.9 Hz, 9H); 0.66 (q, J = 7.9 Hz, 6H). 13C NMR (125 MHz, CDCl3) δC: 147.7; 144.7; 133.3; 132.9; 128.5; 125.4; 122.3; 116.6; 106.7; 90.9; 16.8; 7.7; 4.7. MS EI (m/z): [M]+ calcd for C28H33NS2Si: 475.18182; found: 475.181501. Anal. calcd for C28H33NS2Si: C, 70.68; H, 6.99; N, 2.94; found: C, 67.56; H, 6.73; N, 3.46.Synthesis of 4-ethynyl-N,N-bis(4-(methylthio)phenyl)aniline (12)
11 (575 mg, 1.2 mmol) was placed in a round bottom flask with 12 mL of CH2Cl2. Then, tetrabutylammonium fluoride 1 M in THF (1.7 mL, 1.7 mmol) was added and the mixture was stirred for 2 hours under N2 at r.t. After removing the solvent, the crude product was purified by column chromatography (SiO2, Hexanes/CHCl3 3:1) to afford the product as yellow oil (371 mg, 92% yield). 1H NMR (500 MHz, CDCl3) δH: 7.32 (d, J = 8.7 Hz, 2H); 7.18 (d. J = 8.7 Hz, 4H); 7.02 (d, J = 8.7 Hz, 4H); 6.94 (d, J = 8.7 Hz, 2H); 3.02 (s, 1H); 2.74 (s, 6H). 13C NMR (125 MHz, CDCl3) δC: 148.2; 144.7; 133.3; 133.1; 128.6; 125.6; 122.0; 115.1; 84.0; 76.5; 16.8. MS EI (m/z): [M]+ calcd for C22H19NS2: 361.01534; found: 361.095338. Anal. calcd for C22H19NS2: C, 73.09; H, 5.30; N, 3.87; found: C, 70.23; H, 5.32; N, 4.10.Synthesis of 4,4′-(buta-1,3-diyne-1,4-diyl)bis(N,N-bis(4-(methylthio)phenyl)aniline) (MeS-DATPA)
12 (371 mg, 1 mmol) was placed in a round bottom flask with 50 mL of CH2Cl2. TMEDA (4.5 mL, 30 mmol) and copper(I) chloride (3 g, 30 mmol) were added and the mixture was stirred vigorously for 4 hours under air with a CaCl2 tube on top. Then, a silica plug was run in CH2Cl2 and the solvent removed. The crude product was purified by column chromatography (SiO2, Hexanes/CHCl3 3:2 up to 2:3) to afford the product as yellow solid (270 mg, 80% yield). 1H NMR (500 MHz, CDCl3) δH: 7.33 (d, J = 8.8 Hz, 4H); 7.19 (d, J = 8.6 Hz, 8H); 7.03 (d, J = 8.7 Hz, 8H); 6.92 (d, J = 8.8 Hz, 4H); 2.48 (s, 12H). 13C NMR (125 MHz, CDCl3) δC: 148.4; 144.4; 133.6; 133.6; 128.5; 125.9; 121.5; 114.5; 82.1; 73.8; 16.7. MS EI (m/z): [M]+ calcd for C44H36N2S4: 720.17558; found: 720.175898. Anal. calcd for C44H36N2S4: C, 73.29; H, 5.03; N, 3.89; found: C, 73.25; H, 5.10; N, 3.93. m.p. = 218 °C.Methods
Chemical characterization
1H and 13C NMR spectra were recorded on a Bruker Advance 500 spectrometer (500 MHz for 1H and 125 MHz for 13C). The deuterated solvents are indicated; chemical shifts, δ, are given in ppm, using the solvent residual signal as an internal standard (1H, 13C). MS were recorded on ThermoElectron MAT 900 using electron impact (EI) ionization technique. Elemental analyses were carried out by Stephen Boyer at London Metropolitan University using a Carlo Erba CE1108 Elemental Analyzer. A copy of 1H and 13C NMR spectra have been reported for samples 9 to 12, where elemental analysis exceeded 0.4% on carbon (ESI†).Electrochemical characterization
All cyclic voltammetry measurements were carried out in freshly distilled CH2Cl2 using 0.3 M [TBA][PF6] electrolyte in a three-electrode system, with each solution being purged with N2 prior to measurement. The working electrode was a Pt disk. The reference electrode was Ag/AgCl and the counter electrode was a Pt rod. All measurements were made at room temperature using a μAUTOLAB Type III potentiostat, driven by the electrochemical software GPES. Cyclic voltammetry (CV) measurements used scan rates of 100 mV s−1; square wave voltammetry (SWV) was carried out at a step potential of 4 mV, square wave amplitude of 25 mV, and a square wave frequency of 15 Hz, giving a scan rate of 40 mV s−1. Ferrocene was used as the internal standard in each measurement.Optical characterization
Solution UV-Visible absorption spectra were recorded using a Jasco V-670 UV/Vis/NIR spectrophotometer controlled with SpectraManager software. Photoluminescence (PL) spectra were recorded with a Fluoromax-3 fluorimeter controlled by the ISAMain software. All samples were measured in a 1 cm cell at room temperature with dichloromethane as solvent. Concentration of 2 × 10−5 M and 5 × 10−6 M were used for solution UV/Visible and PL, respectively. Time-resolved and time-integrated PL spectra of film samples were acquired using a time-correlated single photon counting (TCSPC) setup (FluoTime 300, PicoQuant GmbH). Samples were photoexcited using a 507 nm laser head (LDH-P-C-510, PicoQuant GmbH) pulsed at 40 MHz, with a pulse duration of 117 ps and fluence of ∼0.1 nJ cm−2. Fits were carried out using commercial fitting software (FluoFit v4.5.3, PicoQuant GmbH).Crystallographic details
Crystallographic data were collected using Agilent Technologies SuperNova with Cu Kα (λ = 1.54178 Å) radiation at 120(2) K. Single crystals suitable for X-ray diffraction (XRD) were prepared by recrystallization in hot 1-azohexane for H-DATPA and slow evaporation in CH2Cl2 for MeO-DATPA. A summary of data collection and structure refinement is reported in Table 1. The crystal structures were deposited at CCDC with deposition number 921284 and 925482 for H-DATPA and MeO-DATPA, respectively.| H-DATPA | MeO-DATPA | |
|---|---|---|
| Chemical formula | C40H28N2 | C44H36N2O4 |
| Formula weight | 536.64 | 656.78 |
| Data collection | CrysAlisPro | CrysAlisPro |
| Structure solution | Sir9225 | Superflip26 |
| Structure refinement | SHELXTL | CRYSTALS |
| Color, habit | Yellow, rod | Yellow, block |
| Crystal size (mm) | 0.39 × 0.09 × 0.05 | 0.30 × 0.24 × 0.13 |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | P21/n | Pca21 |
| a (Å) | 8.05690(10) | 18.74123(11) |
| b (Å) | 9.2642(2) | 9.94915(7) |
| c (Å) | 18.9932(4) | 18.52083(11) |
| α (°) | 90 | 90 |
| β (°) | 90.551(2) | 90 |
| γ (°) | 90 | 90 |
| V (Å3) | 1417.60(5) | 3453.38(4) |
| Z | 2 | 4 |
| ρ(calc.) (g cm−3) | 1.257 | 1.263 |
| μ (mm−1) | 0.559 | 0.642 |
| N° of rflcn/unique | 8178/3729 | 42111/28155 |
| θ range (°) | 4.7 to 73.7 | 4.7 to 76.5 |
| Compl. to θmax (%) | 97.3 | 99.8 |
| R1 | 0.0427 | 0.0273 |
| wR2 | 0.0941 | 0.0592 |
| GooF | 1.030 | 1.012 |
Computational details
The molecular structures were optimized first in vacuum without any symmetry constrains, followed by the addition of CH2Cl2 solvation via a conductor-like polarizable continuum model (C-PCM).27 The presence of local minimum was confirmed by the absence of imaginary frequencies. All calculations were carried out using the Gaussian 09 program28 with the Becke three parameter hybrid exchange, Lee Yang–Parr correlation functional (B3LYP) level of theory. All atoms were described by the 6-31G(d) basis set. All structures were input and processed through the Avogadro software package.29Charge transport parameters
Charge transport in the HTMs has been investigated according to a published procedure.30 Poly(3,4-ethylendioxythiophene)–poly(styrene sulfonate) (PEDOT:PSS) was spin-coated onto indium tin oxide substrate (ITO) and dried at 140 °C for 30 min in vacuum. The purpose of PEDOT:PSS layer (40 nm) was to reduce the roughness of ITO as well as to improve the work function, achieving enhanced hole-only device properties. The HTMs were spin-coated onto PEDOT:PSS from chloroform solution (40 mg mL−1) in a nitrogen atmosphere. Finally, Au contacts (400 nm thick) were applied via thermal evaporation through a shadow mask in 2 × 10−6 Torr vacuum. The work function of Au and ITO are close to the HOMO energy level of the HTMs as well as far below the LUMO energy level.30 Therefore, the electron injection barrier is higher than the corresponding hole injection barrier. As a result, the transport is dominated by holes. The J–V characteristics of these samples were measured with a Keithley 2420 Source Meter unit at room temperature.Solar cell fabrication
ss-DSSCs were prepared according to a standard procedure,31 and all solvents used for device fabrication were reagent grade and anhydrous. FTO substrates (15 Ω sq−1, Pilkington) were etched with zinc powder and HCl (2 M aqueous solution) to give the desired electrode patterning. The substrates were cleaned with Hellmanex (2% by volume in water), de-ionized water, acetone, and ethanol. The last traces of organic residues were removed by a 10 minutes oxygen plasma cleaning step. The FTO sheets were subsequently coated with a compact layer of TiO2 (100 nm) by aerosol spray pyrolysis deposition at 270 °C, using oxygen as the carrier gas. Films of 1.5 μm thick mesoporous TiO2 were then deposited by screen-printing a commercial paste (Dyesol 18NR-T). The TiO2 films were slowly heated to 500 °C and allowed to sinter for 30 min in air. Once cooled, the samples were immersed into a 15 mM TiCl4 aqueous solution for 45 min at 70 °C and then heated to 500 °C for another sintering step of 45 min. After cooling to 70 °C, the substrates were immersed in a 500 μM dye solution, in 1:1 mixture of acetonitrile and tert-butyl alcohol, for one hour. The dye employed in this study was D102, previously reported by Horiuchi et al.23 After the dyed films were rinsed in acetonitrile, the hole conductor matrix was applied by spin-coating at 1000 rpm for 45 s in air. The solutions for spin coating consisted of 80 mM of hole transporter, 15 mM of lithium bis(trifluoromethylsulfonyl)imide salt and 70 mM of 4-tert-butylpyridine in anhydrous chlorobenzene. After drying overnight, back contacts were applied by thermal evaporation of 200 nm of silver.Solar cells characterization32
For measuring the device merit parameters, simulated AM 1.5 sunlight was generated with a class AAB ABET solar simulator calibrated to give simulated AM 1.5, 100 mW cm−2 irradiance, using an NREL-calibrated KG5 filtered silicon reference cell, with less than 1% mismatch factor; the current–voltage curves were recorded with a sourcemeter (Keithley 2400, USA). The solar cells were masked with a metal aperture defining the active area (0.12 cm2) of the solar cells. All devices were stored in air and in dark for 24 hours prior to testing.Conductivity measurements
Devices for measuring the conductivity of the hole transporter in a dye-sensitized mesoporous TiO2 film were prepared according to a published procedure.31 The preparation (on glass substrates) was identical to that used for the ss-DSSCs, except for the absence of the TiO2 compact layer. The electrode pattern was designed for two point contact measurements with a channel length (direction of current flow) of 200 μm, a channel width of 6.53 cm, and a film thickness of 1 μm. The metal-hole transporter contact resistance was measured to be several orders of magnitude lower than the bulk resistance as estimated by four point conductivity measurement (see ESI†). The spin-coating solutions were identical to those used to prepare the solar cells. Linear current–voltage curves were obtained for the conductivity measurements by testing in dark condition with a sourcemeter (Keithley 2400, USA) in a two-point contact setup. All devices were stored in air and in dark for 24 hours prior to testing.Results and discussion
The HTMs were synthesized according to the procedure described in Scheme 1. A triphenylamine unit containing para-substituted methyl and methoxy functional groups was synthesized by copper-catalyzed Ullmann coupling.33 The thiomethoxy triphenylamine derivative was synthesized by reacting dibromotriphenylamine with sodium thiomethoxide,34 followed by a bromination with NBS. Then, a silyl-protected acetylene bridge was attached via the Sonogashira cross-coupling reaction and was subsequently deprotected with fluoride.35 Finally, homocoupling of the terminal alkyl group was achieved via copper and molecular oxygen.36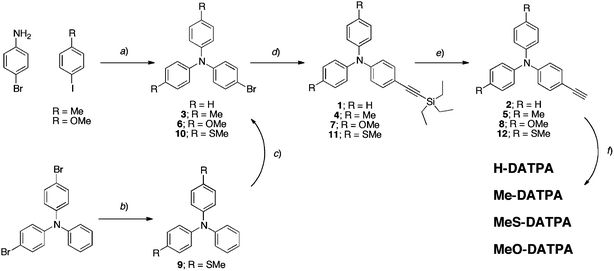 | ||
| Scheme 1 Synthetic procedure for the HTMs preparation. Reaction conditions: (a) CuI, bipy, KOtBu, toluene; (b) NaSMe, DMF; (c) NBS, DMF; (d) triethylsilylacetylene, (PPh3)2PdCl2, PPh3, CuI, piperidine, toluene; (e) 1 M TBAF, CH2Cl2; (f) CuCl, TMEDA, O2, CH2Cl2. | ||
Single crystals suitable for XRD analysis were collected to resolve the crystal packing as shown in Fig. 2. H-DATPA and MeO-DATPA are each arranged in a herringbone pattern, with one molecule in the asymmetric unit and a regular stacking distance between the molecules. The distances between the planes, defined over the central diacetylene unit and the directly attached phenyl rings, are 3.61 and 3.44 Å respectively. The shortest intermolecular N–N distances between molecules of adjacent stacks were found to be 6.03 Å for H-DATPA and 5.80 Å for MeO-DATPA. These values are indicative of effective stacking of π-orbitals distributed, as calculated from DFT (see next section), on the triphenylamine units and diacetylene bridge. We note that both within the stack and between stacks however, shorter interactions were observed for MeO-DATPA. Close π-stacked molecular arrangements can generate high charge transport rate in small molecule HTMs.37 Indeed, we will show later that the shorter N–N distance in MeO-DATPA, thus a closer π–π stacking, corresponds to higher hole mobility.
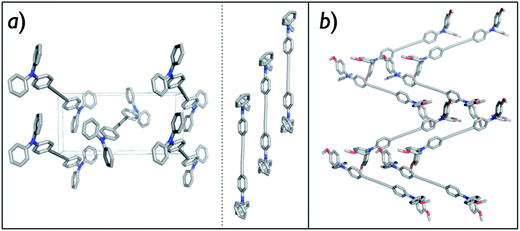 | ||
| Fig. 2 Crystal packing of H-DATPA from a-axis top view together with the π–π stacking (a) and MeO-DATPA (b). Hydrogen atoms have been removed for clarity. | ||
A high level of transparency in the visible to NIR region for the HTM is a key parameter for ss-DSSC application.38 The absorption spectra (solid lines, Fig. 3) are similar for each HTM derivative; the absorption peaks (λmax) are located in the spectral window between 375 and 390 nm, with a half-maximum peak-width of approximately 70 nm. To study the DATPA derivatives in ss-DSSCs, we employed an indoline dye (D102), the absorption spectra of which spans the region 400–650 nm (ESI†),23,24 which makes this sensitizer ideal to test our new HTMs. In addition, photoluminescence spectra were also recorded (dashed line, Fig. 3). The measurements show a clear red-shift and broadening of the PL spectrum with the series, which could be explained as an effective S1 energy stabilization due to the presence of electron-donating groups of various strengths.39,40 From the intersection of emission and absorption spectra we can obtain the E0–0′ transition and, therefore, we can estimate the optical band gap. In addition, the DATPA emission peaks are in the visible region where D102 strongly absorbs, which could enable additional light absorption, via Förster resonant energy transfer from the HTM to D102.41,42 However, we can likely neglect this contribution to the photocurrent since the HTM predominantly absorbs in the UV region, which is strongly filtered by the TiO2 (see ESI†).
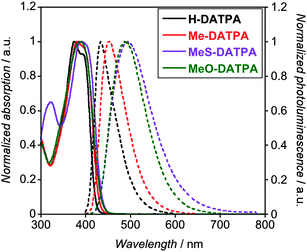 | ||
| Fig. 3 Normalized UV-Visible absorption (solid line) and photoluminescence (dashed line) of H-DATPA (black line) Me-DATPA (red line), MeS-DATPA (purple line) and MeO-DATPA (green line). | ||
The HTM oxidation potential and energy level alignment with the HOMO level of the dye are crucial parameters for constructing high-performance ss-DSSCs. In previous studies, Durrant and co-workers demonstrated that hole transfer from the dye to the HTM (dye regeneration) is not limited by kinetic competition with interface recombination processes.43–45 Rather, dye regeneration depends solely on the oxidation potential offset that drives the hole from the dye to the HTM,46–48 which can be estimated by cyclic voltammetry as described in the experimental section.49 In Fig. 4 we report the oxidation peaks for the DATPA series and Spiro-OMeTAD obtained by square-wave voltammetry. As expected, increasing the substituent electron-donating character on DATPA (H– < Me– < MeS– < MeO–) we observed a strong shift in the oxidation peaks to less positive potentials. However, the first oxidation for Spiro-OMeTAD is still another 300 mV less positive than the MeO-DATPA. All DATPA derivatives show evidence of two oxidation peaks corresponding to one electron oxidation process for each triphenylamine unit. For MeO- and MeS-DATPA, the cyclic voltammetry trace (see ESI†) cannot clearly elucidate the two-oxidation processes, since the oxidation potentials for each electron are similar. Therefore, an electrochemical pulse technique, such as square-wave voltammetry used here, should be used in order to elucidate both oxidation processes. For Spiro-OMeTAD, we observed three oxidation processes, involving one electron for the first and second oxidation and two electrons for the last oxidation process, as reported previously.7,50 We converted the oxidation potentials (V) to EHOMO (eV) by following the procedure described by Thompson et al. which employs an empirical linear translation equal to EHOMO (eV) = −1.4 ECV (V) − 4.6.49 The extracted values are listed in Table 2. It is worth pointing out that we have also measured the oxidation potential of D102 under the same conditions used for the HTMs, in order to make a reasonable estimation of the dye – HTM energy offset (see ESI†). The D102 first oxidation potential in solution was found at more negative potential than H-DATPA and Me-DATPA (see Table 2), which should make these HTMs unable to regenerate the dye. However, the cyclovoltammetry performed for D102 anchored on mesoporous TiO2 gives 0.26 V positive shift compared to the valued collected in solution. Taking in account the new oxidation value, all DATPA derivatives should be able to regenerate the oxidized dye, as we will discuss afterwards.
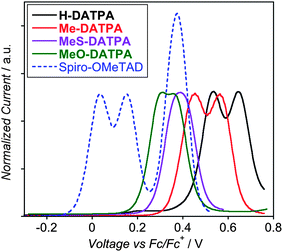 | ||
| Fig. 4 Square-wave voltammetry of H-DATPA (black line), Me-DATPA (red line), MeS-DATPA (purple line), MeO-DATPA (green line) and Spiro-OMeTAD (blue dashed line) plotted vs. ferrocene/ferrocenium. | ||
| λmax (nm) | λem (nm)a | Egap (eV)b | EOX (V)c | EHOMO (eV)d | ELUMO (eV)e | |
|---|---|---|---|---|---|---|
| a Excitation at λmax.b From the intersection of absorption and emission spectra.c From SWV and CV measurements and referenced to ferrocene.d EHOMO (eV) = −1.4 ECV (V) − 4.6.e ELUMO = EHOMO + Egap. | ||||||
| H-DATPA | 376 | 434 | 3.00 | +0.52 | −5.33 | −2.33 |
| Me-DATPA | 382 | 453 | 2.93 | +0.45 | −5.23 | −2.33 |
| MeS-DATPA | 391 | 495 | 2.85 | +0.38 | −5.14 | −2.29 |
| MeO-DATPA | 389 | 490 | 2.86 | +0.30 | −5.02 | −2.16 |
| Spiro-OMeTAD | 385 | 424 | 3.05 | +0.03 | −4.64 | −1.59 |
| D102 in solution | 510 | 640 | 2.19 | +0.42 | −5.18 | −2.99 |
| D102 onto TiO2 | 505 | 670 | 2.07 | +0.58 | −5.41 | −3.34 |
To elucidate the electronic properties of DAPTA derivatives, DFT calculations were performed with Gaussian 09, B3LYP 6-31G(d) level of theory. Absolute HOMO energy values from DFT calculations differ slightly from the experimental (ca. 0.4 eV); however, the trend is reproduced faithfully (see ESI†). Fig. 5 shows the position of the HOMO–LUMO energy levels and their electron density on the molecules. The HOMO is delocalized through the π-orbitals of the triphenylamine units and diacetylene bridge for each derivative. The lowering of the HOMO energy level matches the electron-donating strength for each group, which is in good agreement with the oxidation potentials as extracted from square wave voltammetry. The LUMO energy level, as also reported in Table 2, seems to be less strongly influenced by the substituents on the triphenylamine units. This can be explained by considering that the LUMO electron density is somewhat localized on the diacetylene bridge, with a strong π anti-bonding character, and is therefore not significantly influenced by the functional groups on the para position on the triphenylamine units.
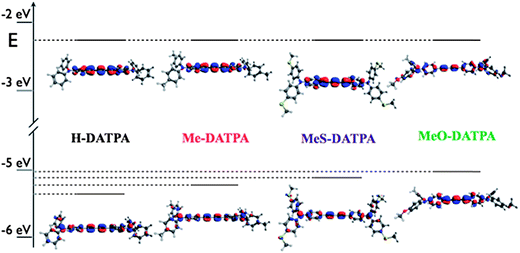 | ||
| Fig. 5 Molecular orbital distribution of HOMO (bottom) and LUMO (top) for HTMs at B3LYP/6-31G(d) level of theory (isodensity = 0.04). | ||
Another significant parameter to consider for designing new HTMs for ss-DSSCs is charge transport.9–11 The device preparation to estimate the charge transport parameters for the reported HTMs is described in the experimental section. Fig. 6 shows the J–V characteristics for the hole-only diodes at room temperature. The curves show two regions of conduction, which is Ohmic conduction (with slope ∼1) at low voltage and non Ohmic conduction (with slope >2) at high voltage. The Ohmic region has been attributed to the background impurity conduction where the injected hole carrier density is smaller than the intrinsic carrier density in the samples.30
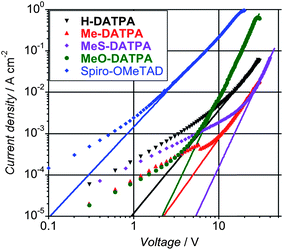 | ||
| Fig. 6 J–V characteristics of devices experimental (symbols) and calculated (solid lines) for H-DATPA (black), Me-DATPA (red), MeS-DATPA (purple), MeO-DATPA (green) and Spiro-OMeTAD (blue) using the procedure previously reported.30 | ||
The non Ohmic behavior at high fields has been analyzed in terms of space charge limited current (SCLC) and fitted as previously reported to extract the charge mobility (Table 3, see ESI† for more details). DATPA derivatives show charge mobility about two orders of magnitude lower than Spiro-OMeTAD.
| Mobility wihout Li doping (cm2 V−1 s−1) | Conductivity after LiTFSI doping (S cm−1) | |
|---|---|---|
| H-DATPA | 3.0 × 10−6 | 3.9 × 10−5 |
| Me-DATPA | 2.8 × 10−6 | 4.0 × 10−5 |
| MeS-DATPA | 4.0 × 10−6 | n/a |
| MeO-DATPA | 6.0 × 10−6 | 5.1 × 10−5 |
| Spiro-OMeTAD | 3.6 × 10−4 | 5.0 × 10−5 |
It has been widely demonstrated that lithium bis(trifluoromethylsulfonyl)-imide (LiTFSI) needs to be added in the HTM to efficiently generate photocurrent in ss-DSSCs.51,52 Furthermore, we have recently demonstrated that LiTFSI is also an strong and stable p-dopant for HTMs.11,53 Therefore, to compare the charge transport in conditions similar to device operation, we measured the effective conductivity after the addition of LiTFSI for the HTM infiltrated in a mesoporous TiO2 film, as described in the Experimental section.31 The measured values for the LiTFSI-doped HTMs are listed in Table 3. The conductivity for MeO-DATPA almost identical to Spiro-OMeTAD, and the remaining DATPA derivatives measured are all in the same range. We were unable to measure the conductivity of devices with MeS-DATPA because the hole transport films were inhomogeneous upon inclusion of LiTFSI. It is surprising that the SCLC mobilities can be so different, yet the conductivities of the doped films are comparable. This may be due to a larger density of low energy charge trap sites in the DATPA HTMs which are filled upon doping resulting in more comparable mobility and conductivity to Spiro-OMeTAD. Regardless of mechanism, the comparable conductivity upon doping is encouraging for use in the solar cells.
We prepared a set of devices with DATPA derivatives and Spiro-OMeTAD as HTMs to measure their respective performance. Fig. 7a shows the characteristic current–voltage (J–V) curves of the device with maximum power conversion efficiency out of a series of four repeats for each HTM. The characteristics have been measured under AM 1.5 simulated sun light of 100 mW cm−2 equivalent solar irradiance. Table 4 lists the solar cell performance parameters for the J–V curves reported in Fig. 7a.
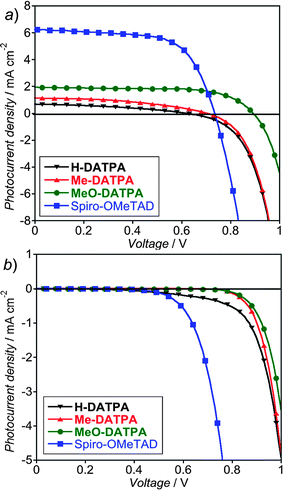 | ||
| Fig. 7 Photocurrent–voltage curves for devices employing DATPA derivatives and Spiro-OMeTAD as HTM under AM 1.5 simulated sunlight of 100 mW cm−2 equivalent solar irradiance (a) and in the dark (b). The reported J–V curves are from the device of maximum power conversion efficiency out of a series of four repeats for each HTM. | ||
| JSC (mA cm−2) | Eff (%) | VOC (V) | ff | |
|---|---|---|---|---|
| H-DATPA | 0.67 | 0.15 | 0.62 | 0.37 |
| Me-DATPA | 1.13 | 0.34 | 0.70 | 0.43 |
| MeO-DATPA | 1.93 | 1.16 | 0.89 | 0.67 |
| Spiro-OMeTAD | 6.23 | 2.96 | 0.74 | 0.64 |
The short circuit photocurrent density (JSC) shows a superlinear increase as HTM oxidation potential decreases. In Fig. 8a we plot the JSC as a function of the Dye – HTM energy offset, where we have determined the D102 HOMO level by voltammetry of D102-sensitized TiO2 nanoparticles. Confirming what was previously observed by Kroeze et al.,47 there is an exponential improvement in JSC with this energy offset.48
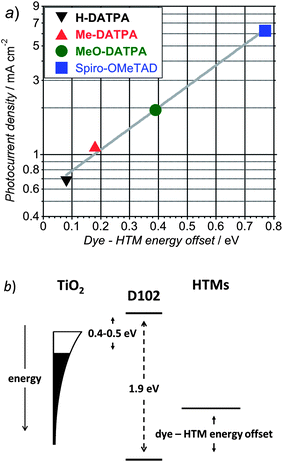 | ||
| Fig. 8 Dependence of the generated photocurrent at short circuit condition versus Dye – HTM energy offset (a). Schematic diagram of the energy levels at dye-sensitized TiO2/organic HTM heterojunction (b).54 | ||
To investigate the hole transfer between D102 and the various HTMs (dye regeneration) we have monitored the quenched emission of the dye adsorbed onto non-injecting metal oxide, such as Al2O3.5 Mesoporous Al2O3 films were prepared in condition similar the TiO2 (see Experimental), then they were sensitized with D102 and the pores infiltrated with the same HTMs used for the devices reported in Fig. 7. The time-integrated and time-resolved photoluminescence spectra in Fig. 9 show an increased quenching of the dye that correlates directly with the increasing dye – HTM offset and photocurrent shown in Fig. 8. This quenching is due to a higher hole transfer yield for the systems with the larger offset, where the hole distribution lies more strongly on the HTM than on the dye.44,46 We should also note a mismatch for H-DATPA and Me-DATPA, which is likely to be due to not uniform HTM film formation, as we also discuss later for the device reported in Fig. 7.
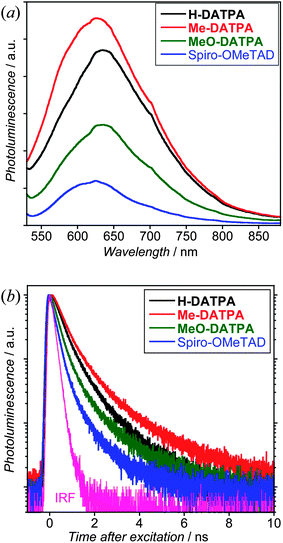 | ||
| Fig. 9 Monitoring the quenching of the PL from D102 due to hole transfer to the HTM. (a) Time-integrated PL spectra from film samples excited at 507 nm. (b) Time-resolved PL decays when monitoring the emission from the samples at 640 nm. Fits to the data were obtained by convoluting biexponential functions with the instrument response function (IRF). The fits reveal initial dominant ultrafast components that cannot be accurately resolved by the system and a second component with time constants of 0.61, 0.45, 0.35 and 0.24 ns for Me-DATPA, H-DATPA, MeO-DATPA and Spiro-OMeTAD, respectively. | ||
In contrast to the JSC, there is no consistent trend with the VOC, MeO-DAPTA has a higher VOC than Spiro-OMeTAD, but the other derivatives have a lower VOC. The VOC is generated by the splitting between the quasi Fermi level for electrons in the TiO2 and for holes in the HTM. It is influenced by many factors, including (i) fixed energy level offsets between the materials, i.e. the difference between TiO2 CB energy and the HTM HOMO energy level, (ii) Any abrupt shift in surface potential at any of the heterojunctions, the TiO2-Dye-HTM interface for instance, and (iii) the charge density build up in the photoactive film (n), which is controlled by the charge generation (G) and recombination rate (krec), where under steady state dn/dt ∼ G − nkrec = 0. In addition, any imperfection in the device can result in dark “leakage” current, which manifests itself electronically as a low shunt resistance with the impact of reducing the VOC. The shunt resistance is often dependent upon whether the solar cell is illuminated or not, adding further variability to the open-circuit voltage. The lower photocurrents, consistent with lower charge generation (hole-transfer) partly explain the lower than expected VOC. From the dark J–V curves, we can see that H-DATPA has a shunting issue, and Me-DATPA exhibits “photo-shunting”. In addition, we can also postulate that if the HOMO level of the HTM moves significantly deeper than the silver electrode work function at this interface, then pinning of the quasi Fermi level for holes, to the silver work function may occur, nullifying any potential VOC increase.55 Correctly choosing the metal/HTM contact may be a requirement to significantly enhance the open-circuit voltage of ss-DSSCs.
What is surprising is that we seem to require up to 0.8 eV offset between the dye and HTM HOMO levels to drive efficient hole-transfer, or at least operation in the solar cell. Fig. 8b shows a schematic diagram of the energy levels at the dye-sensitized TiO2/organic HTM, according to the report of Bisquert et al.54 For the devices studied here using Spiro-OMeTAD, the VOC is 0.74 V. If we assume the determined offset between the dye and Spiro-OMeTAD HOMO levels of 0.8 V is correct, then with an optical band gap of 1.9 eV,24 the energy offsets between the D102 LUMO (or excited state energy) and the maximum quasi-Fermi level for electrons (E*fn) in the TiO2 under full sun illumination is 0.4–0.5 eV. This loss-in-potential at the TiO2 side is reasonably consistent with recent observations with perovskite absorbers, where entirely removing the TiO2 and replacing it with porous alumina resulted in 200 to 300 meV increase in open-circuit voltage in much thinner devices.5 The most efficient solid-state DSSCs employing organic dyes have a total loss-in-potential (difference between the optical band gap and open-circuit voltage) of 0.89 eV,2,8 in comparison to the 1.16 eV for the system studied here. Clearly minimizing this loss is central to further enhancements in solar cell performance since it puts a ceiling on the maximum possible efficiency.56 In principle, the dye regeneration process is a single electron transfer, and should only require a small offset in HOMO levels to occur efficiently. Indeed, although the measured PL decays in Fig. 9 show significant speeding up in hole-transfer rate with shifting of HOMO level, even the slowest quenching system (half life ∼1 ns) is still significantly faster than the nominal lifetime of the oxidized dye (μs). The hole-transfer from the oxidized dye to the hole-transporter should thus compete extremely favorably with recombination of the conduction band electrons with the oxidized dye. There hence appears to be a significant missing bit of information concerning this charge generation mechanism in the ss-DSSC, and understanding firstly why we require such a large offset, and secondly what dye-HTM properties are important for minimizing this offset whilst retaining good operation in the solar cells should be a key focus to significantly advance this technology.
Conclusions
We have presented the synthesis and characterization of triphenylamine derivatives with different electron donating groups to use as hole transport materials in ss-DSSCs. Each of these molecules is comprised of two triphenylamine moieties bridged with a diacetylene group. We systematically changed the energy and the distribution of molecular orbitals by increasing the electron-donating ability of peripheral substituents (–H < –Me < –SMe < –OMe). Our key findings are that the photocurrent generation appears to be exponentially dependent upon dye-HTM HOMO–HOMO energy offset, but the open-circuit voltage is a complicated convolution of many influences, requiring more effort to optimize for maximizing the performance of high-voltage ss-DSSCs. A clear requirement to make these simple triphenylamine HTMs as efficient as, or even more efficient than Spiro-OMeTAD, is to shift the oxidation potential to less positive values, and possibly optimized for each individual dye. However, this will be at the cost of open-circuit voltage. More generally, this study clearly identifies a significant energy loss due to dye regeneration in the current state of the art solid-state DSSCs. Understanding the requirement for and minimizing this loss is critical for the future advancement of solid-state DSSCs.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) APEX project for financial support. We thank Dr Gary Nichol (University of Edinburgh) for the crystal structure data collection, refinement and discussion. We thank Dr James Ball (University of Oxford) for the conductivity measurements.References
- M. Graetzel, R. A. J. Janssen, D. B. Mitzi and E. H. Sargent, Nature, 2012, 488, 304–312 CrossRef CAS.
- B. E. Hardin, H. J. Snaith and M. D. McGehee, Nat. Photonics, 2012, 6, 162–169 CrossRef CAS.
- X. Ren, S. Jiang, M. Cha, G. Zhou and Z. S. Wang, Chem. Mater., 2012, 24(17), 3493–3499 CrossRef CAS.
- X. Lu, Q. Feng, T. Lan, G. Zhou and Z. S. Wang, Chem. Mater., 2012, 24(16), 3179–3187 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS.
- J. M. Ball, M. M. Lee, A. Hey and H. Snaith, Energy Environ. Sci., 2013 10.1039/c3ee40810h.
- U. Bach, D. Lupo, P. Comte, J. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS.
- J. Burschka, A. Dualeh, F. Kessler, E. Baranoff, N. L. Cevey-Ha, C. Yi, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2011, 133(45), 18042–18045 CrossRef CAS.
- F. Fabregat-Santiago, J. Bisquert, L. Cevey, P. Chen, M. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 131, 558–562 CrossRef.
- A. Abate, T. Leijtens, S. Pathak, J. Teuscher, R. Avolio, M. E. Errico, J. Kirkpatrik, J. M. Ball, P. Docampo and I. McPherson, Phys. Chem. Chem. Phys., 2013, 15(7), 2572–2579 RSC.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- J. B. Park, J. Graciani, J. Evans, D. Stacchiola, S. Ma, P. Liu, A. Nambu, J. F. Sanz, J. Hrbek and J. A. Rodriguez, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4975–4980 CrossRef CAS.
- Y. H. Jang, X. Xin, M. Byun, Y. J. Jang, Z. Lin and D. H. Kim, Nano Lett., 2011, 12, 479–485 CrossRef.
- C. Y. Hsu, Y. C. Chen, R. Y. Y. Lin, K. C. Ho and J. T. Lin, Phys. Chem. Chem. Phys., 2012, 14(41), 14099–14109 RSC.
- A. Abate, A. Petrozza, G. Cavallo, G. Lanzani, F. Matteucci, D. W. Bruce, N. Houbenov, P. Metrangolo and G. Resnati, J. Mater. Chem. A, 2013 10.1039/c3ta10990a.
- G. Resnati, P. Metrangolo, A. Abate and F. Matteucci, WO Patent 2,010,142,445, 2010.
- M. Juozapavicius, B. C. O'Regan, A. Y. Anderson, J. V. Grazulevicius and V. Mimaite, Org. Electron., 2012, 13(1), 23–30 CrossRef CAS.
- T. Leijtens, I. K. Ding, T. Giovenzana, J. T. Bloking, M. D. McGehee and A. Sellinger, ACS Nano, 2012, 6, 1455–1462 CrossRef CAS.
- D. Y. Chen, W. H. Tseng, S. P. Liang, C. I. Wu, C. W. Hsu, Y. Chi, W. Y. Hung and P. T. Chou, Phys. Chem. Chem. Phys., 2012, 14(33), 11689–11694 RSC.
- V. Coropceanu, M. Malagoli, D. da Silva Filho, N. Gruhn, T. Bill and J. Brédas, Phys. Rev. Lett., 2002, 89, 275503 CrossRef CAS.
- C. Lambert, W. Gaschler, E. Schmälzlin, K. Meerholz and C. Bräuchle, J. Chem. Soc., Perkin Trans. 2, 1999, 577–588 RSC.
- T. Horiuchi, H. Miura and S. Uchida, Chem. Commun., 2003, 3036–3037 RSC.
- L. Schmidt-Mende, U. Bach, R. Humphry-Baker, T. Horiuchi, H. Miura, S. Ito, S. Uchida and M. Grätzel, Adv. Mater., 2005, 17, 813–815 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision A.1, Gaussian Inc., Wallingford, CT, 2009, vol. 2, p. 4 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- M. T. Khan, A. Kaur, S. Dhawan and S. Chand, J. Appl. Phys., 2011, 109, 114509 CrossRef.
- H. J. Snaith and M. Grätzel, Appl. Phys. Lett., 2006, 89, 262114 CrossRef.
- H. Snaith, Energy Environ. Sci., 2012, 5, 6513–6520 Search PubMed.
- A. A. Kelkar, N. M. Patil and R. V. Chaudhari, Tetrahedron Lett., 2002, 43, 7143–7146 CrossRef CAS.
- B. Chen, S. Lee, D. Venkataraman, F. J. DiSalvo, E. Lobkovsky and M. Nakayama, Cryst. Growth Des., 2002, 2, 101–105 CAS.
- M. Hoffmann, C. J. Wilson, B. Odell and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 3122–3125 CrossRef CAS.
- M. Hoffmann, J. Kärnbratt, M. H. Chang, L. M. Herz, B. Albinsson and H. L. Anderson, Angew. Chem., 2008, 120, 5071–5074 CrossRef.
- A. Cappelli, M. Paolino, G. Grisci, G. Giuliani, A. Donati, R. Mendichi, A. C. Boccia, C. Botta, W. Mróz and F. Samperi, J. Mater. Chem., 2012, 22, 9611–9623 RSC.
- G. Y. Margulis, B. E. Hardin, I. Ding, E. T. Hoke and M. D. McGehee, Adv. Energy Mater., 2013 DOI:10.1002/aenm.201300057.
- L. A. Estrada and D. C. Neckers, Org. Lett., 2011, 13, 3304–3307 CrossRef CAS.
- J. S. Yang, S. Y. Chiou and K. L. Liau, J. Am. Chem. Soc., 2002, 124, 2518–2527 CrossRef CAS.
- J. H. Yum, B. E. Hardin, S. J. Moon, E. Baranoff, F. Nüesch, M. D. McGehee, M. Grätzel and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2009, 48, 9277–9280 CrossRef CAS.
- N. Humphry-Baker, K. Driscoll, A. Rao, T. Torres, H. J. Snaith and R. H. Friend, Nano Lett., 2012, 12(2), 634–639 CrossRef CAS.
- U. Bach, Y. Tachibana, J. E. Moser, S. A. Haque, J. R. Durrant, M. Grätzel and D. R. Klug, J. Am. Chem. Soc., 1999, 121, 7445–7446 CrossRef CAS.
- S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538–547 CrossRef CAS.
- J. Nelson, S. A. Haque, D. R. Klug and J. R. Durrant, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 205321 CrossRef.
- S. A. Haque, T. Park, A. B. Holmes and J. R. Durrant, ChemPhysChem, 2003, 4, 89–93 CrossRef CAS.
- J. E. Kroeze, N. Hirata, L. Schmidt-Mende, C. Orizu, S. D. Ogier, K. Carr, M. Grätzel and J. R. Durrant, Adv. Funct. Mater., 2006, 16, 1832–1838 CrossRef CAS.
- T. Daeneke, A. J. Mozer, Y. Uemura, M. Fekete, N. Koumura, U. Bach, L. Spiccia, Y. Tachibana and S. Makuta, J. Am. Chem. Soc., 2012, 134(41), 16925–16928 CrossRef CAS.
- B. W. D'Andrade, S. Datta, S. R. Forrest, P. Djurovich, E. Polikarpov and M. E. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef CAS.
- S. Fantacci, F. De Angelis, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 115, 23126–23133 CAS.
- U. B. Cappel, A. L. Smeigh, S. Plogmaker, E. M. J. Johansson, H. Rensmo, L. Hammarström, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2011, 115, 4345–4358 CAS.
- A. Abrusci, R. S. Santosh Kumar, M. Al-Hashimi, M. Heeney, A. Petrozza and H. J. Snaith, Adv. Funct. Mater., 2011, 21, 2571–2579 CrossRef CAS.
- Y. Qi, T. Sajoto, M. Kröger, A. M. Kandabarow, W. Park, S. Barlow, E. G. Kim, L. Wielunski, L. Feldman and R. A. Bartynski, Chem. Mater., 2009, 22, 524–531 CrossRef.
- J. Bisquert, F. Fabregat-Santiago, I. Mora-Seró, G. Garcia-Belmonte, E. M. Barea and E. Palomares, Inorg. Chim. Acta, 2008, 361, 684–698 CrossRef CAS.
- C. Tengstedt, W. Osikowicz, W. R. Salaneck, I. D. Parker, C. H. Hsu and M. Fahlman, Appl. Phys. Lett., 2006, 88, 053502–053503 CrossRef.
- H. J. Snaith, Adv. Funct. Mater., 2009, 20, 13–19 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional electrochemical measurements of DATPA derivatives and D102; transmittance of mesoporous TiO2 film, absorption and emission spectra of D102 anchored onto TiO2, metal – hole transport contact resistance, extended crystallographic table, calculated HOMO energies for DATPA derivatives, detailed description of mobility measurements and 1H and 13C NMR spectra for compounds 9–12, fluorescence lifetime of D102 on Al2O3 in absence of any of the hole conductors. CCDC 921284 and 925482. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ta11417a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |