 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon buffered-transition metal oxidenanoparticle–graphene hybrid nanosheets as high-performance anode materials for lithium ion batteries†
Xin
Huang
,
Jing
Chen
,
Hong
Yu
,
Ren
Cai
,
Shengjie
Peng
,
Qingyu
Yan
* and
Huey Hoon
Hng
*
School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798. E-mail: alexyan@ntu.edu.sg; ashhhng@ntu.edu.sg
First published on 12th April 2013
Abstract
In this article, we report a simple and general method for the synthesis of carbon buffered-metal oxidenanoparticle (NP)–graphene hybrid 2D nanosheets, which include C-SnO2–rGO and C-Fe2O3–rGO nanosheets. For the preparation of these anodes, tannic acid (TA), a kind of polyphenol extracted from plants, was used as a dispersing agent to introduce a metal precursor on the surface of rGO, and the metal precursor was subsequently converted to the corresponding metal oxide NPs by thermal annealing in a vacuum. During the thermal annealing process, TA was decomposed to form carbon materials, which acted as a buffering matrix to effectively suppress the aggregation and pulverization of the active NPs during the electrochemical performances. It is found that the as-prepared C-SnO2–rGO and C-Fe2O3–rGO nanosheets both exhibited high reversible capacity and rate capability. After 100 discharge/charge cycles, the C-SnO2–rGO nanosheet delivered the reversible capacity of 633.2 mA h g−1 at a current density of 200 mA g−1 with extremely low capacity fading (0.32 mA h g−1 per cycle), and it can deliver discharge capacities of 641.3, 526.5, 452.7, 408.1 and 379.5 mA h g−1 in the 10th cycle at current densities of 200, 400, 800, 1200 and 1600 mA g−1, respectively. Upon return to a cycling rate of 200 mA g−1, the C-SnO2–rGO can maintain a specific capacity of 607.0 mA h g−1 even after 35 cycles. As for the C-Fe2O3–rGO nanosheet, it can deliver 504.1 mA h g−1 at a current density of 500 mA g−1 after 100 cycles, and the corresponding discharge capacities in the 10th cycle at current densities of 1000, 1500 and 2000 mA g−1 are 365.9, 319.0 and 288.6 mA h g−1, respectively.
1 Introduction
There is great interest in developing lithium ion batteries (LIBs) because of the steadily growing demand for portable electronic devices. However, the graphite that is currently used as an anode in commercial LIBs has a low energy density (372 mA h g−1),1 which is insufficient to meet the demand for batteries with high energy density. Thus, exploring anode materials with high energy density to replace graphite is of key importance for the development of LIBs.Transition metal oxides such as tin oxides,2,3iron oxides4 and cobalt oxides5 have attracted tremendous attention as promising anode materials for the next generation of LIBs due to their high specific capacity and energy density. However, these anode materials would generally suffer large volume changes and stresses during lithium insertion/extraction processes, which cause serious cracking and pulverization of the electrode, and eventually lead to poor cycling performances. To solve these problems, one effective approach is to fabricate nanostructured materials modified with carbon. Downsizing particle size to the nanoscale can not only facilitate lithium ions to diffuse into the electrode matrix but also accommodate the physical strains associated with volume change.6–8 The use of carbon can effectively increase the electrode conductivity and prevent the detachment and agglomeration of pulverized active materials during cycling, thus leading to enhanced cycle life of the batteries.9–11 To date, various nanostructured metal oxide–carbon nanocomposites have been prepared. Among them, graphene-based meal oxide nanocomposites have been shown to be the most promising anode materials owing to the unique physiochemical properties of graphene.12
Graphene is a one-atom-thick two-dimensional carbon nanosheet (carbon atoms organized in a honeycomb structure), which has an excellent electrical conductivity (resistivity = 10−6 Ω cm) and high theoretical surface area (2600 cm2 g−1).13–15 Until now, a large number of nanostructured transition-metal oxide–graphene nanocomposites have been extensively investigated,16–21 such as SnO2–graphene, CoO–graphene, CuO–graphene, Fe2O3–grapheme and TiO2–graphene. Indeed, these hybrid nanocomposites exhibit much higher capacity and stability as compared with their bare counterparts. However, majority of these hybrid nanocomposites are prepared by direct host active nanomaterials on the surface of pristine graphene or graphene oxide (GO), followed by reduction of GO to reduced GO (rGO), where the deposited active nanoparticles will suffer irreversible aggregation or detachment during electrochemical cycles owing to the absence of stabilization by functional groups or buffering matrices among the active NPs. For example,22 Li et al. reported a facile and efficient method for the synthesis of SnO2–grapheme nanosheet composites, which is based on the reduction of graphene oxide (GO) by Sn2+ ions. Due to the absence of a buffering carbon matrix, the as-prepared materials suffered a slow capacity fading, from 541.3 to 377.3 mA h g−1 even at a current density of 100 mA g−1. Paek and his co-workers prepared SnO2–graphene nanoporous electrodes (SnO2–GNS).23 After 30 cycles, the charge capacity of SnO2–GNS remained at 570 mA h g−1, accounting only for 70% retention of the reversible capacity. To circumvent these obstacles, the possible solutions include chemical functionalization of graphene or buffering the active nanoparticles (NPs) by carbon materials. Considering that chemical functionalization of graphene may lead to the destruction of a long range π-conjugation, the latter alternative would be a more feasible strategy. Along this line, some important work has been pioneered for different anode materials systems (such as Fe3O4, Ge and Si),24–26 where the anode nanomaterials are confined within carbon shells, and then dispersed onto the flexible and conductive graphene.
In this article, we report a simple and general method for the synthesis of carbon buffered-transition-metal oxide NP–graphene hybrid nanosheets. Tannic acid (TA) is a typical polyphenol extracted from plants, which contains a number of pyrogallol hydroxyls that have strong chelating ability towards various transition metal ions with empty orbitals. Herein, TA was first chelated with transition-metal ions to ensure good dispersion of metal precursors on the surface of rGO. Subsequently, the metal precursors were converted to their corresponding metal oxide NPs via vacuum annealing, during which TA was simultaneously decomposed to carbon materials to buffer the metal oxide NPs. In this way, the metal oxide NPs can be firmly anchored onto the surface of the rGO without risking the destruction of the long range π-conjugation of the rGO. It can be expected that such well designed 2D hybrid nanocomposites can not only facilitate the rapid diffusion of lithium ions to the electrode but also effectively suppress the aggregation and pulverization of the active NPs due to the presence of buffering carbon materials. We chose to anchor carbon buffered-SnO2 NPs and -Fe2O3 NPs on the rGO for their high theoretical capacity. As expected, the as-prepared C-SnO2–rGO and C-Fe2O3–rGO nanosheets exhibited high reversible capacity, good cycling stability, and high rate capability. Considering the wide-ranging chelating ability of TA towards a variety of transition-metal ions, the approach demonstrated here can be extended to the fabrication of other carbon buffered metal oxide–graphene hybrid 2D nanocomposite anodes.
2 Experimental section
Preparation of rGO
GO was prepared from natural graphite by a modified Hummer's method.27,28 Subsequently, GO was dispersed in hydrazine–water solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v) to react at 100 °C for 24 h. The product was isolated by centrifugation and fully washed with deionized water and ethanol. The obtained rGO black powder was dried in a vacuum oven at 50 °C for 24 h.
:2, v/v) to react at 100 °C for 24 h. The product was isolated by centrifugation and fully washed with deionized water and ethanol. The obtained rGO black powder was dried in a vacuum oven at 50 °C for 24 h.
Preparation of C-SnO2–rGO nanosheets
8.0 mg of tannic acid (C76H52O46, Sigma-Aldrich) was dissolved in 1.0 mL of ethanol, and then 8.0 mg of rGO was suspended in the above solution, followed by ultrasonication for 4 h. After that, 1.0 mL of ethanol containing 104.2 mg of tin(IV) chloride hydrate (SnCl4·xH2O, Alfa Aesar) was dropwise added into the above suspension. The resultant mixture was stirred for 2 h, and then vacuum-dried at 50 °C. The collected samples were vacuum annealed at 400 °C for 1.0 h (or at 500 °C for 2.0 h). The obtained C-SnO2–rGO nanosheet was thoroughly washed with deionized water and ethanol, followed by vacuum drying at 50 °C.Preparation of C-Fe2O3–rGO
8.0 mg of rGO was mixed with 24 mL of ethanol, which contains 8.0 mg of tannic acid. After ultrasonication for 4 h, 1.0 mL of ethanol containing 32.4 mg of ferric chloride (FeCl3, Alfa Aesar) was dropwise added into the above suspension, followed by constant stirring for 12 h. The resultant materials were collected by centrifugation and thoroughly washed with deionized water and ethanol. After vacuum drying at 50 °C, the intermediate materials were further vacuum annealed at 500 °C for 2.0 h. The obtained C-Fe2O3–rGO nanosheet was thoroughly washed with deionized water and ethanol, and vacuum-dried at 50 °C.Electrochemical measurement
70 wt% of the C-SnO2–rGO nanosheet (or C-Fe2O3–rGO nanosheet), 20 wt% of conductive carbon black, and 10 wt% of polyvinylidene fluoride (PVDF) binder were fully mixed into N-methyl-2-pyrrolidinone (NMP). The resultant slurry was coated onto Cu foils, and vacuum-dried at 50 °C to completely remove the solvent. The electrochemical properties of the obtained working electrodes were measured using two-electrode CR2032 (3 V) coin-type cells with lithium foil serving as both counter and reference electrodes at ambient temperature. The electrolyte was 1 M LiPF6 in a 50:50 (w/w) mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC). Cell assembly was carried out in an argon-filled glove box with both moisture and oxygen contents below 1.0 ppm. Galvanostatic discharge/charge tests were performed using a NEWARE battery tester.
Other characterizations
Ultraviolet-visible (UV-vis) spectra analyses were conducted using a UV-vis spectrometer (Shimadzu UV-2501PC). Thermogravimetry analyses (TGA, Q500) were carried out in the temperature range 33–900 °C at a heating rate of 15 °C min−1 in N2. The X-ray diffraction patterns of the samples were recorded using a Bruker D8 Advance diffractometer using Cu Kα radiation. The morphology of the samples was characterized with a field emission scanning electron microscope (FESEM) system (JEOL, Model JSM-7600F) and a transmission electron microscope (TEM) system (JEOL, Model JEM-2010F) operating at 200 kV. Raman spectra were obtained with a WITec CRM200 confocal Raman microscopy system.3 Results and discussion
Due to the presence of pyrogallol hydroxyls in its molecule, TA is able to chelate with transition-metal ions with empty d-orbitals by donating a lone pair of electrons of the two adjacent phenolic oxygen molecules, which results in the formation of stable five-membered chelating rings.29–31 To verify the chelating reactions of TA toward Sn4+ and Fe3+, UV-visiblespectra of TA before and after the reaction with Sn4+ and Fe3+ were recorded. As shown in ESI S1,† pure TA shows an intensive characteristic adsorption peak at 278 nm. After mixing with Sn4+ in ethanol, the specific adsorption peak of tannic acid exhibits a red-shift from 278 nm to 303 nm, which confirms the chelating interactions between tannic acid and Sn4+. As for the chelating interactions between TA and Fe3+, a specific adsorption peak of TA–Fe3+ was observed at 600 nm after the mixing of TA with Fe3+.Moreover, the reaction is extremely fast, which is accompanied by an immediate color change from light yellow to dark blue. Actually, the stability constant of TA–Fe3+ is high up to 1030–1050,32 and the chelating reaction between TA and Fe3+ has already been utilized as an effective staining method for mammalian tissue.33Scheme 1 shows the proposed preparation mechanism of the C-SnO2–rGO and C-Fe2O3–rGO nanosheets. For the preparation of the C-SnO2–rGO nanosheet, TA plays the role as a dispersing agent as well as the source of the carbon buffering matrix. TA–Sn4+ precursors were first impregnated on rGO. During this process, TA acts as a dispersing agent to ensure good dispersion of TA–Sn4+ on the surface of rGO owing to the noncovalent π–π stacking interactions between the aromatic rings of TA and rGO.34 Subsequently, the Sn4+ precursors were converted to SnO2 NPs by thermal annealing at high temperature in a vacuum. According to TGA in a N2 flow (ESI, S2†), the weight loss of TA is higher than 60% when the temperature is beyond 400 °C. Hence, the TA molecules that are chelated with Sn4+ precursors will decompose to a carbon matrix when thermally annealed at high temperature in a vacuum, which can act as a buffering matrix to effectively suppress the aggregation of the SnO2 NPs. Moreover, the high temperature annealing can ensure firm anchoring of the carbon buffered-SnO2 NPs on the surface of rGO. In this way, a C-SnO2–rGO nanosheet with high stability was prepared. The C-Fe2O3–rGO nanosheet shares the same preparation mechanism with the C-SnO2–rGO nanosheet.
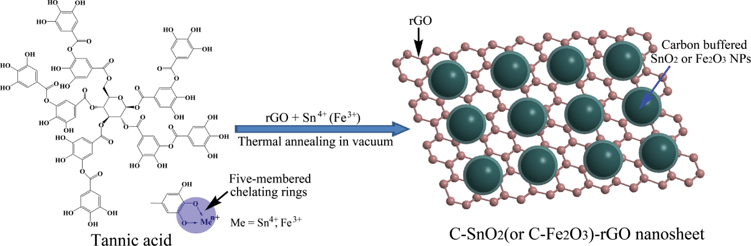 | ||
| Scheme 1 The proposed preparation mechanism of the C-SnO2–rGO and C-Fe2O3–rGO nanosheets. | ||
We first prepared the C-SnO2–rGO nanosheet by vacuum annealing the precursor at 500 °C, the corresponding XRD pattern of which, ESI S3,† exhibits intensive characteristic peaks of SnO2 (JCPDS no. 41-1445) at 25.5° (110), 33.8° (101), 51.6° (211) and 66° (301).22 In general, tin oxide with high crystallinity is more likely to suffer structural collapse during charge/discharge processes as compared with the amorphous counterpart.16 In order to decrease the crystallinity of the SnO2 NPs, the annealing temperature was reduced to 400 °C. As shown in ESI S3,† the obtained sample still exhibits the characteristic peaks of SnO2 but these peaks are considerably broadened, and the peak intensity is much weak in comparison to those samples at 500 °C, which therefore indicate that the as-prepared C-SnO2 NPs have lower crystallinity (higher content of amorphous phase) and smaller particle size. The XRD patterns of the C-Fe2O3–rGO nanosheet at 500 °C are shown in ESI S3,† which show the characteristic peaks of α-Fe2O3 with high intensity,35 thus suggesting that the C-buffered α-Fe2O3 NPs have been successfully anchored onto the rGO.
The scanning electron microscopy (SEM) images of the C-SnO2–rGO at 400 °C and C-Fe2O3–rGO at 500 °C are shown in Fig. 1. It is observed that the SnO2 NPs and Fe2O3 NPs are successfully anchored onto the surface of the rGO nanosheet. For the C-SnO2–rGO nanosheet, the C-SnO2 NPs exhibit a homogeneous dispersion on the surface of the rGO without significant aggregation, and the particle size of the C-SnO2 NPs is quite small, which is in the range of 5–15 nm. In general, small NPs have a high tendency to self-aggregate, but the carbon buffering matrix derived from the decomposition of TA effectively suppresses SnO2 NPs from aggregating. The C-Fe2O3–rGO nanosheet shows a similar morphology. In Fig. 1c, the morphology of rGO nanosheets is clearly observed, where small Fe2O3 NPs (10–20 nm) are anchored. At higher magnification, it is found that the Fe2O3 NPs anchored onto the surface of the rGO nanosheet have a closely packed arrangement but serious aggregation is rarely found. This should also be attributed to the formation of a carbon buffering matrix among the Fe2O3 NPs.
 | ||
| Fig. 1 Scanning electron microscopy (SEM) images of the C-SnO2–rGO (a and b) and the C-Fe2O3–rGO nanosheets (c and d) with different magnifications. | ||
The transmission electron microscopy (TEM) images of the C-SnO2–rGO nanosheet with different magnifications are given in Fig. 2a and b. Although a strong ultrasonic treatment was applied during preparation of the TEM samples, it is seen that the entire rGO nanosheet is still fully decorated by small SnO2 NPs. This indicates that the C-SnO2 NPs are firmly anchored onto the surface of the rGO nanosheet. In ESI S4,† C-SnO2–rGO at 500 °C shows similar TEM images with the sample at 400 °C but these SnO2 NPs have a more packed arrangement on the surface of the rGO, which is possibly caused by the higher thermal annealing temperature. The high resolution TEM (HR-TEM) image of the C-Fe2O3–rGO at 500 °C shows clear lattices, which suggest that SnO2 NPs with high crystallinity are formed on the rGO. These results are consistent with the XRD analyses.
 | ||
| Fig. 2 Transmission electron microscopy (TEM) images of the C-SnO2–rGO at 400 °C (a and b) and the C-Fe2O3–rGO at 500 °C (c and d) with different magnifications. | ||
In Fig. 2c, the C-Fe2O3–rGO nanosheet shows similar TEM images with C-SnO2–rGO at 500 °C. The Fe2O3 NPs still exhibit a packed arrangement on the surface of the rGO. In Fig. 2d, the HR-TEM image of the Fe2O3–rGO nanosheet shows clear lattices, which confirms the formation of Fe2O3 NPs with high crystallinity. The corresponding Raman spectra of C-SnO2–rGO at 400 °C and C-Fe2O3–rGO at 500 °C are shown in Fig. 3. Compared with rGO, C-SnO2–rGO and C-Fe2O3–rGO nanosheets both contain D and G bands of the rGO,36 which suggest that no serious damage was incurred to the extended π-conjugation of rGO when preparing the nanocomposites, and this will be beneficial for achieving good anode performances.
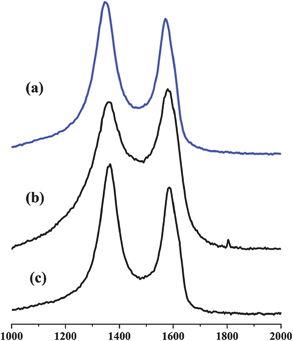 | ||
| Fig. 3 Raman spectra of the rGO (a), C-SnO2–rGO nanosheet at 400 °C (b) and C-Fe2O3–rGO at 500 °C (c). | ||
The Li storage properties of the C-SnO2–rGO and C-Fe2O3–rGO nanosheets were examined using coin-type half-cells with a Li counter electrode and reference electrode. Fig. 4a and b show the galvanostatic discharge and charge voltage profiles generated by the C-SnO2–rGO prepared at 500 °C and 400 °C in the 1st, 2nd and 100th cycles at a current density of 200 mA g−1 in the voltage window of 0.005–3.0 V. The discharge and charge capacities of the C-SnO2–rGO at 500 °C are 1085.5 and 615.9 mA h g−1 in the 1st cycle, respectively, which indicate that the corresponding coulombic efficiency is 56.74%, and it can deliver a reversible discharge capacity of 621.3 mA h g−1 in the 2nd cycle. As for the C-SnO2–rGO at 400 °C, its discharge and charge capacities are 997.6 and 656.9 mA h g−1 in the 1st cycle, respectively, with an initial coulombic efficiency of 65.85%, and a reversible discharge capacity of 664.6 mA h g−1 is delivered in the 2nd cycle. According to the literature,37,38 Sn-based anode materials usually exhibit a large initial irreversible loss, which is often related to the formation of a solid electrolyte interface (SEI) layer on the anode surface and the decomposition of the electrolyte. In the 100th cycle, the discharge voltage profile of the C-SnO2–rGO at 500 °C is dropped more rapidly compared to the 1st cycle. In comparison, the C-SnO2–rGO at 400 °C delivered a discharge capacity of 633.2 in the 100th cycle, and the corresponding discharge voltage profile almost overlaps with that of the 1st cycle.
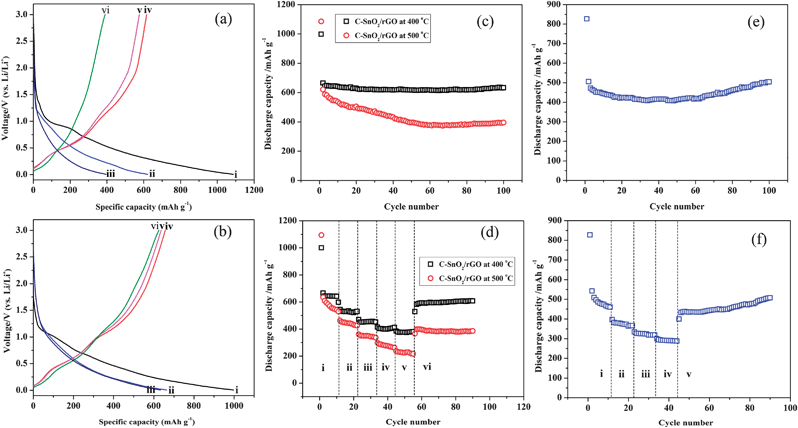 | ||
| Fig. 4 Discharge/charge profiles of the C-SnO2–rGO nanosheet at 500 °C (a) and 400 °C (b) at a current density of 200 mA g−1 in a voltage window of 0.005–3.00 V: the discharge curves in the (i) 1st, (ii) 2nd and (iii) 100th cycles, and the charge curves in the (iv) 1st, (v) 2nd and (vi) 100th cycles. Discharge capacities against cycle numbers for C-SnO2–rGO nanosheets at a current density of 200 mA g−1 in the voltage window of 0.005–3.00 V (c). The rate capabilities of the C-SnO2–rGO nanosheets at different current densities (d): (i) 200, (ii) 400, (iii) 800, (iv) 1200, (v) 1600 and (vi) 200 mA g−1. Discharge capacities against cycle numbers for the C-Fe2O3–rGO nanosheet at a current density of 500 mA g−1 in the voltage window of 0.005–3.00 V (e). The rate capability of the C-Fe2O3–rGO nanosheet at different current densities (f): (i) 500, (ii) 1000, (iii) 1500, (iv) 2000 and (v) 500 mA g−1. | ||
Fig. 4c presents the cyclic performance of the C-SnO2–rGO at a current density of 200 mA g−1 in the voltage window of 0.005–3.0 V. The C-SnO2–rGO at 500 °C exhibits a slow but constant reversible capacity fading along with the cycling, which delivers a discharge capacity of 395.5 mA g−1 in the 100th cycle, with a capacity fading of 2.28 mA h g−1 per cycle on average. The C-SnO2–rGO prepared at 400 °C shows superior cycling stability compared to the sample prepared at 500 °C. This nanocomposite worked pretty stably with the capacity of 633.2 mA h g−1 in the 100th cycle, which retains almost 95.28% of the 2nd cycle capacity, with a negligible capacity fading (0.32 mA h g−1 per cycle on average), thus exhibiting excellent cycle performance. The cycling stability of the C-SnO2–rGO at 400 °C was also obtained in the voltage window of 0.02–1.5 V at a current density of 400 mA g−1 (ESI S5†). In this narrow voltage window, the anode materials still deliver a high reversible capacity of 400.3 mA h g−1 in the 40th cycle, suggesting its potential application in full cell batteries.
The rate capability is an important parameter for lithium ion batteries, and thus, the electrochemical performances of the C-SnO2–rGO at 500 °C and 400 °C are measured at current densities of 200, 400, 800, 1200, 1600 and 200 mA g−1, respectively. As shown in Fig. 4d, the C-SnO2–rGO at 500 °C delivers discharge capacities of 540.1, 428.0, 339.8, 264.1 and 218.1 mA h g−1 in the 10th cycle at current densities of 200, 400, 800, 1200 and 1600 mA g−1, respectively. When the current density is decreased from 1600 to 200 mA g−1, the discharge capacity could increase back to ∼400 mA h g−1. As for the C-SnO2–rGO at 400 °C, it maintains a steady discharge capacity at every current density, and the corresponding discharge capacities in the 10th cycle are 641.3, 526.5, 452.7, 408.1 and 379.5 mA h g−1, respectively. Upon return to a cycling rate of 200 mA g−1, the specific capacity of the anode is increased to 581.7 mA h g−1, and it can maintain a stable discharge capacity of 607.0 mA g−1 even after 35 cycles. As a consequence, the C-SnO2–rGO at 400 °C is promising to be stably operated at high discharge rate. Obviously, the C-SnO2–rGO nanosheet with low crystallinity prepared at 400 °C exhibits better cycling stability and rate capability than the sample with higher degree of crystallinity prepared at 500 °C. According to the literature,39,40 it is likely that the C-SnO2–rGO with low degree of crystallinity was less dense than its counterpart with higher crystallinity, which is associated with a homogenous volume expansion and contraction, thus eliminating the existence of two phase regions and improving the cycling stability. Consequently, the excellent electrochemical performances of the C-SnO2–rGO nanosheet at 400 °C should be attributed to three factors: (1) the presence of a carbon buffering matrix considerably suppresses the fraction of SnO2 NPs to lose their electrical contact with rGO. (2) The rGO nanosheet provides good electronic conductivity and buffering effect to some extent. (3) The SnO2 NPs with low crystallinity accommodate the substantial volume changes to some extent.
The cycling stability and rate capability of the C-Fe2O3–rGO nanosheet are shown in Fig. 4e and f. In Fig. 4e, the C-Fe2O3–rGO shows good stability at a current density of 500 mA g−1. In the 100th cycle, the discharge capacity of the anode is 504.1 mA h g−1, which accounts for 99.62% of the capacity in the 2nd cycle, thus exhibiting an excellent cycling stability. As seen in Fig. 4f, the C-Fe2O3–rGO delivers discharge capacities of 463.0, 365.9, 319.0 and 288.6 mA h g−1 in the 10th cycle at current densities of 500, 1000, 1500 and 2000 mA g−1, respectively, and the cycling performance is very stable at each discharge rate. The discharge capacity of the C-Fe2O3–rGO can maintain at 508 mA h g−1 in the 45th cycle when the current density is decreased from 2000 to 500 mA g−1, which also exhibited an excellent rate capability.
4 Conclusion
We have developed a simple and general method for the synthesis of carbon buffered-transition metal oxide NP–graphene hybrid 2D nanosheets, where tannic acid is used as a dispersing agent as well as the source of buffering carbon. Due to the presence of a carbon buffering matrix, the SnO2 NPs and Fe2O3 NPs can be stably anchored onto the surface of rGO without obvious aggregation. Moreover, the carbon buffering matrix can effectively accommodate the substantial volume changes of the active NPs, and accordingly, the as-prepared C-SnO2–rGO and C-Fe2O3–rGO nanosheets both exhibited excellent cycling stability and rate capability. It should be noted that this strategy could be extended to the synthesis of other hybrid 2D nanosheets due to the strong chelating ability of TA towards a variety of transition-metal ions.Acknowledgements
This work was supported by the Singapore Ministry of Education (MOE2010-T2-1-017), A*STAR SERC grant 1021700144, NRF2009EWT-CERP001-026 (Singapore), Singapore National Research Foundation under the CREATE program: EMobility in Megacities and Singapore MPA 23/04.15.03 RDP 020/10/113 grant.Notes and references
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395 CrossRef CAS.
- C. A. Bonino, L. W. Ji, Z. Lin, Ozan, Toprakci, X. W. Zhang and S. A. Khan, ACS Appl. Mater. Interfaces, 2011, 3, 2534 CAS.
- Y. Yu, C.-H. Chen and Y. Shi, Adv. Mater., 2007, 19, 993 CrossRef CAS.
- M. V. Reddy, T. Yu, C.-H. Sow, Z. X. Shen, C. T. Lim, G. V. Subba Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2792 CrossRef CAS.
- Y. M. Sun, X. L. Hu, W. Luo and Y. H. Huang, J. Phys. Chem. C, 2012, 116, 20794 CAS.
- W. J. Zhang, J. Power Sources, 2011, 196, 13 CrossRef CAS.
- X. J. Zhu, Y. W. Zhu, S. Murali, M. D. Stollers and R. S. Ruoff, ACS Nano, 2011, 5, 3333 CrossRef CAS.
- Y. G. Guo, J. S. Hu and L. J. Wan, Adv. Mater., 2008, 20, 2878 CrossRef CAS.
- W. M. Zhang, J. S. Hu, Y. G. Guo, S. F. Zheng, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20, 1160 CrossRef CAS.
- H. Q. Li and H. S. Zhou, Chem. Commun., 2012, 48, 1201 RSC.
- Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P. Chen, G. M. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187 CrossRef CAS.
- X. Huang, Z. Y. Zeng, Z. X. Fan, J. Q. Liu and H. Zhang, Adv. Mater., 2012, 24, 5979 CrossRef CAS.
- H. X. Chang and H. K. Wu, Adv. Funct. Mater., 2012, 23, 1984 CrossRef.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef CAS.
- D. Chen, L. Tang and J. Li, Chem. Soc. Rev., 2010, 39, 3157 RSC.
- X. F. Li, X. B. Meng, J. Liu, D. S. Geng, Y. Zhang, M. N. Banis, Y. L. Li, J. L. Yang, R. Y. Li, X. L. Sun, M. Cai and M. W. Verbrugge, Adv. Funct. Mater., 2012, 22, 1647 CrossRef CAS.
- F. D. Wu and Y. Wang, J. Mater. Chem., 2011, 21, 6636 RSC.
- J. Y. Xiang, J. P. Tu, L. Zhang, Y. Zhou, X. L. Wang and S. J. Shi, J. Power Sources, 2010, 195, 313 CrossRef CAS.
- W. W. Zhou, J. X. Zhu, C. W. Cheng, J. P. Liu, H. P. Yang, C. X. Cong, C. Guan, X. T. Jia, H. J. Fan, Q. Y. Yan, C. M. Li and T. Yu, Energy Environ. Sci., 2011, 4, 4954 CAS.
- D. H. Wang, R. Kou, D. Choi, Z. G. Yang, Z. M. Nie, J. Li, L. V. Saraf, J. G. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587 CrossRef CAS.
- D. H. Wang, D. Choi, J. Li, Z. G. Yang, Z. M. Nie, R. Kou, D. H. Hu, C. M. Wang, L. V. Saraf, J. G. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907 CrossRef CAS.
- Y. M. Li, X. J. Lv, J. Lu and J. H. Li, J. Phys. Chem. C, 2010, 114, 21770 CAS.
- S. M. Paek, E. J. Yoo and I. Honma, Nano Lett., 2009, 9, 72 CrossRef CAS.
- B. J. Li, H. Q. Cao, J. Shao and M. Z. Qu, Chem. Commun., 2011, 47, 10374 RSC.
- D. J. Xue, S. Xin, Y. Yan, K. C. Jiang, Y. X. Yin, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 2512 CrossRef CAS.
- B. Wang, X. L. Li, X. F. Zhang, B. Luo, M. H. Jin, M. H. Liang, S. A. Dayeh, S. T. Picraux and L. J. Zhi, ACS Nano, 2013, 7, 1437 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- Y. X. Xu, H. Bai, G. W. Lu, C. Li and G. Q. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS.
- S. Quideau, D. Deffieux, C. D. Deffieux and L. Pouysegu, Angew. Chem., Int. Ed., 2011, 50, 586 CrossRef CAS.
- G. Tondi, C. W. Oo, A. Pizzi, A. Trosa and M. F. Thevenon, Ind. Crops Prod., 2009, 29, 336 CrossRef CAS.
- X. M. Zhan and X. Zhao, Water Res., 2003, 37, 3905 CrossRef CAS.
- I. A. T. Khan and Z. T. Maqsood, Sci. Iran., 2007, 14, 106 CAS.
- T. D. Pizzolato, Bull. Torrey Bot. Club, 1977, 104, 277 CrossRef.
- P. Petrov, F. Stassin, C. Pagnoulle and R. Jerome, Chem. Commun., 2003, 2904 RSC.
- C. T. Cherian, J. Sundaramurthy, M. Kalaivani, P. Ragupathy, P. Suresh Kumar, V. Thavasi, M. V. Reddy, C. H. Sow, S. G. Mhaisalkar, S. Ramakrishna and B. V. R. Chowdari, J. Mater. Chem., 2012, 22, 12198 RSC.
- G. K. Ramesha and S. Sampath, J. Phys. Chem. C, 2009, 113, 7985 CAS.
- W. J. Cui, F. Li, H. J. Liu, C. X. Wang and Y. Y. Xia, J. Mater. Chem., 2009, 19, 7202 RSC.
- X. X. Ji, X. T. Huang, J. P. Liu, J. Jiang, X. Li, R. M. Ding, Y. Y. Hu, F. Wu and Q. Li, Nanoscale Res. Lett., 2010, 5, 649 CrossRef CAS.
- J. P. Maranchi, A. F. Hepp and P. N. Kumta, Electrochem. Solid-State Lett., 2003, 6, A198 CrossRef CAS.
- J. T. Yin, M. Wada, K. Yamamoto, Y. Kitano, S. Tanase and T. Sakai, J. Electrochem. Soc., 2006, 153, A472 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: UV-visspectra, TGA and TEM analysis. See DOI: 10.1039/c3ta10986k |
| This journal is © The Royal Society of Chemistry 2013 |