 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacroporous poly(dicyclopentadiene) γFe2O3/Fe3O4 nanocomposite foams by high internal phase emulsion templating†
Sebastijan Kovačič*a, Nadejda B. Matskob, Gregor Ferkc and Christian Slugovc*a
aGraz University of Technology, Institute for Chemistry and Technology of Materials, Stremayrgasse 9, A 8010 Graz, Austria. E-mail: s.kovacic@tugraz.at; slugovc@tugraz.at; Tel: +43 316 873 32280
bGraz Centre for Electron microscopy (FELMI-ZFE), Stremayrgasse 17, A 8010 Graz, Austria
cUniversity of Maribor, Faculty of Chemistry and Chemical Engineering, Smetanova 17, SI-2000 Maribor, Slovenia
First published on 9th May 2013
Abstract
The high internal phase emulsion (HIPE) templating approach to macroporous poly(dicyclopentadiene) γFe2O3/Fe3O4 nanocomposite foams via ring opening metathesis polymerisation was elaborated and the influence of the formulation of the HIPE on structural and mechanical properties of the magnetic composite foams of 80% nominal porosity was studied. HIPEs solely stabilized with the nanoparticles resulted in considerably shrunken monolithic specimens characterized by an open cellular morphology with cavities bigger than 265 μm. Nanoparticles were situated in the bulk and on the surface of the polymeric foam skeleton. Precise control over the feature sizes could not be obtained in this case. In contrast, HIPE formulations co-stabilized with a surfactant yielded samples of good casting quality characterized by a fully open cellular morphology in all cases. The cavity and the window size could be controlled by the amount of surfactant in the emulsion. A low surfactant loading of 1.5 v% with respect to the monomer yielded diameters of the cavities of the order of 20 μm interconnected with windows with diameters in the order of 4 μm, while 10 v% surfactant resulted in smaller cavities (10 μm) and windows (2 μm). All these feature sizes are hardly affected by the nanoparticle loading which was varied from 1 to 30 wt%. Surfactant stabilized and cured HIPEs featured the nanoparticles predominantly on the surface of the cavities. Mechanical properties of the composite foams were assessed by stress–strain tests and revealed a strengthening of the foams prepared with 10 v% surfactant upon addition of the nanoparticles. Indicative of the strengthening is an increase of the Young's modulus from 13 ± 2 MPa in the case of a sample without nanoparticles to 104 ± 4 MPa in the case of the composite foam with 15 wt% nanoparticles. This trend was accompanied by a decrease of the elongation at break from 21 ± 4 to less than 1%. Specimens prepared with 1.5 v% surfactant are ductile and gave the same high Young's modulus (104 ± 9 MPa) irrespective of the nanoparticle loading and became stronger upon raising the nanoparticle amount reaching an ultimate strength of 3.4 ± 0.4 MPa at an elongation at break of 13 ± 4%.
Introduction
Curing of surfactant stabilized high internal phase emulsions (HIPEs)1 containing a monomer phase as the minority component and a non-polymerizable phase as the majority component yields macroporous polymeric foams of a typical hierarchically interconnected pore structure consisting of large cavities (in the range of 1–100 μm) connected via smaller windows (typically one fourth of the cavity size).2 These foams offer low density, high porosity and in many cases a fully interconnected pore structure, features which make them attractive in many diverse applications2 ranging from adsorption materials3 to scaffolds for tissue engineering4 or separators in lithium-ion batteries.5 To meet the requirements for many applications the foams have to be properly functionalized,2 which can be done using two principal functionalization strategies. Either the functional unit can be incorporated (e.g. using an appropriately functionalized co-monomer) into the HIPE before curing or a post-polymerisation modification of the foam can be performed.6 A special case of adding function to (or changing properties of) the porous scaffold is to make inorganic–organic hybrid materials.7 Again, there are several ways to prepare such hybrid-foams. The first comprises imbibing of the preformed (organic) foam with a solution containing soluble precursors and subsequent deposition of the inorganics.8 A second possibility is to have the inorganics, e.g. nano-particulate matter, in one of the liquids used for the HIPE preparation and subsequent curing of the HIPE.9,10 The latter strategy has been mainly used to improve mechanical and thermal properties of the porous materials7,11 but is also eminently suitable for introducing function (e.g. conductivity or catalytic activity) to the porous support.7,12Nano- or micrometer-scaled particles have also the ability to stabilize an emulsion, being a valuable alternative to surfactants, and composite materials derived from so-called Pickering emulsions,13i.e. emulsions which are solely stabilized by nanoparticles, have been intensely studied in the last few years.14 Suitable particles migrate to the interface between two liquid phases and form there a rigid structure slowing down coalescence.15 Important particle properties influencing the Pickering emulsion stability are the size and shape as well as the surface energy.16 Furthermore, the concentration of the particles in the emulsion plays an important role.17 Upon curing of Pickering HIPEs typically closed cell foam structures featuring large cavities (200–800 μm) are obtained. Accordingly the desirable features of HIPE derived foams, i.e. fully interconnected open cellular architectures with feature sizes in the low μm range, cannot be realized with this approach. However, using nanoparticles and additionally conventional surfactants, control over the openness and the feature sizes of the foam could be improved.18
Recently we disclosed porous foams prepared by curing surfactant stabilized dicyclopentadiene (DCPD)/water HIPEs by Ring Opening Metathesis Polymerisation (ROMP).19 Such derived foams offer unique mechanical properties (high toughness and strength) and at the same time a convenient possibility to perform post-polymerisation functionalization (because of the high amount of double bonds present in the foam scaffold) rendering them promising for many practical applications.
Herein we wish to report on the preparation, characteristics and possible applications of composite foams based on poly(dicyclopentadiene) (pDCPD) and magnetic oleic acid coated γFe2O3/Fe3O4 nanoparticles (FeOx-NPs) focusing on the foam morphologies which can be achieved. Particularly, the influence of additional surfactant on the foam morphology and the mechanical properties of the foams will be discussed and a way to achieve control over the location of the FeOx-NPs and the cavity and window sizes at the same time will be disclosed.
Experimental section
Materials
Dicyclopentadiene (DCPD, Aldrich), Pluronic®L-121 (poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol), Aldrich), the initiator (H2IMes)(PCy3)Cl2Ru(3-phenyl-indenylid-1-ene) (M2, Umicore, H2IMes = N,N-bis(mesityl) 4,5-dihydroimidazol-2-yl, PCy3 = tricyclohexylphosphine) and toluene (p.a. Aldrich) were used as received. γFe2O3/Fe3O4 nanoparticles (FeOx-NP, containing 13 wt% of oleic acid) were prepared according to the literature.20General preparation of the foams
The monomer DCPD (9.8 mmol, 1.30 g), the appropriate amount of Pluronic® L121 (cf.Table 1) and the appropriate amount of FeOx-NP (cf.Table 1) were placed in a 3 neck round-bottomed flask equipped with a mechanical stirrer and a dropping funnel. The mixture was stirred at 400 rpm for 5 min and upon continuous stirring at 35 °C deionised water (5.5 mL) was added dropwise over about 1 h. The temperature was lowered to room temperature and the stirring was prolonged for 1 h. Afterwards the initiator M2 (1.3 mg, 0.0007 mmol with respect to DCPD; in the cases where no FeOx-NPs were present, 0.0014 mol M2 was used), dissolved in toluene (0.25 mL), was added and the emulsion was stirred for further 5 min. Subsequently, the emulsion was transferred to appropriate moulds (poly(styrene) containers, steel moulds or glass vials) and the filled moulds were transferred into a preheated oven operating under air. Curing of the emulsions at 80 °C for 4 h resulted in the formation of brown rigid monoliths in all cases where FeOx-NPs were added. The specimens were purified by Soxhlet extraction with acetone for 24 h and subsequently dried in a desiccator under vacuum (10 mbar) until the weight was constant.| Sample | FeOx-NP [wt%] | Surf [v%] | dcav ± σa [μm] | dwin ± σa [μm] | |
|---|---|---|---|---|---|
| a Determined from SEM pictures of broken samples according to ref. 21; window and cavity size distributions can be found in the ESI. | |||||
| System I | pDCPD-1w | 1 | — | n.d. | n.d. |
| pDCPD-5w | 5 | — | 265 ± 100 | 24 ± 10 | |
| pDCPD-10w | 10 | — | 950 ± 360 | 110 ± 60 | |
| pDCPD-15w | 15 | — | 450 ± 170 | 70 ± 30 | |
| pDCPD-20w | 20 | — | 280 ± 110 | 34 ± 20 | |
| System II | pDCPD-1w-1.5v | 1 | 1.5 | 27 ± 10 | 3.6 ± 0.7 |
| pDCPD-5w-1.5v | 5 | 1.5 | 31 ± 16 | 5.1 ± 2.0 | |
| pDCPD-10w-1.5v | 10 | 1.5 | 30 ± 14 | 3.9 ± 1.0 | |
| pDCPD-15w-1.5v | 15 | 1.5 | 23 ± 10 | 3.5 ± 1.5 | |
| pDCPD-20w-1.5v | 20 | 1.5 | 23 ± 12 | 3.8 ± 1.0 | |
| pDCPD-1w-10v | 1 | 10 | 10 ± 4 | 2.0 ± 0.5 | |
| pDCPD-5w-10v | 5 | 10 | 9 ± 5 | 1.5 ± 0.7 | |
| pDCPD-10w-10v | 10 | 10 | 9 ± 6 | 2.3 ± 0.7 | |
| pDCPD-15w-10v | 15 | 10 | 7 ± 3 | 1.4 ± 0.5 | |
| pDCPD-20w-10v | 20 | 10 | 12 ± 7 | 1.6 ± 0.9 | |
| pDCPD-30w-10v | 30 | 10 | 5.2 ± 1.7 | 1.3 ± 0.4 | |
| pDCPD-1.5v | — | 1.5 | 10 ± 3 | 2.0 ± 0.8 | |
| pDCPD-10v | — | 10 | 5 ± 2 | 1.0 ± 0.5 | |
Characterization of the foams
TGA measurements were performed with a Netzsch Simultaneous Thermal Analyzer STA 449C (crucibles: aluminium from Netzsch). An oxygen flow of 50 mL min−1 was used in combination with a protective flow of helium of 8 mL min−1. The heating rate until a final temperature of 550 °C was 10 °C min−1.Morphology investigations were done by scanning electron microscopy (SEMs were taken on a JWS-7515, JEOL Ltd. microscope). Micrographs were taken at several magnifications from 2500 to 7000 fold, at 7 mm working distance and at 20 kV acceleration voltage. The samples were broken and mounted on a carbon tab and a thin layer of gold was sputtered onto the samples. Evaluation of the feature sizes was done according to ref. 21.
For TEM and AFM analysis, samples were embedded in Araldite–Epon embedding mixture which was composed of 49% w/w Araldite–Epon stock solution, 49% w/w Hardener DDSA (Fluka) and 2% w/w Accelerator DMP-30 (Fluka). Infiltration was performed stepwise (impregnation at room temperature for 24 h, and polymerisation at 50 °C for 72 h.). The resin embedded specimens were mounted in special holders which at the same time fit the microtome and are suitable for the examination of the block face by AFM. Ultrathin sections (10–50 nm) were obtained using a Leica Ultracut E microtome (Leica, Austria) equipped with a diamond knife (Diatome, Switzerland). Sections for TEM analysis were collected on both: carbon and formvar coated 400-mesh copper TEM grids and examined using Philips CM 20 (Philips/FEI, Eindhoven) electron microscopes at 200 kV accelerating voltage. No staining has been applied. All EFTEM experiments were acquired in TEM mode. For the calculation of elemental distribution images, the jump ratio method was used. Experimental conditions for the acquisition of jump ratio map: energy slit width 50 eV, acquisition time 30 s for Fe L ionization edge (733 eV). The block faces of specimens after cryo-ultramicrotomy were investigated under ambient conditions using a Dimension 3100 AFM/SPM (Veeco, USA) atomic force microscope. AFM images were collected in tapping mode using silicon nitride cantilevers with natural frequencies in the 300 kHz range (force constant 20 N m−1, tip radius 10 nm (NT-MDT, Russia)). AFM image processing was performed using Nanoscope v720 software (Veeco, USA).
For mechanical testing the HIPEs were prepared as described and then transferred to stainless steel templates (shouldered tensile test specimens, of 9 mm width at breaking point, thickness of 9 mm, 35 mm gage length, 65 mm distance between shoulders and 140 mm overall length). Stretching tests were carried out at room temperature on an electromechanical universal testing machine (AGS-X by Shimadzu) equipped with a 50 kN load cell. Samples were tested at a test rate of 1 mm min−1. The mean Young's moduli were calculated from data obtained from the initial linear slope of the stress–strain plot with two to four samples of the same composition.
Porosity of the specimens was measured according to a literature protocol.22 Samples were cut with a knife into pieces with dimensions of about 0.9 × 0.4 × 0.4 cm, weighed and immersed into 1-pentanol for 24 h. Then the samples were removed from the liquid, the surfaces were dried using a filter paper and weighed again.
Results and discussion
Stability of the emulsions
HIPEs were prepared by slowly adding 5.5 mL of deionized water to 1.3 mL of DCPD at 35 °C under vigorous stirring with a mechanical stirrer. Two systems were studied. In system I only oleic acid covered γFe2O3/Fe3O4 nanoparticles (FeOx-NP) were used for stabilizing the emulsion and two FeOx-NP loadings were tested. Accordingly, 1 wt% FeOx-NP (with respect to DCPD) further on denoted as DCPD-1w or 20 wt% of FeOx-NP (DCPD-20w) was added to the DCPD before the addition of water, resulting in a clear brown dispersion. After the addition of water was finished, the emulsions were further stirred at 35 °C for 1 h, poured into poly(styrene) tubes and transferred to an oven which was preheated to 80 °C. Subsequently the consistency of the (not agitated) emulsion was monitored. The elevated temperature was chosen, because curing of the emulsions will be later on carried out under that condition. DCPD-1w showed considerable coalescence within the first hour but no creaming or sedimentation occurred in 24 h, which is in good agreement with the results of Bismarck et al. who studied styrene based HIPEs which were stabilized with 1.5 wt% of similar NPs.14e In their case all emulsions were also stable against coalescence for more than 3 months at room temperature. The fast coalescence in our case is attributed to the higher frequency of droplet collision at 80 °C.23 Nevertheless a considerable stabilization of the DCPD/water HIPE was achieved in DCPD-1w as evidenced by a control experiment assessing the stability of a non-stabilized emulsion under these conditions. DCPD/water HIPE completely phase separates in only 15 s at approx. 35 °C. DCPD-20w behaved differently. After the first hour at 80 °C some creaming and particularly sedimentation were observed which resulted in complete phase separation after 24 h at 80 °C. Accordingly, in this case, the sedimentation (or creaming) tendency is not decreased by the higher particle concentration as it was found in other cases.24 The fast sedimentation in our case might be best explained by the partial dispersion of FeOx-NPs in the DCPD phase (density = 0.98 g cm−3) which increases the density of this phase and causes eventually sedimentation.In system II, FeOx-NPs and surfactant (Pluronic®L-121) were used as stabilizers of the HIPEs. Pluronic®L-121 is a non-ionic poloxamer particularly suitable for the stabilization of DCPD/water HIPEs.19 Four formulations (containing 1 wt% FeOx-NP and 1.5 v% surfactant (DCPD-1w-1.5v) or 10 v% Pluronic®L-121 (DCPD-1w-10v) and 20 wt% FeOx-NP with 1.5 v% (DCPD-20w-1.5v) and 10 v% Pluronic®L-121 (DCPD-1w-20v)) were examined. The HIPE formulation DCPD-1w-1.5v showed some creaming after 3 h at 80 °C and pronounced creaming after 24 h. Accordingly, DCPD-1w-1.5v is considerably more stable than DCPD-1w. Increasing the surfactant amount to 10 v% in DCPD-1w-10v further increased the HIPE stability as creaming after 24 h at 80 °C was less pronounced as in the case of DCPD-1w-1.5v. Increasing the FeOx-NP loading to 20 wt% yielded the most stable formulations. Some sedimentation was observed in the case of DCPD-20w-1.5v after 24 h and DCPD-20w-10v did not show any sign of HIPE instability during storing for 24 h at 80 °C. Accordingly, the addition of surfactant is distinctly improving the kinetic stability of the HIPEs which is neither a surprising nor an unprecedented effect. Studies describing a so-called synergistic effect of nanoparticles and surfactants have been published recently.25
The assessment of the kinetic stability of the HIPE formulations revealed the reasonableness of using them for the preparation of foams via ROMP of DCPD/water HIPEs under the conditions established so far.19
Curing of the emulsions
Several HIPE formulations as presented in Table 1 were now cured upon addition of the initiator M2 (0.0067 mol% with respect to DCPD) transferring the HIPE into moulds and heating the samples for 4 h at 80 °C. In all cases where FeOx-NPs were used brown rigid monoliths were obtained, which were removed from the mould, extracted for 24 h with acetone in a Soxhlet apparatus and dried in a vacuum. Interestingly, the catalyst amount of 0.014 mol% used in previous studies19 led to a premature curing of the formulation including FeOx-NPs. Presumably the initiator is activated by abstraction of the phosphine ligand by the FeOx-NPs and a reasonable pot life of the emulsion, allowing the transfer to the moulds within 5 min after initiator addition was achieved by a reduction of the catalyst loading by the factor of 2.Visual inspection of the specimens readily revealed a good casting quality for all samples prepared with additional surfactant. The casting quality of specimens prepared without surfactant (pDCPD-1w–pDCPD-20w) was rather poor in terms that the volume of foams collapsed to approx. 75% (to 50% in the case of pDCPD-1w) of the original volume and the foams exhibited big cavities apparent to the naked eye. Evaluation of the microstructure of the composite foam was then performed with scanning electron microscopy (SEM, see Fig. 1), transmission electron spectroscopic (TEM) techniques and atomic force microscopy (AFM) (cf. below). The porosity of the specimens was determined22 to be 78 ± 4% for the series prepared with 1.5 v% surfactant, and 70 ± 4% for the series prepared with 10v% surfactant (cf. ESI†).
 | ||
| Fig. 1 SEM pictures of composite foams prepared without surfactant (upper row), prepared with 1.5 v% surfactant (middle row) and with 10 v% surfactant (bottom row) containing 5 wt% (1st column), 10 wt% (2nd column), 15 wt% (3rd column) and 20 wt% FeOx-NPs (4th column). | ||
During extraction hardly any FeOx-NPs were washed out as revealed by only slightly light brown coloured extracts. The extracts were dried in a vacuum and weighed. In cases where no or low surfactant amount was used the weight of the residue was negligible (approx. 2 wt% of the sum of the masses of DCPD, FeOx-NP and surfactant used). In the case of samples prepared with 10 v% surfactant the mass of the residue was higher. Investigation of the residues with NMR spectroscopy revealed the presence of DCPD-oligomers, oleic acid and cross-metathesis products of oleic acid in all cases. The extracts of pDCPD-1w-10v–pDCPD-30w-10v contained the same species and additionally the surfactant, which is responsible for the higher residue masses obtained in these cases. These results suggest that a great proportion of the FeOx-NP is contained in the samples.
Results from thermogravimetric investigations of aged, i.e. oxidized (cf. ESI† and ref. 19), composite foams corroborated the presence of a high proportion of the FeOx-NPs in the samples. Actually, slightly increased values for the weights of residues were determined for unknown reasons. However, the residual mass after having heated the samples in a pure oxygen atmosphere with a heating rate of 10 °C min−1 to 550 °C increased from 5% in the case of pDCPD-5w-10v to 22.3% in the case of pDCPD-30w-10v (cf.Fig. 2). A reference sample containing no FeOx-NPs lost 95% of its weight under these conditions and reached a weight loss of 99.3% after heating for further 15 min at 550 °C. In the presence of the FeOx-NPs the final mass was reached at lower temperatures e.g. at approx. 420 °C in the case of pDCPD-30w-10v.
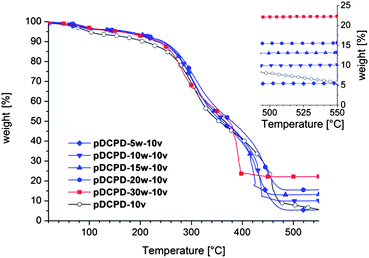 | ||
| Fig. 2 TGA of the composite foams performed in an oxygen atmosphere; the inset shows the masses of the residues upon burning. | ||
Characterization of the morphology of the composite foams
For morphology investigations aged samples were used because oxidation made them more brittle and specimens could be broken or cut which is a prerequisite for the following characterizations. SEM was used to visualize and evaluate the foam morphology. All samples except pDCPD-1w, which was not investigated because it underwent major phase separation during ROMP and was not possible to break, exhibited a macroporous open-cell structure with different cavity and window dimensions. Specimens derived from Pickering HIPEs (system I) exhibited very large cavities (mean diameters were >265 μm, cf.Fig. 1 and Table 1) typical for composite foams prepared by this approach.14 It should be noted that no cavity size trend could be established. As supported by the emulsion stability study presented above, FeOx-NPs are not suitable for guaranteeing a kinetically stable HIPE under curing conditions and cavity sizes are strongly dependent on the curing conditions, i.e. the foam morphology cannot be properly controlled. In all samples window formation took place being most pronounced in pDCPD-15w where windows with a diameter of 70 ± 30 μm were formed all over the cavities. This is uncommon for Pickering HIPE derived composite foams which exhibit mostly closed-cell morphologies.14Upon addition of 1.5 v% surfactant to the HIPE formulation (system II) the feature size of the cavities and the windows was distinctly decreased. The mean diameter of the cavities is about 23–31 μm and the cavity size distribution is rather broad (typical standard deviations of the cavity size diameters are 10–16 μm, cf.Table 1). Window sizes range on average from 3.5 to 5.1 μm (sigma = 0.7–2 μm). Upon variation of the FeOx-NP content no significant change of cavity or window sizes could be observed. However, when comparing these data with the cavity and window size of a sample prepared with surfactant but without FeOx-NP (pDCPD-1.5v) an influence of FeOx-NPs was apparent. The specimens pDCPD-1.5v were characterized by a cavity diameter of 10 ± 3 μm and window diameter of 2.0 ± 0.8, i.e. 2–3 fold smaller than in the case of the presence of FeOx-NPs. The same phenomenon was observed for the series containing 10 v% surfactant. Here average cavity diameters of 7 to 12 μm and average window diameters of 1.4 to 2.0 were found for the sample with FeOx-NP loading of 1–20 wt% (cf.Table 1), whereas the control sample without FeOx-NPs (pDCPD-10v) is characterized by mean diameters of 5 μm for cavities and 1 μm for windows. Accordingly, feature sizes of the composite foams can be intentionally adjusted upon variation of the surfactant loading. Feature sizes are bigger in cases where FeOx-NPs are used and the amount of FeOx-NPs does not have a big impact on the foam morphology.
Having established the skeleton morphology, further investigations clarifying the distribution of the FeOx-NPs within the composite materials were done by TEM and AFM analysis on representative samples. In system I (the Pickering HIPE derived foams) FeOx-NPs were found on the surface and in the bulk of the foam skeleton. FeOx-NPs located in the bulk are mainly concentrated in islands of about 2 μm in size (biggest dimension) and only minor amounts are evenly distributed within the pDCPD matrix (cf.Fig. 3). A closer evaluation of those islands revealed a hollow structure reminiscent of collapsed cavities. Most probably those islands originated from FeOx-NP surrounded water droplets formed at the early stage of the HIPE history. During coalescence of the HIPE the water of these droplets was expelled and gathered in the big “reservoirs” i.e. the big cavities observed in the composite foams (see Fig. 3 – first row) leaving the FeOx-NPs behind. This theory is supported by the TEM and AFM pictures of samples from system II. In place of the many foams prepared with FeOx-NPs and surfactant, the results for the samples pDCPD-15w-1.5v and pDCPD-15w-10v are discussed in detail. As evident from TEM and energy filtered TEM (EFTEM) investigations of pDCPD-15w-10v the vast majority of all FeOx-NPs can be found close to the surface of the cavities (cf.Fig. 4). As the TEM images of the thin section might be misleading because they are essentially 2D projections of the objects, i.e.FeOx-NPs and the pDCPD matrix are superimposed upon each other in the direction of the electron beam, additionally an AFM analysis was performed. From this it is clearly evident that the FeOx-NPs are embedded in the pDCPD matrix just beneath the actual surface. This observation might explain why hardly any FeOx-NPs were washed out during the purification of the foams by Soxhlet extraction with acetone. Furthermore, it is worth noting, that in all cases the original size and shape of the FeOx-NPs present in the composite foam is apparently conserved.20 In the case of pDCPD-15w-1.5v a part of the FeOx-NPs is found in the bulk of the skeleton (Fig. 5).
 | ||
| Fig. 3 Elastic filtered TEM and AFM phase (phase variation: 0–25°) images of pDCPD-15w. | ||
 | ||
| Fig. 4 Elastic filtered TEM pictures of pDCPD-15w-10v (first row and second row on the left) and pDCPD-15w-1.5v (third row on the right), the EFTEM jump ratio map of Fe L of pDCPD-15w-10v (iron rich regions appear bright; second row right) and the AFM phase (phase variation 0–10°) image of the block face of pDCPD-15w-10v (third row on the left). | ||
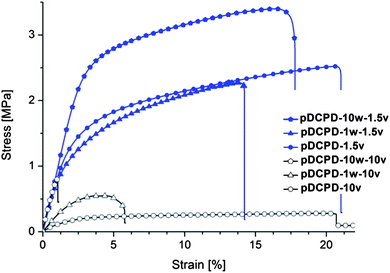 | ||
| Fig. 5 Stress–strain diagram from mechanical testing of foams. | ||
Mechanical characterization
The mechanical characteristics of the composite foams prepared according to system II were determined by tensile stress–strain tests of non-oxidized specimens (see ESI† for elemental analyses and experimental details). Addition of FeOx-NPs to HIPEs prepared with 10 v% surfactant results in an increase of the Young's modulus (E-module), an increase of the offset yield point at 0.2% strain (Rp0.2%) and a decrease of the elongation at break (cf.Table 2). pDCPD-10w-10v is already very brittle but a high Young's modulus of 107 ± 4 MPa is obtained. Accordingly, the E-module is increased by a factor of approx. 8 upon addition of 10 wt% FeOx-NPs yielding a specimen of pronounced brittleness. Composite foams prepared with 1.5 v% surfactant are ductile exhibiting a pronounced plasticity characterized by a Young's modulus of about 100 MPa and an elongation at break of more than 10%. While the addition of 1 wt% FeOx-NPs to the emulsion does not significantly alter the mechanical properties, pDCPD-10w-1.5v is significantly stronger as revealed from an ultimate strength of more than 2 MPa compared to about 1.2 and 0.9 MPa for pDCPD-1.5v and pDCPD-1w-1.5v. Accordingly, FeOx-NPs increase the strengths and the brittleness of specimens prepared with a high surfactant amount and increase the strength and toughness of specimen prepared with a low surfactant amount. Based on earlier work19c the overall favourable mechanical properties of the composite foams prepared from low surfactant loading can be attributed to undamaged polymer walls, which are further enforced upon the presence of the FeOx-NPs. In the case of the specimens prepared with 10 v% surfactant, surplus surfactant is located within the foam walls and upon extraction of the surfactant cracks are formed which are held responsible for the poorer mechanical properties. The increase of the Young's modulus upon addition of FeOx-NPs might be explained by an interaction of the surfactant and the FeOx-NPs decreasing the surfactant amount available for stabilizing the DCPD/water HIPE and facilitating the dispersion of the FeOx-NPs in the pDCPD phase.18 This theory is further supported by two experimental observations: (a) FeOx-NPs were observed in the TEM picture of pDCPD-15w-10v (cf.Fig. 4) in the bulk phase but not in the corresponding pictures of pDCPD-15w-1.5v, and (b) upon addition of FeOx-NPs the cavity and window sizes of the composite foams are bigger compared to the corresponding feature sizes in specimens prepared without FeOx-NPs (cf.Table 1).| Sample | E-module [MPa] | Rp0.2 [MPa] | ε @ break [%] | Ult. strength [MPa] |
|---|---|---|---|---|
| a Measured by stress–strain tests. | ||||
| pDCPD-1.5v | 108 ± 5 | 1.24 ± 0.2 | 18 ± 3 | 2.5 ± 0.2 |
| pDCPD-1w-1.5v | 104 ± 9 | 0.9 ± 0.2 | 12 ± 2 | 2.3 ± 0.2 |
| pDCPD-10w-1.5v | 105 ± 9 | 2.2 ± 0.2 | 13 ± 4 | 3.4 ± 0.4 |
| pDCPD-10v | 13 ± 2 | 0.11 ± 0.1 | 21 ± 4 | 0.3 ± 0.1 |
| pDCPD-1w-10v | 25 ± 2 | 0.32 ± 0.1 | 6 ± 1 | 0.5 ± 0.1 |
| pDCPD-10w-10v | 107 ± 4 | — | <1 | 0.8 ± 0.1 |
The FeOx-NP composite foams are still magnetic as expected and all samples are attracted to a permanent magnet. Additionally one exemplary sample (pDCPD-15w-1.5v) was tested in an inductive heating experiment. The specimen was exposed to an alternating magnetic field (field strength of 16 kA m−1) and the temperature rise was determined to be 18 °C. Accordingly, inductive heating of such composite foams is in principle feasible. However, as FeOx-NPs are prone to oxidation thereby loosing the self-heating capability, a possible exploitation of the self-heating capability of the composite foams disclosed here is limited.
Finally we briefly investigated the scope of the composite foams to serve as precursors for the preparation of macroporous Fe2O3 samples. For this purpose the residue from the TGA experiment with sample pDCPD-30w-10v was investigated with SEM. As evident from Fig. 6 the residue exhibited an open cellular structure with a cavity size of 3.7 ± 1.4 μm and a window size of 1.4 ± 0.4 μm reminiscent of the composite foam before burning. However, the resulting marcroporous α-Fe2O3 (identified by XRD, cf. ESI†) is brittle and the original macroscopic structure disintegrated upon touching. Such prepared α-Fe2O3 might find application as an anode material in lithium-ion batteries26 or as a photocatalyst for water splitting.27 Further investigations on the scope of macroporous α-Fe2O3 and the preparation of further macroporous metal oxides by this unprecedented approach are currently on-going in our laboratory.
 | ||
| Fig. 6 SEM pictures of pDCPD-30w-10v before (left) and after thermal treatment under pure oxygen (right) showing macroporous α-Fe2O3. | ||
Conclusions
γFe2O3/Fe3O4 poly(dicyclopentadiene) nanocomposite foams were prepared using either a purely particle stabilized or/and particle–surfactant co-stabilized high internal phase emulsion of 20 v% dicyclopentadiene in 80 v% water as the template. While emulsions stabilized only with particles exhibited undesirably large cavities partly interconnected with windows surfactant co-stabilised emulsions yielded open cellular nanocomposite foams with desirably small feature sizes. Accordingly, purely particle-stabilized HIPEs are not kinetically stable under curing conditions and the use of additional surfactant, even at low loadings of 1.5 v%, is drastically improving the kinetic stability of the HIPE allowing for an adjustment of the feature sizes by varying the surfactant amount. Moreover, it was shown, that mainly the surfactant loading is determining the cavity and window sizes, while the nanoparticle amount (varied from 1–30 wt%) hardly changes the feature sizes. Surfactant stabilized and cured HIPEs featured the nanoparticles predominantly on the surface of the cavities. Evaluation of the mechanical properties of the magnetic composite foams revealed a strengthening of the foams prepared with 10 v% surfactant upon addition of nanoparticles as evident from an increase of the Young's modulus from 13 ± 2 MPa in the case of a sample without nanoparticles to 104 ± 4 MPa in the case of the composite foam with 15 wt% nanoparticles. This trend was accompanied by a loss in ductility. In contrast, specimens prepared with 1.5 v% surfactant were ductile irrespective of the nanoparticle loading and became stronger upon raising the nanoparticle amount reaching an ultimate strength of 3.4 ± 0.4 MPa at an elongation at break of 13 ± 4%. With the disclosure of these results it became obvious that the HIPE templating of DCPD is a powerful method for the preparation of nanocomposite foams with controlled morphology and outstanding mechanical properties. Moreover, such foams can be used to prepare macroporous α-Fe2O3 upon heating in oxygen atmosphere.Acknowledgements
Financial support by the European Community (CP-FP 211468-2 EUMET) and VARTA Micro Innovation GmbH is gratefully acknowledged.Notes and references
- (a) K. J. Lissant, in “Surfactant Science series”, “Emulsion and Emulsion Technology”, Part 1, ed. K. J. Lissant, Marcel Deckker Inc., N.Y., 1974, vol. 6 Search PubMed; (b) J. Esquena and C. Solans, in Emulsions and emulsion stability, ed. J. Sjöblom, Marcel Dekker Inc., Amsterdam, 2006, pp. 245–261; (c) H. M. Princen, J. Colloid Interface Sci., 1983, 91, 160 CrossRef CAS.
- For recent reviews see: (a) I. Pulko and P. Krajnc, Macromol. Rapid Commun., 2012, 33, 1731 CrossRef CAS; (b) D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959 CrossRef CAS; (c) N. R. Cameron, P. Krajnc and M. S. Silverstein, Colloidal templating, in Porous Polymers, ed. M. S. Silverstein, N. R. Cameron and M. A. Hillmyer, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011 Search PubMed.
- (a) Y. Liu, Z. Wu, X. Chen, Z. Shao, H. Wang and D. Zhao, J. Mater. Chem., 2012, 22, 11908 RSC; (b) M. Oschatz, L. Borchardt, I. Senkovska, N. Klein, M. Leistner and S. Kaskel, Carbon, 2013, 56, 139 CrossRef CAS.
- (a) S. D. Kimmins and N. R. Cameron, Adv. Funct. Mater., 2011, 21, 211 CrossRef CAS; (b) Z. Li, M. Xiao, J. Wang and T. Ngai, Macromol. Rapid Commun., 2012, 33, 169 Search PubMed.
- S. Kovačič, H. Kren, P. Krajnc, S. Koller and C. Slugovc, Macromol. Rapid Commun., 2013, 34, 581 CrossRef.
- (a) L. Kircher, P. Theato and N. R. Cameron, Polymer, 2013, 54, 1755 CrossRef CAS; (b) L. Kircher, P. Theato and N. R. Cameron, Functionalization of porous polymers from high-internal-phase emulsions and their applications, in Functional polymers by post-polymerization modification: concepts, practical guidelines and applications, ed. P. Theato and H.-A. Klok, Wiley-VCH, Weinheim, D, 2012 Search PubMed.
- For reviews see: (a) N. Brun, S. Ungureanu, H. Deleuze and R. Backov, Chem. Soc. Rev., 2011, 40, 771 RSC; (b) L. Chen, D. Rende, L. S. Schadler and R. Ozisik, J. Mater. Chem. A, 2013, 1, 3837 RSC.
- (a) I. J. Brown, D. Clift and S. Sotiropoulos, Mater. Res. Bull., 1999, 7, 1055 CrossRef; (b) H. Maekawa, J. Esquena, S. Bishop, C. Solans and B. F. Chmelka, Adv. Mater., 2003, 15, 591 CrossRef CAS; (c) H. Zhang, I. Hussain, M. Brust and A. I. Cooper, Adv. Mater., 2004, 16, 27 CrossRef CAS; (d) H. Zhang, G. C. Hardy, Y. Z. Khimyak, M. J. Rosseinsky and A. I. Cooper, Chem. Mater., 2004, 16, 4245 CrossRef CAS; (e) A. Desforges, R. Backov, H. Deleuze and O. Mondain-Monval, Adv. Funct. Mater., 2005, 15, 1689 CrossRef CAS; (f) M. G. Schwab, I. Senkovska, M. Rose, M. Koch, J. Pahnke, G. Jonschker and S. Kaskel, Adv. Eng. Mater., 2008, 10, 1151 CrossRef CAS; (g) T. Hasell, H. Zhang and A. I. Cooper, Adv. Mater., 2012, 24, 5732 CrossRef CAS.
- (a) M. A. Bokhari, G. Akay, S. Zhang and M. A. Birch, Biomaterials, 2005, 26, 5198 CrossRef CAS; (b) W. P. Steckle, J. R. Schoonover, N. E. Lanier and A. Nobile, J. Mater. Sci., 2006, 41, 4055 CrossRef CAS; (c) H. Zhang, G. C. Hardy, M. J. Rosseinsky and A. I. Cooper, Adv. Mater., 2003, 15, 78 CrossRef CAS.
- (a) H. Tai, A. Sergienko and M. S. Silverstein, Polymer, 2001, 42, 4473 CrossRef CAS; (b) M. S. Silverstein, H. Tai, A. Sergienko, Y. Lumelsky and S. Pavlovsky, Polymer, 2005, 46, 6682 CrossRef CAS; (c) J. Normatov and M. S. Silverstein, Chem. Mater., 2008, 20, 1571 CrossRef CAS; (d) A. Menner, K. Haibach, R. Powell and A. Bismark, Polymer, 2006, 47, 7628 CrossRef CAS.
- K. Haibach, A. Menner, R. Powell and A. Bismarck, Polymer, 2006, 47, 4513 CrossRef CAS.
- S. Wang, Z. Zhang, H. Liu, W. Zhang, Z. Qian and B. Wang, Colloid Polym. Sci., 2010, 288, 1031 CAS.
- S. U. Pickering, J. Chem. Soc., 2001, 91, 907 Search PubMed.
- (a) A. Menner, V. Ikem, M. Salgueiro, M. S. P. Shaffer and A. Bismarck, Chem. Commun., 2007, 4274 RSC; (b) V. O. Ikem, A. Menner and A. Bismarck, Angew. Chem., Int. Ed., 2008, 47, 8277 CrossRef CAS; (c) V. O. Ikem, A. Menner and A. Bismarck, Langmuir, 2010, 26, 8836 CrossRef CAS; (d) I. Gurevitch and M. S. Silverstein, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1516 CrossRef CAS; (e) A. Vilchez, C. Rodriguez-Abreu, J. Esquena, A. Menner and A. Bismarck, Langmuir, 2011, 27, 13342 CrossRef CAS; (f) T. Li, H. Lui, S. Yang, Z. Li, J. Zhang and X. Zhou, J. Mater. Chem., 2011, 21, 12865 RSC; (g) I. Gurevitch and M. S. Silverstein, Macromolecules, 2011, 44, 3398 CrossRef CAS.
- S. Melle, M. Lask and G. G. Fuller, Langmuir, 2005, 21, 2158 CrossRef CAS.
- J. Frelichowska, M. A. Bolzinger and Y. Chevalier, J. Colloid Interface Sci., 2010, 351, 348 CrossRef CAS.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21 CrossRef CAS.
- (a) V. O. Ikem, A. Menner, T. S. Horozov and A. Bismarck, Adv. Mater., 2010, 22, 3588 CrossRef CAS; (b) S. Wang, Z. Zhang, H. Liu, W. Zhang, Z. Qian and B. Wang, Colloid Polym. Sci., 2010, 288, 1031 CrossRef CAS; (c) V. O. Ikem, A. Menner and A. Bismarck, Soft Matter, 2011, 7, 6571 RSC; (d) L. L. C. Wong, V. O. Ikem, A. Menner and A. Bismarck, Macromol. Rapid Commun., 2011, 32, 1563 CrossRef CAS; (e) S. Kovačič, G. Ferk, M. Drofenik and P. Krajnc, React. Funct. Polym., 2012, 72, 955 CrossRef.
- (a) S. Kovačič, P. Krajnc and C. Slugovc, Chem. Commun., 2010, 46, 7504 RSC; (b) S. Kovačič, K. Jerabek, P. Krajnc and C. Slugovc, Polym. Chem., 2012, 3, 325 RSC; (c) S. Kovačič, N. B. Matsko, K. Jerabek, P. Krajnc and C. Slugovc, J. Mater. Chem. A, 2013, 1, 487 RSC.
- (a) S. Gyergyek, D. Makovec, A. Mertelj, M. Huskic and M. Drofenik, Colloids Surf., A, 2010, 366, 113 CrossRef CAS; (b) S. Gyergyek, D. Makovec and M. Drofenik, J. Colloid Interface Sci., 2011, 354, 498 CrossRef CAS.
- A. Barbetta and N. R. Cameron, Macromolecules, 2004, 37, 3188 CrossRef CAS.
- N. H. Idris, M. M. Rahman, J.-Z. Wang and H.-K. Liu, J. Power Sources, 2012, 201, 294 CrossRef CAS.
- L. Lobo and A. Svereika, J. Colloid Interface Sci., 2003, 261, 498 CrossRef CAS.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 2539 CrossRef CAS.
- (a) B. P. Binks, A. Desforges and D. G. Duff, Langmuir, 2007, 23, 1098 CrossRef CAS; (b) B. P. Binks, J. A. Rodrigues and W. J. Frith, Langmuir, 2007, 23, 3626 CrossRef CAS; (c) B. P. Binks and J. A. Rodrigues, Langmuir, 2007, 23, 7436 CrossRef CAS; (d) R. Pichot, F. Spyropoulos and I. T. Norton, J. Colloid Interface Sci., 2009, 329, 284 CrossRef CAS; (e) E. Dickinson, Curr. Opin. Colloid Interface Sci., 2010, 15, 40 CrossRef CAS.
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988 CrossRef CAS.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photographs taken while studying the emulsion stability, photographs illustrating the mould quality, elemental analyses, description for calculating the original nanoparticle content from TGA experiments, results of porosity measurements, additional SEM, TEM and AFM pictures, pore and window size distribution plots, EDX results, inductive heating and magnetization plot, XRD of porous α-Fe2O3. See DOI: 10.1039/c3ta11402c |
| This journal is © The Royal Society of Chemistry 2013 |